Author byline as per print journal: Jon SB de Vlieger, PhD; Professor Stefan Mühlebach, PhD; Vinod P Shah, PhD; Scott E McNeil, PhD; Professor Gerrit Borchard, PharmD, PhD; Vera Weinstein, PhD; Beat Flühmann, PhD; Sesha Neervannan, PhD; Professor Daan JA Crommelin, PhD
|
Abstract: |
Submitted: 16 September 2015; Revised: 15 October 2015; Accepted: 19 October 2015; Published online first: 2 November 2015
The concept of non-biological complex drug (NBCD) products has been presented and discussed on several occasions in the GaBI Journal [1–4]. The growing interest in this topic in academic, industrial and regulatory circles led to the establishment of an Editorial Section on Non-Biological Complex Drugs in both the GaBI Journal and GaBI Online starting in the third quarter of 2015.
In 2011, the first publication on NBCDs appeared [5] as a result of a workshop held in Leiden, The Netherlands, in 2009. For the first time this class of drug products was identified and recognized. These products are more complex than small, low molecular drugs and as complex or even more complex than biologicals; sharing many of the characteristics of the latter category but not being derived from living sources. As a consequence, the authors argued that for the equivalence testing of NBCD follow-on products a regu latory pathway should be developed that was similar to the pathways developed by the European Medicines Agency (EMA) and later the US Food and Drug Administration (FDA) for approving biosimilars. This proposal was backed up by evidence provided by a (still growing) number of published studies on NBCD follow-on versions that were authorized using a ‘standard’ but inadequate generic assessment protocol.
A working group hosted by the Dutch Top Institute (TI) Pharma in Leiden was set up to raise awareness of the challenges NBCD products present worldwide, to stimulate the publication of scientific reports and discussions and to provide a rigorous, sciencebased, regulatory policy for NBCD products [6]. These efforts led to a number of publications and a book exclusively dedicated to the NBCD concept and its regulation. These publications provide a definition for NBCD products, discuss different classes of NBCDs, propose an overarching regulatory philosophy for evaluating NBCD follow-on versions and, finally, outline what issues are still unresolved [7–9].
This paper to the GaBI Journal further explains:
A definition of NBCD products that was published earlier by the NBCD working group reads: ‘A Non-Biological Complex Drug is a medicinal product, not being a biological medicine, where the active substance is not a homo-molecular structure, but consists of different (closely related and often nanoparticulate) structures that can’t be isolated and fully quantitated, characterized and/or described by stateof-the-art (physico)chemical analytical means and where the clinical meaning of the observed differences is not known. The composition, quality and in vivo performance of NBCDs are highly dependent on manufacturing processes of both the active ingredient as well as in most cases the formulation’ [7].
Present product ‘families’ and beyond
At present the following NBCD product groups or ‘families’ have been identified and discussed in the literature: liposomes, polymeric micelles, glatiramoids, iron-carbohydrate complexes, albuminanticancer drug nanoparticles and nanocrystals. The rapidly growing group of nanomedicines will add many NBCDs to this list [9–10]. Interestingly, there are also medicinal products such as the low molecular weight heparins (LMWHs) showing similar complexity but falling under different regulatory policies, i.e. by EMA LMWHs are seen as biologicals and by FDA as non-biologicals.
Over the last few years a number of studies on these NBCD product families have been published. This list is growing and expanding beyond only parenteral drugs. Recently, the present science base, including the: (1) chemistry and structure; (2) manufacturing; (3) (physico)chemical characterization; (4) pharmacology; and (5) regulatory status, of these product groups was reviewed in a book [9]. The availability of these data in the public domain should contribute to sciencebased discussions and could serve as a model to be followed for consideration of other NBCD product families. In addition, questions regarding interchangeability and substitutability of NBCD follow-on versions have important implications for the handling of such medicinal products by healthcare professionals. Requests are being made by these healthcare professionals for education on the topic of NBCDs and for further, tailor-made, reliable information for patients.
Other candidate NBCD product families that are waiting to be identified include emulsions (parenteral or ocular), dry powder inhalers and oral bioactive polymers such as complex phosphate binders.
The regulatory landscape
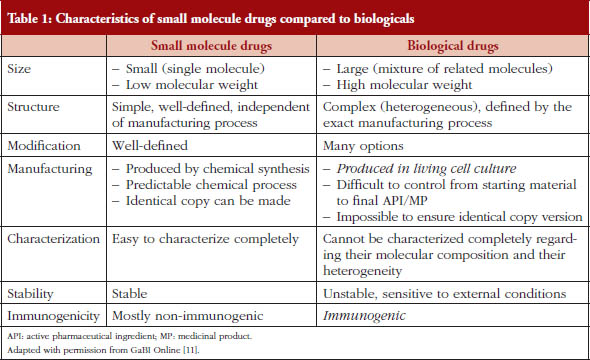
In earlier publications in GaBI Online the differences between the characteristics of small, low molecular weight molecules and biologicals were listed, see Table 1 [11]. If the items in the ‘biological drugs’ column (in italics) that relate to the biological source of the product and immunogenicity are removed, what remains demonstrates that there is a striking resemblance between the characteristics of biologicals and NBCD products.
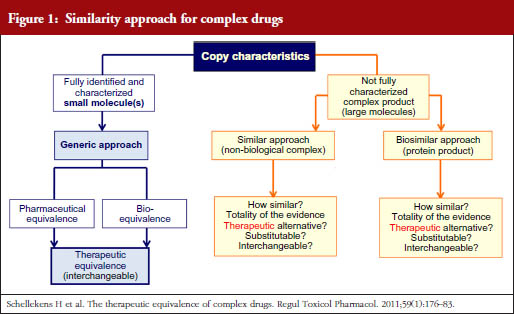
Because of this similarity in product characteristics, the NBCD Working Group has proposed on several occasions that regulators should follow the same regulatory pathway for NBCD follow-on products as for biosimilars. This proposal is schematically shown in Figure 1 where ‘Totality of evidence’ is the key phrase when assessing therapeutic equivalence of NBCD innovator and follow-on products.
Neither FDA nor EMA uses special NBCD regulatory schemes. These agencies use existing pathways for the introduction of innovative and follow-on NBCD products. FDA uses the 505(b)(1)/505(b)(2) and 505(j) pathways for innovator products and follow-on versions, respectively. However, both FDA and EMA are paying increasing attention to regulatory issues related to NBCD families [12–13].
Generally speaking, there are two clearly distinct regulatory documents for these NBCD product families. On the one hand, FDA and/or EMA published ‘draft guidance’ and/or ‘reflection papers’ on new products such as liposomes [14], polymeric micelles [15], and surface coatings [16]. On the other hand, both agencies issued documents on the development of follow-on versions of NBCD products such as EMA documents on iron-based nano-colloidal products [17–19] and on existing products [20], and FDA on iron complexes [21], ciclosporin emulsions [22] and on liposome follow-on products [23–24]. Interestingly, FDA has awarded funding to characterize and clinically compare originator and follow-on versions after approving these products [25–26]. It is clear that the present arsenal of regulatory documents from these agencies will be expanded in the years to come. Hopefully, it is not too late to reach global agreement through the World Health Organization (WHO) or perhaps the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) initiatives. On the pharmacopoeial side, interest is growing in dealing with NBCD families. For example, a European Directorate for the Quality of Medicines and HealthCare (EDQM) working group is developing a monograph on ‘iron sucrose concentrated solution’ as an example for non-biological complexes and the United States Pharmacopeia (USP) is currently evaluating similar actions [27]. The British Pharmacopoeia (BP) has published a new monograph on iron sucrose injections, which is basically in line with the USP existing monograph.
One interesting feature of the European legal landscape is that certain NBCD product families are not approved through the centralized procedure (EMA). Instead, approval follows either the purely national procedures or the mutual recognition procedure under the aegis of national competent authorities. This is true for iron sucrose products and for oral bioactive polymer phosphate binders [28]. Considering the complexity of these products and the problems encountered in certain countries with generic versions of NBCD products (see below), approval thro ugh the centralized procedure and the CHMP team would be preferred.
For all NBCD product families where follow-on versions are on the market, a growing number of studies have become available in the public domain demonstrating examples of follow-on products that were approved by (national) competent authorities that differed structurally and/or in clinical practice from the originator products [9]. Often these differences were clinically relevant. Examples include publications by Rottembourg et al. [29]; Martin-Malo et al. [30]; Stein et al. [31]; Lee et al. [32] and Agüera et al. [33] for iron sucrose similars; and Weinstein et al. [34] and Towfic et al. [35] for glatiramer acetate follow-on products. These (clinical) examples raise questions to the regulatory science community in the countries mentioned in these publications about how appropriate the current systems are in ensuring equivalence in NBCD product quality, efficacy and safety. Is the current approach rigorous enough?
These examples provide lessons that should be communicated throughout the scientific community as well as to medical/pharmaceutical practitioners. More over, in the years to come, study of these examples can also help competent authorities to establish appropriate, science-based approval procedures for these complex drug products.
Expanding: The number of NBCD product families will continue to grow. It is time to pay attention to these (new) families and discuss their specific characteristics and their implications for the regulatory process at an early stage. Much information is often already available but there is a need to go through the archives and analyse these data so that scientific deficiencies are brought to the forefront.
Outstanding issues: for a list of outstanding issues concerning NBCD products one can refer to the list drawn up for biologicals. For example, labelling, comparability and attribute drift, NBCD-questionables (cf. bio-questionables [36]), extrapolation [37], interchangeability, substitution, and last but not least: a single global approach (WHO in the lead?) [38].
Facts please: For a fact-based debate on NBCD products we need to stimulate publications in the public domain. This will strengthen the science base for decisionmaking. Transparency of the regulatory process is another essential element for such a discussion. We hope that all parties (academic, industry and regulatory) involved in this debate will continue, and even step up their efforts to provide this science base.
Goal: We feel that the GaBI Journal and the GaBI Online platform offer excellent opportunities to stimulate awareness around the critical issues related to both new, innovative NBCD products and the introduction of follow-on versions. The growing science base for NBCD product legislation, e.g. in Europe and the US, and hopefully in other countries in the future, needs a non-biased publication outlet. With the GaBI publication platform we have the ambition to become the central and preferred publication hotspot for this complex topic.
Jon SB de Vlieger, PhD
Dutch Top Institute Pharma, PO Box 142, NL-2300 AC Leiden, The Netherlands
Professor Stefan Mühlebach, PhD
Vifor Pharma Ltd, 61 Flughofstrasse, PO Box, CH-8152 Glattbrugg, Switzerland; Department of Pharmaceutical Sciences, Pharmacenter, University of Basel, 50 Klingelbergstrasse, CH-4056 Basel, Switzerland
Vinod P Shah, PhD
NBCD Steering Committee member, Pharmaceutical Consultant, 11309 Dunleith Place, North Potomac, MD 20878, USA
Scott E McNeil, PhD
Director, Nanotechnology Characterization Laboratory, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research Inc, PO Box B, Frederick, MD 21702-1201, USA
Professor Gerrit Borchard, PharmD, PhD
Professor, Biopharmaceutical Sciences, School of Pharmaceutical Sciences, University of Geneva, University of Lausanne 30, Quai Ernest-Ansermet, CH-1211 Geneva 4, Switzerland
Vera Weinstein, PhD
Teva Pharmaceutical Industries Ltd, Discovery and Product Development, Global Research and Development, Netanya, Israel
Beat Flühmann, PhD
Vifor Fresenius Medical Care Renal Pharma Ltd, 37 Rechenstrasse, PO Box, CH-9001 St Gallen, Switzerland
Sesha Neervannan, PhD
Senior Vice President, Pharmaceutical Development Brands R & D, Allergan Plc, RD2-3A, 2525 Dupont Drive, Irvine, CA 92612, USA
Emeritus Professor Daan JA Crommelin, PhD
Department of Pharmaceutical Sciences, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, The Netherlands
Competing interest: All authors are members of the steering committee of the Non-Biological Complex Drug (NBCD) Working Group, hosted at the Dutch Top Institute Pharma (TI Pharma), Leiden, The Netherlands (http://www.tipharma.com/NBCD).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Mühlebach S, et al. The authorization of nonbiological complex drugs (NBCDs) follow-on versions: specific regulatory and interchangeability rules ahead? Generics and Biosimilars Initiative Journal (GaBI) Journal. 2013;2(4):204-7. doi:10.5639/gabij.2013.0204.054
2. Borchard G. Complex molecules – current developments. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):54-5. doi:10.5639/gabij.2014.0302.016
3. Walson PD, Mühlebach S, Flühmann B. First Asia-Pacific educational workshop on non-biological complex drugs (NBCDs), Kuala Lumpur, Malaysia, 8 October 2013. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):30-3. doi:10.5639/gabij.2014.0301.010
4. Nicholas MJ. Clinical development, immunogenicity, and interchangeability of follow-on complex drugs. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):71-8. doi:10.5639/gabij.2014.0302.020
5. Schellekens H, Klinger E, Mühlenbach S, Brin JF, Storm G, Crommelin DJ. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
6. TI Pharma. Non Biological Complex Drugs Working Group [homepage on the Internet]. 2015 [cited 2015 Oct 15]. Available from: http://www.tipharma.com/nbcd
7. Crommelin DJ, de Vlieger JS, Weinstein V, Mühlebach S, Shah VP, Schellekens H. Different pharmaceutical products need similar terminology. AAPS J. 2014;16(1):11-4.
8. Schellekens H, et al. How to regulate non-biological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2014;16(1):15-21.
9. Crommelin DJA, de Vlieger, JSB. Non-biological complex drugs. The science and the regulatory landscape. Advances in the Pharmaceutical Sciences series. NY: AAPS/Springer; 2015.
10. Mühlebach S, Borchard G, Yildiz S. Regulatory challenges and approaches to characterize nanomedicines and their follow-on similars. Nanomedicine (Lond) 2015; 10(4):659-74.
11. GaBI Online – Generics and Biosimilars Initiative. Small molecule versus biological drugs [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Oct 15]. Available from: www.gabionline.net/Biosimilars/Research/Small-molecule-versus-biological-drugs
12. Ehmann F, et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine (Lond). 2013;8(5): 849-56.
13. Spicher K, et al. Differences in tissue distribution of iron from various clinically used intravenous iron complexes in fetal avian heart and liver. Regul Toxicol Pharmacol. 2015;73(1):65-72.
14. U.S. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Guidance for industry. Liposome drug products chemistry, manufacturing, and controls; human pharmacokinetics and bioavailability; and labeling documentation. Draft guidance. 2002 [homepage on the Internet]. 2002 Jul 29 [cited 2015 Oct 15]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070570.pdf
15. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products. 17 January 2013 [homepage on the Internet]. 2013 Jan 23 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/02/WC500138390.pdf
16. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Reflection paper on surface coatings: general issues for consideration regarding parenteral administration of coated nanomedicine products. 22 May 2013 [homepage on the Internet]. 2013 Jul 19 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/08/WC500147874.pdf
17. European Medicines Agency. Committee for Human Medicinal Products (CHMP). Reflection paper on non-clinical studies for generic nanoparticle iron medicinal product applications. 17 March 2011 [homepage on the Internet]. 2011 Apr 1 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/04/WC500105048.pdf
18. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Overview of comments received on reflection paper on the data requirements for intravenous iron-based nano-colloidal products developed with reference to an innovator medicinal product. 26 March 2015 [homepage on the Internet]. 2015 Mar 26 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/ document_library/ Overview_of_comments/2015/03/WC500184921.pdf
19. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Reflection paper on the data requirements for intravenous iron-based nano-colloidal products developed with reference to an innovator medicinal product. 26 March 2015 [homepage on the Internet]. 2015 Mar 26 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/03/WC500184922.pdf
20. European Medicines Agency. Assessment report for: iron containing intravenous (IV) medicinal products. 13 September 2013 [homepage on the Internet]. 2013 Sep 27 [cited 2015 Oct 15]. Available from: www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/IV_iron_31/WC500150771.pdf
21. U.S. Food and Drug Administration. Draft guidance on iron sucrose [homepage on the Internet]. 2013 Nov 1 [cited 2015 Oct 15]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm297630.pdf
22. U.S. Food and Drug Administration. Draft guidance of cyclosporine [homepage on the Internet]. 2013 Jun 20 [cited 2015 Oct 15]. Available from http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358114.pdf
23. U.S. Food and Drug Administration. Draft guidance on doxorubicin hydrochloride [homepage on the Internet]. 2014 Dec 12 [cited 2015 Oct 15]. Available from http://www.fda.gov/downloads/Drugs/…/Guidances/UCM199635.pdf
24. European Medicines Agency. Committee for Human Medicinal Products (CHMP). Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal products. 21 February 2013 [homepage on the Internet]. 2013 Mar 13 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf
25. Federal Business Opportunities. Therapeutic equivalence of generic iron complex product [homepage on the Internet]. [cited 2015 Oct 15]. Available from: https://www.fbo.gov/index?s=opportunity&mode=form&id=592788989854da145c8e7b6d103c898d&tab=core&tabmode=list&
26. National Institutes of Health. Michel SL, Kane MA, Polli JE. Evaluation of iron species in healthy subjects treated with generic and reference sodium ferric gluconate [homepage on the Internet]. [cited 2015 Oct 15]. Available from: http://grantome.com/grant/NIH/U01-FD005266-01
27. U.S. Pharmacopeial Convention. Monographs in need of modernization (Last updated 26 November 2014) [database on the Internet]. [cited 2015 Oct 15]. Available from: http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/2014-11-26_monographs_needing_modernization.xlsx
28. CBG-MEB. [Openbaar verslag 797e Collegevergadering verslag]. Dutch [homepage on the Internet]. [cited 2015 Oct 15]. Available from: http://www.cbgmeb.nl/documenten/vergaderstukken/2014/01/09/collegevergadering-797-verslag
29. Rottembourg J, Kadri A, Leonard E, Dansaert A, Lafuma A. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10):3262-7.
30. Martin-Malo A, Merino A, Carracedo J, et al. Effects of intravenous iron on mononuclear cells during the haemodialysis session. Nephrol Dial Transplant. 2012;27(6):2465-71.
31. Stein J, Dignass A, Chow KU. Clinical case reports raise doubts about the therapeutic equivalence of an iron sucrose similar preparation compared with iron sucrose originator. Curr Med Res Opin. 2012;28(2):241-3.
32. Lee ES, Park BR, Kim JS, Choi GY, Lee JJ, Lee IS. Comparison of adverse event profile of intravenous iron sucrose and iron sucrose similar in postpartum and gynecologic operative patients. Curr Med Res Opin. 2013;29(2):141-7.
33. Agüera ML, et al. Efficiency of original versus generic intravenous iron formulations in patients on haemodialysis. Plos One. 2015;10(8):e0135967.
34. Weinstein V, Nicholas JM, Schwartz R, Grossman I, Zeskind B. Glatiramoids. In: Crommelin DJA, de Vlieger J, editors. Non-biological complex drugs. Advances in the pharmaceutical sciences series. NY: AAPS/Springer; 2015: p. 107-49.
35. Towfic F, et al. Comparing the biological impact of glatiramer acetate with the biological impact of a generic. PLoS One. 2014;9(1): e83757.
36. Halim LA, et al. How bio-questionable are the different recombinant human erythropoietin copy products in Thailand? Pharm Res. 2014;31(5):1210-18.
37. Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191-6.
38. Crommelin DJA, et al. The similarity question for biologicals and non-biological complex drugs. Eur J Pharm Sci. 2015;76:10-7.
|
Author for correspondence: Jon SB de Vlieger, PhD; Dutch Top Institute Pharma, PO Box 142, NL-2300 AC Leiden, The Netherlands |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/non-biological-complex-drugs-nbcds-and-their-follow-on-versions-time-for-an-editorial-section.html
Author byline as per print journal: Jon SB de Vlieger1, PhD; Professor Daan JA Crommelin2, PhD; Beat Flühmann3, PhD; Professor Imre Klebovich4, PharmD, PhD, DSc; Professor Stefan Mühlebach3,5, PhD; Vinod P Shah6, PhD
|
Abstract: |
Submitted: 28 August 2019; Revised: 9 September 2019; Accepted: 10 September 2019; Published online first: 23 September 2019
The 3rd International Symposium on Scientific and Regulatory Advances in Biological and Non-Biological Complex Drugs: A to Z in Bioequivalence was organized in Budapest, Hungary on 12–14 November 2018 by the Department of Pharmaceutics of Semmelweis University with other Hungarian science organizations under the auspices of the International Pharmaceutical Federation (FIP), European Federation of Pharmaceutical Sciences (EUFEPS) and the NBCD Working Group hosted by Lygature [1]. The aim of the conference was to provide an update on the progress in the field by experts and to discuss recent developments in the regulatory status of nanomedicinal drug products in moderated panels with stakeholders. A widespread international group of experts from originator and generic drug companies, regulatory authorities, academia and contract research organizations participated in the meeting and discussion sessions. The conference introduced and discussed in detail the US Government Accountability Organization (GAO) report [2] on non-biological complex drugs (NBCDs) together with the US Food and Drug Administration (FDA) draft guidance on drug products containing nanomaterials [3] and the possibility of global harmonization of regulatory policies regarding NBCD products. This report highlights the main conclusions of the meeting and lists a number of topics that would benefit from further debate within the scientific and regulatory community.
The meeting was structured to discuss the following key questions:
– What is equivalence and why does it need our attention?
– Will recent technology developments solve the challenges in determining Critical Quality Attributes (CQAs) of complex drug products?
– Can the successful biosimilar development approach inspire the development of nanosimilars?
– Is there a possibility for global harmonization of regulatory policies for NBCD products and other complex drug products?
In the sections below, we summarize the discussions held during the meeting, in an attempt to answer these key questions.
What is equivalence and why does it need our attention?
Generic drug manufacturers are generally seeking therapeutic equivalence (TE) designation for their drug, as this is a prerequisite for substitution and/or interchangeability with the reference drug product (Orange Book in the US [4). Therapeutic equivalence means that the product is pharmaceutically equivalent (PE) and bioequivalent (BE) to the brand-name product, i.e. PE + BE = TE. Drug products are considered pharmaceutically equivalent if they contain the same active ingredient(s) in the same dosage form, are taken via the same route of administration and are identical in dose. Bioequivalence can be demonstrated using pharmacokinetic, or pharmacodynamic approaches, comparative clinical trials or an in vitro bioequivalence method. Already for decades, these approaches are successfully applied to small molecule drug products for drug approval. Because of the complexity of biological and NBCD products, developers encounter challenges in applying the TE concept.
For follow-on versions of biological products (biosimilars), extensive comparative characterization and preclinical studies are required followed by a limited set of clinical studies to demonstrate safety and efficacy. This may demonstrate similarity but not ‘automatically’ therapeutic equivalence. This biosimilar approach has been introduced and regularly evaluated and updated by the European Medicines Agency (EMA) and is currently considered an established, science-based regulatory approach. FDA has introduced its biosimilar approach as well, which essentially follows the EMA principles.
Determination of TE, through PE and BE, of NBCD products, including nanomedicines, can be a significant challenge. The first discussion on this issue in Budapest in 2014 identified the problem and indicated that CQAs are key to determine TE [5]. The preferred regulatory pathways to establish TE for NBCDs was debated during a number of meetings, including at the New York Academy of Sciences in 2016 [6] and subsequently at the second conference in Budapest in 2016 [7]. Because of the difficulties involved in assessing equivalence with the originator NBCD products, concerns have been raised about safety and efficacy that might appear only after the follow-on versions are introduced on the market.
During the discussions, the topic of consistent terminology has also been raised. Earlier in 2014, a terminology paper was released by the NBCD Working Group [8]. Since then, several new items popped up and the most recent discussion on terminology has evolved around using the terms ‘generic’, ‘follow-on’ and ‘similar’. In a recent publication, Marden et al. make an important statement that terminology in the regulatory field should be used correctly and consistently [9]. As the word ‘generic’ implies therapeutic equivalence and substitutability, this report refers to follow-on products and similars for complex drug products that cannot be fully characterized by physicochemical characterization. The term complex generic is mainly used by FDA and EMA tends to use follow-on, and/or similar.
Will recent technology developments solve the challenges in determining Critical Quality Attributes of complex drug products?
Identification and a thorough (physicochemical) characterization of CQAs is an important step towards the development of a follow-on or similar complex drug product. According to ICH ‘a CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality’. In other words, a critical attribute is a property of which its variation will adversely impact the quality and the clinical profile of the product. At present, both EMA and FDA have adopted a stepwise approach in similarity determination of biosimilars. The development of a biosimilar product therefore requires a thorough comparative in vitro (physico-)chemical characterization of (critical) quality attributes of the test product with the reference product. These thorough analytical comparability studies are followed by non-clinical and clinical studies.
Acknowledging the present challenges to assess CQAs for NBCD products and their design space (DS) that would lead to PE, attention should be paid to further research on elucidating mechanisms of action and on the development and validation of relevant analytical techniques.
This is broadly recognized as, in the meeting, recent developments in state-of-the-art technologies were presented. More importantly, examples were presented how these technologies were applied in the process of characterizing complex drug products. Although the relevance of application of such advanced technologies is to be seen on a product-by-product basis, the progress in technology development for sure impacts the efficiency of characterization activities and brings CQA/DS information closer by.
Can the successful biosimilar development approach inspire the development of nanosimilars?
In a report on an earlier meeting in the SRACD Conference series, the similarities between the complexity of biological and non-biological complex drug products and the implications for TE assessment [5] were pointed out. These discussions continued and the idea of applying the biosimilar paradigm to other complex drug products gained traction. Very concretely, following a publication by authors from EMA, a nanosimilar evaluation process was suggested using the ‘totality of evidence’ concept with a stepwise procedure [10]. This stepwise procedure includes physicochemical structural characterization, animal toxicity studies, in vivo comparative equivalence studies (PK/PD).
For example, in a series of reflection papers for intravenous iron-based nano-colloidal products EMA (proposes a stepwise procedure and ‘totality of evidence’ approach [11].
The success of a new drug modality concept is depending on more factors than appropriate regulatory frameworks alone. In the end, the adoption of new technology and its related products needs multi-stakeholder support. A major effort by different stakeholders was made to explain the scientific background of the biosimilar concept and clinical experience with these biosimilars to healthcare professionals [12]. The EMA brochure on biosimilars from 2017 is a good example of such an effort [13]. Stakeholders in the nanomedicine field should use this successful example of a policy to inform healthcare professionals about the specific challenges encountered with nanosimilars, now and in the future. A first step has already been made by Astier et al. by publishing a list of factors to consider when selecting a nanosimilar [14].
Is there a possibility for global harmonization of regulatory policies for NBCD products and other complex drug products?
The success of harmonization efforts can be judged when comparing the content of guidance documents and reflection papers by the regulatory authorities. A prominent example of guidance documents that could be harmonized are those concerning the approval of follow-on doxorubicin liposome formulations.
Liposomes are one of the most important classes of NBCD products that are used since years in patients. The complexities of liposomal drug products are recognized and dealt with in the FDA and EMA guidance reflection documents. Major efforts were undertaken through the Global Bioequivalence Harmonization Initiative, with participation of EMA and FDA, to align the approaches by the two regulators. It is important to differentiate between nice-to-know and need-to-know requirements. At present, and despite the harmonization exercise, FDA and EMA have taken different decisions on essentially the same follow-on formulation of Doxil®. Moreover, very recently, a case study was published describing the authorization of a generic version Copaxone® in Europe and the US. Whilst EMA requested a full clinical assessment programme, US FDA approved a follow-on version of Copaxone without any clinical data supporting therapeutic equivalence. The approval of these follow-on versions evaluated through different approaches by both agencies leaves open questions on the equivalence of these drug products [15].
In response to concerns raised by the scientific community, the US Government Accountability Office (GAO) was asked to assess FDA’s process of reviewing follow-on versions of NBCD products. In the conclusion and recommendations section, the GAO report underlined the scientific challenges involved when demonstrating equivalence between brand and generic NBCD products [2]. One of the main conclusions was that although all stakeholders agreed that demonstration of equivalence of follow-on NBCDs is scientifically challenging, no agreement could be found on how to move forward and whether the current legislation is sufficient. See Table 1 for a list of conclusions distilled from the full report.
Of note, shortly after the release of the GAO report, FDA issued a draft guidance for industry in December 2017 on ‘Drug products including biological products that contain nanomaterials’ [3]. The agency identified 11 key factors for the assessment of drug products containing nanomaterials. To discuss this draft guidance, an AAPS Guidance Forum workshop was held in September 2018 in Washington DC and the report of that workshop was published earlier in 2019 [16].
In Europe, the debate on whether similar versions of NBCD products should follow the centralized approval procedure are ongoing. A recent approval of an iron carbohydrate similar through the hybrid 10(3) pathway instead of earlier 10(1) generic drug approvals indicates that the field is moving. An overview of all NBCD products and their follow-on versions approved in Europe was recently published by Klein et al., presenting an interesting view on how these products were approved over the years and where the regulatory system may be improved [17].
Just before resigning from office, the former FDA Commissioner Scott Gottlieb voiced his opinion in support of an ICH approach in the area of complex generic drug products [18]. It is to be seen how this will develop, but the scientific and regulatory community should be ready to engage in discussions to increase coherence in the regulatory approach for this type of products.
The meeting successfully brought together experts from all relevant stakeholders, including regulatory authorities, academic institutes, generic and innovative pharmaceutical companies. Participants came from all over the world (five continents, 44 countries) and actively participated in the debates. It is important to note that compared to previous SRACD meetings, significant progress was made in formulating answers to the above four questions. Progress may have been made, but there was also a clear call for action to take the international discussions around alignment and harmonization to the next level.
This paper may include opinions of the authors and not necessarily the opinions of their employers. Authors JdV, DC, BF, IK, SM, VS are members of the Non-biological Complex Drugs Working Group, hosted at Lygature, The Netherlands (www.lygature.org/NBCD).
The authors declare that there is no funding received to prepare this manuscript.
Competing interests: BF and SM are employees of Vifor Pharma Ltd, Glattbrugg, Switzerland.
Provenance and peer review: Not commissioned; externally peer reviewed.
1Foundation Lygature, 6 Jaarbeursplein, NL-3521 AL Utrecht, The Netherlands
2Department of Pharmaceutics, Utrecht University, Utrecht, The Netherlands
3Vifor Pharma Ltd, Glattbrugg, Switzerland
4Semmelweis University, Department of Pharmaceutics, Budapest, Hungary
5Department Pharmaceutical Sciences, Unit of Clinical Pharmacy and Epidemiology, University of Basel, Basel, Switzerland
6VPS Consulting LLC, North Potomac, MD, USA
References
1. Klebovich I, Shah VP, Mühlebah S, Editors. SRACD 2018, 3rd International Symposium on Scientific and Regulatory Advances in Biological and Non-Biological Complex Drugs: A to Z in Bioequivalence; 12-14 November 2018; Budapest, Hungary. Final Program & Book of Abstracts, ISBN 978-615-5270-50-5; Veszprém, Hungary: OOK-Press Ltd.
2. United States Government Accountability Office. Generic Drugs. FDA should make public its plans to issue and revise guidance on nonbiological complex drugs. December 2017. [homepage on the Internet]. [cited 2019 Sep 9]. Available from: https://www.gao.gov/assets/690/689047.pdf
3. U.S. Food and Drug Administration. Drug products, including biological products, that contain nanomaterials. Guidance for Industry. December 2017 [homepage on the Internet]. [cited 2019 Sep 9]. Available from: https://www.fda.gov/media/109910/download
4. U.S. Food and Drug Administration. Approved drug products with therapeutic equivalence evaluations (Orange Book) [homepage on the Internet]. [cited 2019 Sep 9]. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book
5. Crommelin DJ, Shah VP, Klebovich I, McNeil SE, Weinstein V, Flühmann B, et al. The similarity question for biologicals and non-biological complex drugs. Eur J Pharm Sci. 2015;76:10-7.
6. Hussaarts L, Mühlebach S, Shah VP, McNeil S, Borchard G, Flühmann B, et al. Equivalence of complex drug products: advances in and challenges for current regulatory frameworks. Ann N Y Acad Sci. 2017;1407(1):39-49.
7. Crommelin DJ. Meeting report on International Symposium on Scientific and Regulatory Advances in Complex Drugs (SRACD 2016). Eur J Pharma Sci. 2017;96:I-II.
8. Crommelin DJ, de Vlieger JS, Weinstein V, Mühlebach S, Shah VP, Schellekens H. Different pharmaceutical products need similar terminology. AAPS J. 2014; 16(1):11-4.
9. Marden E, Ntai I, Bass S, Flühmann B. Reflections on FDA draft guidance for products containing nanomaterials: is the abbreviated new drug application (ANDA) a suitable pathway for nanomedicines? AAPS J. 2018;20(5):92.
10. Ehmann F, Sakai-Kato K, Duncan R, Hernán Pérez de la Ossa D, Pita R, Vidal JM, et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine (Lond). 2013;8(5):849-56.
11. European Medicines Agency. Reflection paper on the data requirements for intravenous iron-based nano-colloidal products developed with reference to an innovator medicinal product. 26 March 2015 [homepage on the Internet]. [cited 2019 Sep 9]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-data-requirements-intravenous-iron-based-nano-colloidal-products-developed_en.pdf
12. Boone N, van der Kuy H, Scott M, Mairs J, Krämer I, Vulto A, Janknegt Rob. How to select a biosimilar? Eur J Hosp Pharm Sci Pract. 2013;20:275-86.
13. European Medicines Agency. Biosimilars in the EU. Information guide for healthcare professionals [homepage on the Internet]. [cited 2019 Sep 9]. Available from: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
14. Astier A, Barton Pai A, Bissig M, Crommelin DJA, Flühmann B, Hecq JD, et al. How to select a nanosimilar. Ann N Y Acad Sci. 2017;1407(1):50-62.
15. Rocco P, Eberini I, Musazzi UM, Franzè S, Minghettia P. Glatiramer acetate: a complex drug beyond biologics. Eur J Pharm Sci. 2019;133:8-14.
16. de Vlieger JSB, Crommelin DJA, Tyner K, Drummond DC, Jiang W, McNeil SE. Report of the AAPS Guidance Forum on the FDA Draft Guidance for Industry: “drug products, including biological products, that contain nanomaterials”. AAPS J. 2019;21(4):56.
17. Klein K, Stolk P, De Bruin ML, Leufkens HGM, Crommelin DJA, De Vlieger JSB. The EU regulatory landscape of Non-Biological Complex Drugs (NBCDs) follow-on products: observations and recommendations. Eur J Pharm Sci. 2019;133:228-35.
18. U.S. Food and Drug Administration. Statement from FDA Commissioner Scott Gottlieb, M.D., on 2019 efforts to advance the development of complex generics to improve patient access to medicines [homepage on the Internet]. [cited 2019 Sep 9]. Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-2019-efforts-advance-development-complex-generics
|
Author for correspondence: Jon SB de Vliegr, PhD, Coordinator, NBCD Working Group, p/a Foundation Lygature, 6 Jaarbeursplein, NL-3521 AL Utrecht, The Netherlands |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2019 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-progress-report-on-the-3rd-international-symposium-on-scientific-and-regulatory-advances-in-biological-and-non-biological-complex-drugs-a-to-z-in-bioequivalence.html
Copyright ©2024 GaBI Journal unless otherwise noted.