|
Abstract: |
Submitted: 14 November 2016; Revised: 17 November 2016; Accepted: 18 November 2016; Published online first: 25 November 2016
On 27–28 September 2016, the European Directorate for the Quality of Medicines of HealthCare (EDQM) held a major International Conference in Tallinn, Estonia to celebrate the publication of the 9th edition of the European Pharmacopoeia: ‘European Pharmacopoeia: tackling future challenges of the quality of medicines together’ [1]. Among the topics covered in the programme was a workshop on Setting Pharmacopoeial Standards for biotherapeutic products. This manuscript is prepared based on the presentation entitled ‘The role of European Pharmacopoeia monographs in setting quality standards for biotherapeutic products’ at the workshop.
The European Pharmacopoeia (Ph. Eur.) has driven the development, drafting and publication (what we call ‘elaboration’) of monographs for biotherapeutic products for several decades. The value and utility of these monographs have been questioned in recent years, both in the press [2] and in scientific conferences [3–5, Charton E 2016, personal communication, November 17]. Although the discussions have been open and stimulating, the arguments put forward against pharmacopoeial standards for biotherapeutic products have often been either unjustified or based on an incorrect line of reasoning. The aim of this manuscript is to illustrate past and more recent Ph. Eur. achievements in the field of biotherapeutics and provide users of the Ph. Eur. with responses to the questions raised. In doing so, we hope to lay the foundations for future constructive discussions.
The Ph. Eur. lays down common, compulsory quality standards for all medicinal products in Europe. It is mandatory on the same date in 37 European states and the European Union (EU). Some stakeholders opposed to biotherapeutic product monographs stated that individual monographs might exclude products from the market if the requirements of the monographs are not met [2]. Monographs are public standards; therefore, products that do not comply with the monographs and requirements of the Ph. Eur. are normally excluded from the market. However, these standards are not written in stone and a licensing authority may nonetheless decide to accept such products if their quality, safety and efficacy have been demonstrated. The authority should then request revision of the relevant monograph as per EU Directive 2001/83/EC: ‘In cases where a specification contained in a monograph of the European Pharmacopoeia or in the national pharmacopoeia of a Member State might be insufficient to ensure the quality of the substance, the competent authorities may request more appropriate specifications from the marketing authorisation holder. The competent authorities shall inform the authorities responsible for the pharmacopoeia in question. The marketing authorisation holder shall provide the authorities of that pharmacopoeia with the details of the alleged insufficiency and the additional specifications applied’.
The legislation therefore includes a mechanism to provide the pharmacopoeia authority with information on the quality of products on the market. Obviously, the Ph. Eur. must keep pace with the needs of licensing, control and inspection authorities in the public health sector, with industrial constraints and with technological and scientific advances. Today, with the rapid rise of biotechnological products, the question of how the Ph. Eur. can best fulfil this need for biologicals has become a burning issue.
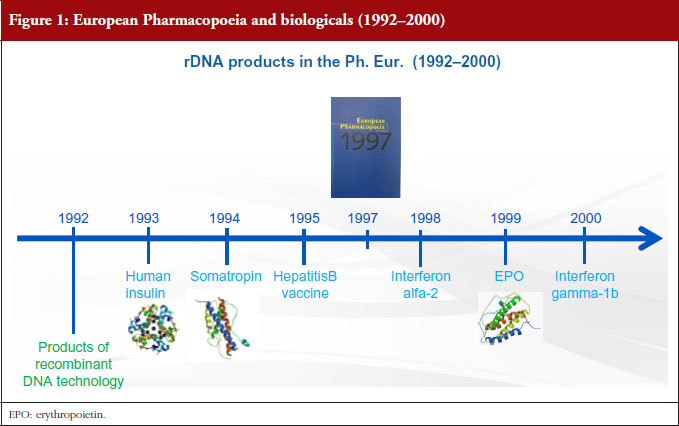
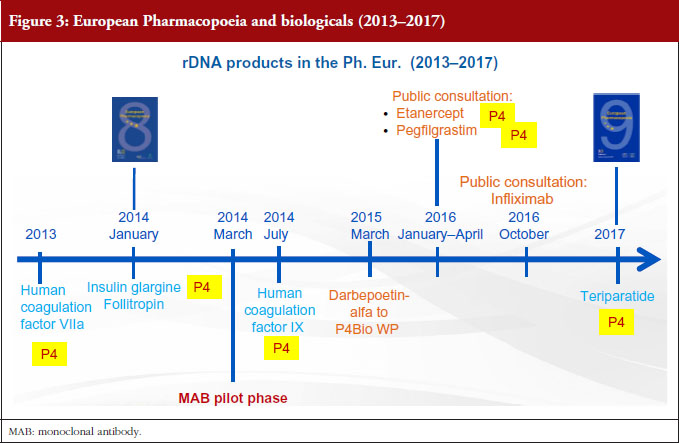
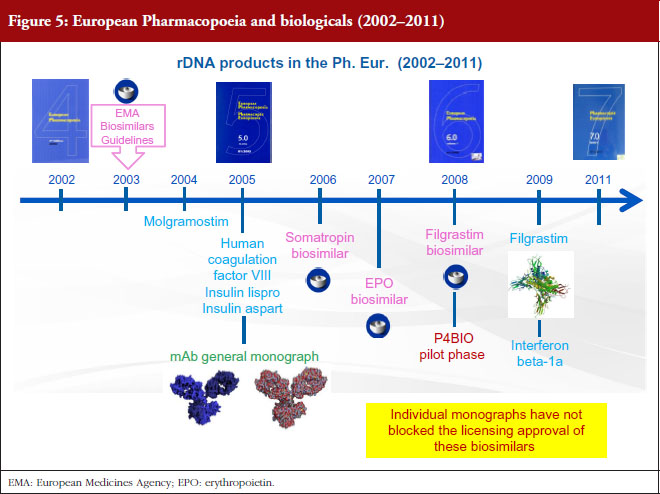
There are two procedures for the elaboration of Ph. Eur. monographs: the multi-source approach and the single source approach. The first of these, the multisource approach, which is also called ‘Procedure 1’ or P1 in our jargon, is the classical procedure, by which the Ph. Eur. takes into account the specifications of more than one marketed product in order to produce a single monograph. In this case, elaboration takes place in collaboration with more than one manufacturer. The members of the groups in charge of the elaboration of these monographs include expert representatives of the regulatory authorities, Official Medicine Control Laboratories (OMCLs), industry and academia. Industry, however, has expressed concerns about this multi-source approach, stating that elaborating a monograph based on several products leads the Ph. Eur. to establish a standard of inferior quality, without consideration of the criticality of quality attributes and preclinical/clinical evidence [2]. The EDQM does not share these concerns. As explained before, Ph. Eur. monographs are based on specifications approved by licensing authorities. The EDQM considers, on the contrary, that monographs in a multi-manufacturer situation have multiple advantages, since comparison of different products and procedures provides a forum for consensus and leads to the elaboration of more robust standards. Examples of this are the insulins and somatropin monographs, published in 1992 and 1993 respectively, hepatitis B vaccine published in 1995, interferon alfa-2 in 1998, erythropoietin in 1999, interferon gamma-1b in 2000, molgramostim in 2004, human coagulation factor VIII rDNA, insulin lispro and insulin aspart in 2005, interferon beta-1a and filgrastim in 2009, see Figures 1 and 2. It was only recently, in 2008, that another procedure, known as ‘P4’, was established for the elaboration of monographs on biotherapeutic products. This is a single-source approach, already successfully applied for many years in the field of chemically defined substances. When a monograph is elaborated under the P4 procedure, the relevant Ph. Eur. group collaborates with the innovator while the substance is still under patent protection to ensure that the monograph is ‘up and running’ at patent expiry. Texts are dealt with by a specific group of experts composed only of representatives of national pharmacopoeia secretariats or regulatory authorities. If we look at what has happened in the last three years, see Figure 3, with the exception of follitropin (published in 2014), all new monographs on biotherapeutics have been elaborated under the P4 procedure, i.e. human coagulation factor VIIa rDNA in 2013, insulin glargine in 2014, human coagulation factor IX rDNA in 2014, teriparatide in 2017.
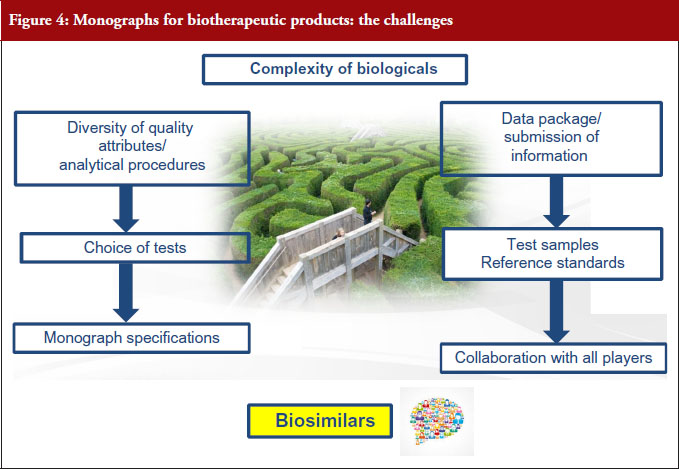
The elaboration of these monographs has not been an easy task. The next part reports the challenges, see Figure 4, we encountered and how they were overcome.
One of the concerns expressed by industry [2] about the elaboration of monographs has been that ‘Due to their inherent complexity and interdependence with their manufacturing processes, the quality and consistency of biologicals can only be defined and ensured through individual and comprehensive process- and product-specific control strategies’. The Ph. Eur. has always recognized the complexity of biologicals and, despite this complexity, has nonetheless been able to develop monographs for biologicals such as vaccines or blood products over the past decades. The experts who elaborate the monographs are fully aware of the fact that biologicals consist of complex mixtures of closely related variants, i.e. naturally occurring heterogeneity in glycosylation or other post-translational modified forms; that the manufacturing process is complex and that changes may lead to distinct quality attributes, e.g. glycosylation, charge heterogeneity, chemical modification. As a result, public standard setting for this class of products is a complex and demanding exercise.
The experts have addressed the complexity of biotherapeutic products by introducing a certain degree of flexibility in the monographs. As a result of this, Ph. Eur. biotherapeutic monographs take into account not only biomolecule complexity but also of the potential diversity in biosimilar compounds resulting from the different manufacturing processes applied. This flexibility is visible in the production section, which has been adapted to reflect process-dependent heterogeneity, e.g. glycosylation. As a reminder, statements under the heading Production draw attention to particular aspects of the manufacturing process but are not necessarily comprehensive [6]. They are mandatory requirements for manufacturers, unless otherwise stated.
To give an example, the Ph. Eur. has successfully elaborated a monograph for recombinant human coagulation factor IX rDNA, which is a complex molecule consisting of more than 400 amino acids produced by mammalian cell lines [7]. To address the complexity of the molecule, a glycan analysis was introduced in the production section together with a number of flexibility statements. In the case of rDNA FIX, the glycan profile depends on the manufacturing process. The test prescribes the use of an in-house reference preparation (available only to the manufacturer). Generic methods of analysis are prescribed, for example, the Ph. Eur. general Glycan analysis of glycoproteins (2.2.59); a specific analytical procedure is given as an example and has no mandatory character; finally, the monograph states that acceptance criteria are to be defined in agreement with the competent authority. We consider that the glycan analysis approach taken in this monograph is a means of improving monograph flexibility under well-defined conditions and that it is compatible with the development of biosimilars. This same approach has recently been taken for other biotherapeutic monographs [8–10] to address structural complexity.
At this juncture, it is important to set the record straight about the complexity of biological products argument that has been used against product-specific monographs. The argument refers to a ‘decision of the EDQM in November 2009 to exclude biological products from the scope of Certificates of Suitability of Monographs of the European Pharmacopoeia (CEP) [11]. CEP is used to certify that a product-specific monograph in the Ph. Eur. is able to adequately control the quality of the manufacturer’s pharmaceutical ingredient obtained by a given manufacturing route. The exclusion of biological products supports the idea that compliance with the monograph is not sufficient to ascertain the quality, safety and efficacy of these products’ [2]. However, this argument is not relevant as the lawmakers’ intention clearly was not to restrict the elaboration of monographs. The EU allows for the quality part of the dossier in a Marketing Authorisation Application (MAA) to include, instead of full documentation on the active substance, either reference to an Active Substance Master file (ASMF) or to a CEP. This possibility is not extended to biologicals because the marketing authorization holder (MAH) must have access to complete information concerning the production of a biological substance, without which they are unable to take responsibility for the final product. This is stated in the EMA guideline on ASMF: ‘The MAH applicant for a biological medicinal product could therefore not comply with the requirement to ‘take responsibility for the medicinal product’ without having full and transparent access to these quality-related data. The use of an ASMF would prevent such access, and should therefore not be allowed for biological active substance’ [12]. This is not comparable to the use of a monograph and therefore has nothing to do with the issue of whether or not a monograph can or cannot address the complexity of biologicals.
A second challenge is that because biologicals are complex, they display a large diversity of quality attributes, which in turn can be analysed using a large variety of methods. The authors of monographs are therefore faced with the difficulty of choosing which tests to include in the monograph. This raises the crucial question of how the information required for a public standard should be defined. The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) gives us the answer to this question: ‘Specifications are chosen to confirm the quality of the drug substance and drug product rather than to establish full characterization and should focus on those molecular and biological characteristics found to be useful in ensuring the safety and efficacy of the product’ [13]. As previously stated, approved specifications form the basis for monograph elaboration. Monographs are therefore drafted using the information submitted to EDQM by the manufacturer of the substance in question. Unfortunately, the manufacturer’s specifications may not be appropriate for a public standard for a variety of reasons. For example, as part of the testing strategy, specific analyses may be omitted from routine testing or may be performed as in-process control tests and, therefore, are no longer included in the specifications. In addition, if the process is shown to reduce levels of specific impurities to within acceptable limits, routine testing for a specific impurity may not be performed. As a result, further tests may be required. Sometimes, a specific quality attribute may be flagged in the Production Section.
Analytical procedures are part of specifications. One real challenge is the verification of the methods before they are included in the Ph. Eur. The EDQM ensures that the robustness and transferability of future Ph. Eur. methods are verified experimentally. Ph. Eur. methods should include criteria to verify method performance and the EDQM makes sure that these criteria are appropriate. Sometimes, the method can enter the pharmacopoeia without further work. But sometimes the methods are out-of-date or insufficiently robust. In such cases, specific additional instructions may be needed, for example, information that was not necessarily included in a SOP (Standard Operating Procedure). Sometimes the experts may recommend tightening the criteria for verification of method performance. When the method proposed by the manufacturer already exists in the Ph. Eur., the monograph may refer to this method, if appropriate. When a monograph for a closely related substance already exists in the Ph. Eur., this is also taken into consideration to ensure that Ph. Eur. texts remain compatible with each other. For certain tests, experimental verification may go beyond the monograph itself, e.g. peptide mapping by LC-MS to confirm marker peaks in complex peptide maps. In extreme cases, the decision may be taken to use an alternative method but in this event, a complete validation is required. All this work obviously requires considerable input from our expert groups and the EDQM laboratory. Close collaboration with the manufacturer is therefore essential in order to find the best way forward for public standard setting.
The EDQM is always extremely grateful to the manufacturers who have chosen to collaborate with the Ph. Eur. on the elaboration of its public standards.
The reader may find it surprising that the development of biosimilars has created yet another challenge to be overcome by the groups responsible for elaborating biotherapeutic product monographs. This is mainly due to the mistaken belief that the Ph. Eur. monographs can be used to demonstrate biosimilarity. The following information will hopefully clarify any misunderstandings or misuse of Ph. Eur. monographs. Firstly, reference standards described in pharmacopoeial monographs and reference products necessary for the demonstration of biosimilarity are frequently confused. Ph. Eur. reference standards are not intended to be used as reference (comparator) products in the context of applications for biosimilars, rather they are supposed to be used within the scope of Ph. Eur. monographs (see Ph. Eur. Chapter 5.12) and within this scope only. Secondly and most importantly, compliance to a monograph does not mean demonstration of biosimilarity. The EMA guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues states that comparison of the biosimilar to a publicly available standard, e.g. a pharmacopoeial monograph, is not sufficient for the purposes of comparability [14]. The unfortunate misuse of monographs in this respect has had a negative impact on the acceptance of monographs for biotherapeutic products by some stakeholders [15].
In current debates, we should bear in mind that the Ph. Eur. is a set of public standards providing harmonized quality requirements for medicinal products throughout Europe: it is used by all. The process by which the product is being approved (generics/biosimilars) is not taken into consideration when drafting a monograph. On the other hand, biosimilars are a type of product that were established to avoid unnecessary repetition of preclinical and clinical studies. The regulatory pathway to be followed is defined in appropriate guidelines. Biosimilars are developed by companies and evaluated by licensing authorities, whether or not a compendial standard exists.
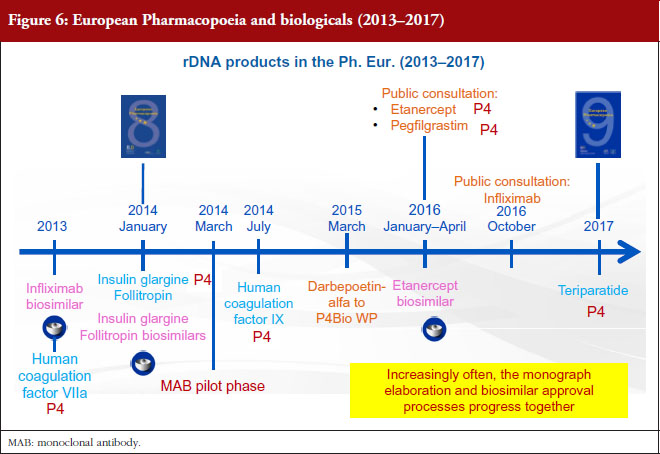
However, there has been some concern that monographs could actually hold up the regulatory process [2]. The EDQM totally refutes this statement: as far as biological products are concerned, the Ph. Eur. is elaborated based on licensed products and since authorization takes place before the monograph is elaborated, there is simply no way the monograph could delay product authorization. Moving on to biosimilars, 18 of the 21 biosimilar products approved in Europe are covered by a Ph. Eur. monograph and there is no evidence to suggest that the monographs have delayed their authorization. If we look at the timeline, see Figure 5, the EMA biosimilar guidelines were established in 2003. The first biosimilar to be approved was somatropin in 2006. The first erythropoietin (EPO) biosimilar was approved in 2007, and since that time, four more EPO biosimilars have been approved. Both somatropin and EPO are covered by monographs. The EPO monograph has even been revised to accommodate the glycan distribution of one of these biosimilars. And then there is filgrastim, for which a monograph was published in 2009. The first filgrastim biosimilar was approved in 2008 and since then, seven other filgrastim biosimilars have been approved. Since all these biosimilars have been approved, one thing is certain: individual monographs have not blocked the licensing approval of these biosimilars. If we take a closer look at the present situation, see Figure 6, it is obvious that increasingly often, the monograph elaboration and biosimilar approval processes progress together. This is the case, for example, for insulin glargine and follitropin.
An etanercept biosimilar was approved in early 2016. At the same time, a monograph for this substance was under enquiry in Pharmeuropa [9]. The outcome of the enquiry has been very positive, with comments received from both regulatory authorities and industry: these comments have been addressed and the monograph is soon to be published in the Ph. Eur. During the same period, a monograph on pegfilgrastim was published and here as well, input from industry has been extremely constructive. The monograph itself is still being discussed as technical issues have arisen. In the meantime, at least three pegfilgrastim biosimilars are under assessment and many biosimilar manufacturers are requesting scientific advice for their products. This is a very strong indication that there is a pressing need for a public standard for pegfilgrastim.
To summarize, it has proven possible to overcome the challenges linked to the complexity of biologicals and to elaborate biotherapeutic product monographs. However, the success of the monograph elaboration process depends on the willingness of manufacturers to provide the information and candidate reference materials required for the process. This has proven to be more problematic since the advent of biosimilars, probably due to misunderstandings about the role of Ph. Eur. monographs in European legislation on biotherapeutic products.
Individual monographs play a major role in ensuring that medicinal products throughout Europe meet the same quality standards, thereby contributing to patient safety. From a quality and standardization standpoint, biotherapeutic substances should not be viewed any differently from any other substance for which a monograph exists. The Ph. Eur. will continue to fulfil its mission with regard to setting quality standards for biologicals; the question is how this role can be played.
The EDQM wishes to warmly thank all the manufacturers who contribute to the elaboration of monographs by sharing information on the quality part of their dossiers to serve as future public standards. The EDQM would also like to take this opportunity to draw attention to the outstanding work carried out by its experts in the complex exercise of monograph elaboration and hopes that, through debate and open dialogue, will identify how best to continue the work of standardization for biotherapeutic products.
Competing interests: None.
Provenance and peer review: Commissioned; internally peer reviewed.
References
1. Council of Europe. European Pharmacopoeia prepares for the future through exchange with stakeholders [homepage on the Internet]. [cited 2016 Nov 17]. Available from: https://www.edqm.eu/en/news/european-pharmacopoeia-prepares-future-through-exchange-stakeholders
2. IFPMA & European Generic Medicines Association. Reflection Paper. The role of product-specific monographs for biotherapeutic products in pharmacopoeias future role of product specific monographs stakeholders. 3 October 2014 [homepage on the Internet]. [cited 2016 Nov 17]. Available from: http://www.ifpma.org/wp-content/uploads/2014/10/IFPMA_EGA_-_Future_Role_of_Product_Specific_Monographs_vFinal_01.pdf
3. Plenary Session: Role of monographs for biotechnological products. CMC Strategy Forum Europe; 23–25 April 2012; Berlin, Germany.
4. Roundtable Session: Product specific monographs. WCBP 2015, 19th Symposium on the Interface of Regulatory and Analytical Sciences for Biotechnology Health Products; 27–29 January 2015; Washington DC.
5. Session: Implications of product specific monographs for biotherapeutic products. WCBP 2016, 20th Symposium on the Interface of Regulatory and Analytical Sciences for Biotechnology Health Products; 26–28 January 2016; Washington DC.
6. Council of Europe. General notices, General chapter 1. Ph. Eur. 8th Edition Strasbourg, France.
7. Human coagulation factor IX (rDNA), concentrated solution, monograph 2522. Ph. Eur. 8th Edition. Strasbourg, France: Council of Europe 2016.
8. Council of Europe. Human coagulation factor VIIa (rDNA), concentrated solution, monograph 2534. Ph. Eur. 8th Edition. Strasbourg, France.
9. Etanercept, monograph 2895. Pharmeuropa. 2016;28(2). Available from: http://pharmeuropa.edqm.eu/home/
10. Infliximab, monograph 2928. Pharmeuropa. 2016;28(4). Available from: http://pharmeuropa.edqm.eu/home/
11. Council of Europe. Public Health Committee. Resolution AP-CSP (07) 1. Certification of suitability to the monographs of the European Pharmacopoeia (revised version). Adopted by the Public Health Committee (Partial Agreement) (CD-P-SP) on 21 February 2007 [homepage on the Internet]. [cited 2016 Nov 17]. Available from: https://www.edqm.eu/medias/fichiers/cep_procedure_revised_version.pdf
12. European Medicines Agency. Guideline on active substance master file procedure. 31 May 2013. CHMP/QWP/227/02 Rev 3/Corr* [homepage on the Internet]. [cited 2016 Nov 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129994.pdf
13. European Medicines Agency. ICH Topic Q 6 B. Specifications: test procedures and acceptance criteria for biotechnological/biologicalproducts. September 1999. CPMP/ICH/365/96 [homepage on the Internet]. [cited 2016 Nov 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002824.pdf
14. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 24 May 2012. EMA/CHMP/BWP/247713/2012 [homepage on the Internet]. [cited 2016 Nov 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf
15. Ian Schofield. Poor biosimilarity practices, impurities and new technologies debated at European Pharmacopoeia Event. The Pink Sheet. 2016 Oct 6.
|
Author: Emmanuelle Charton, PhD, Head of Division, European Pharmacopoeia Department, European Directorate for the Quality of Medicines and HealthCare, 7 Allée Kastner, CS 30026, FR-67081 Strasbourg, France |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/the-role-of-european-pharmacopoeia-monographs-in-setting-quality-standards-for-biotherapeutic-products.html
Copyright ©2024 GaBI Journal unless otherwise noted.