Regulatory requirements for the development and registration of biosimilars in South Africa
|
Abstract:
In South Africa, medicines are registered and regulated in terms of the Medicines and Related Substances Control Act (Act 101 of 1965), as amended, and the regulations to this Act. A guideline which outlines the quality, non-clinical and clinical requirements for the registration of a biosimilar medicine was first published in March 2012. This guideline was amended in August 2014 to include requirements for registering monoclonal antibody biosimilars.
|
Submitted: 23 June 2015; Revised: Not applicable; Accepted: 1 July 2015; Published online first: 14 July 2015
Introduction
The South African guideline for the development and registration of biosimilar medicines [1] defines a biosimilar as a biological medicine that is similar, but not necessarily identical, in terms of quality, safety and efficacy to an already registered reference biological medicine. The reference biological medicine is the innovator medicine that is used as comparator in head-to-head comparative quality, non-clinical and clinical studies with the biosimilar product to demonstrate similarity. A requirement for the reference medicine is that it must be registered in South Africa on the basis of efficacy and safety data; however, samples of the reference product used in comparability studies do not need to be procured from the South African market but can be sourced from a country with which the Medicines Control Council (MCC) is aligned. These include countries generally recognized to have stringent regulatory systems such as those forming part of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) regions as well as Australia, Canada and Switzerland.
The guideline for biosimilars applies only to those biologicals composed of well-characterized recombinant DNA-derived therapeutic proteins. It excludes vaccines, even if these are derived from recombinant DNA technology. Guidance for the development and registration of biosimilar monoclonal antibody (mAb) products is set out in an annex to the main guideline document [1].
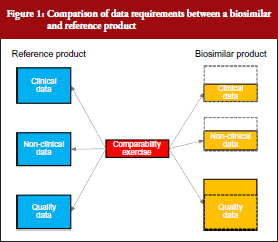
The South African biosimilars guideline is essentially based on the corresponding guidelines of the European Medicines Agency (EMA) [2] and World Health Organization (WHO) [3]. All three guidelines stipulate that biosimilars, being biological molecules with complex structures, would require proof of efficacy and safety in humans prior to registration. The generic medicines registration pathway used for small well-characterized molecules and which require only the demonstration of pharmaceutical equivalence (comparative dissolution in three pH media) and bioequivalence with the innovator (in the case of most solid oral dosage forms) would not be appropriate. Although the extent of clinical and non-clinical studies required for the registration of biosimilars would be less than for innovator medicines, it will to a large degree be dependent on how well the active ingredient has been characterized and its similarity to that of the reference drug substance. Complete physico-chemical characterization of the active ingredient coupled with a high degree of similarity with the reference substance, using state-of-the-art analytical methods, would require substantially reduced clinical and non-clinical data. This is illustrated in Figure 1, which shows that whereas the amount of quality data are greater than for an innovator product (since it includes a comparability study with the reference besides full structural characterization to the same extent as for the reference), the non-clinical and clinical data packages are significantly less. Specific requirements for quality, safety and efficacy are briefly reviewed below.
Quality requirements
A full quality data package detailing the structural characteristics, chemical and physical properties of the biosimilar active pharmaceutical ingredient (API), its validated manufacturing and control processes and stability profile must be submitted. In addition, the formulation, manufacturing, filling and packaging procedures as well as the stability profile of the final product must also be included. These data for the biosimilar are equivalent in amount and extent to that required for the registration of the innovator. However, apart from the quality data on the biosimilar API and final product, the manufacturer of a biosimilar must also submit data on side-by-side comparability studies that show that the biosimilar and reference products are indeed similar in terms of their structures, chemistry and physical properties. This means that for a biological to be considered a biosimilar, it must have an identical primary structure, i.e. amino acid sequence and disulphide bonding, to the innovator reference substance as the primary structure determines the biological function(s) of the substance [4]. Furthermore, it must be shown to be highly similar in terms of post-translational modifications, e.g. glycosylation, three-dimensional structure, e.g. α-helix, β-sheet, and even impurity profiles, although a product with fewer impurities would be preferred [1, 2]. The comparability studies must be carried out in line with the principles of ICH Q6B [5] as well as ICH Q5E [6].
Non-clinical requirements
Non-clinical studies should be conducted before initiation of clinical investigations. The non-clinical studies should be comparative, and be designed to detect differences between the biosimilar and reference medicine. Ideally, a number of endpoints should be monitored such as pharmacodynamic effects as well as toxic effects as determined in at least one repeat dose study. Toxicokinetic measurements should be included and must involve analysis of immunogenicity as the latter may be of value in demonstrating similarity of immune responses to both reference and biosimilar products. Animal immunogenicity studies cannot, however, serve as alternative to immunogenicity studies in humans. In general, other routine toxicological studies such as safety pharmacology, reproduction pharmacology, mutagenicity and carcinogenicity studies are not required for biosimilar medicines.
The trend nowadays is to move away from animal studies; hence, if the minimum required data can be obtained with in vitro biological assays, then these should be used or considered. Justification will, however, be required.
Clinical requirements
Clinical comparability studies should only proceed after acceptable biosimilarity has been established at a physicochemical level. Non-clinical (or in vitro bioassay) comparability data should support the conclusion of biosimilarity based on physicochemical results. The clinical comparability exercise is a stepwise procedure that should begin with pharmacokinetic (PK) and pharmacodynamic (PD) studies followed by clinical efficacy and safety trials. In general, an equivalence trial design is preferred although other designs could be acceptable if properly motivated and justified. Since the clinical data package for a biosimilar is not as extensive as for an innovator medicine, a patient population that is highly sensitive should be selected and the patient population should be large enough to detect meaningful differences in safety, efficacy and immunogenicity.
Clinical studies should be provided for each indication for which the reference product is authorized. However, in cases where the clinical effects have the same underlying mechanism of action, data from a clinical trial of the biosimilar in one indication may be used to support approval of the biosimilar for other indications for which the reference medicine is approved.
Demonstration of comparable efficacy alone does not constitute clinical biosimilarity; equivalent safety and particularly immunogenicity with the reference medicine are also important. Preregistration safety data, derived from the efficacy trials, should be obtained to address the adverse effect profiles of the biosimilar and reference medicines. Preclinical safety data are, however, not sufficient to identify all potential differences and, hence, post-approval pharmacovigilance must be continued accompanied by the submission of regular periodic safety update reports (PSURs) to MCC. This, in fact, is a condition of the registration of all new biologicals, including biosimilars. A suitable Risk Management Plan (RMP) for monitoring immunogenicity, inherent safety concerns and unknown safety signals that could result from the impurity profile and other properties of the biosimilar, should be submitted at the time of application for registration of the biosimilar product.
For most applications for the registration of a biosimilar, comparative clinical trials will be required. However, in certain instances well-designed comparative PK/PD studies may be sufficient to demonstrate clinical comparability where sufficient justification is provided and certain conditions are met. A critical condition is that at least one PD marker is accepted as a surrogate marker for efficacy and that the relationship between dose/exposure to the biosimilar or reference product and this surrogate marker is validated and well known. Examples include absolute neutrophil count to assess the effect of granulocyte colony-stimulating factor and early viral load reduction in chronic hepatitis C to assess the effect of alpha interferon [1].
Finally, since biosimilars, unlike generics, are not considered identical to their innovator reference products, they are neither interchangeable nor substitutable with their reference products or other medicines of the same class. This is in line with similar approaches of other regulatory authorities, such as EMA [2] and Health Canada [7]. Even the WHO guideline stipulates that biosimilars are not interchangeable with their reference products. Interestingly, a few national regulatory authorities in Europe such as The Netherlands [8] and Finland [9] do allow substitution of biosimilars for their reference medicines, albeit under special conditions. In the case of The Netherlands, substitution is allowed but only with adequate clinical monitoring and after the patient has been informed [8]. In Finland, it is permitted but must take place under supervision of a healthcare professional. In France, provisions in the law that allow the substitution of originators by generics have been adapted to extend to biosimilars; however, it has still to be enacted and consequently has not been implemented yet [10].
Current status of biosimilar registrations in South Africa
As of July 2015, no biosimilars have been registered in South Africa. This is despite several biosimilar applications for products containing erythropoietin, filgrastim and insulin having been received by MCC over the past 10 years. None of those which have been reviewed have complied with the registration requirements for a biosimilar medicine. The major deficiencies identified in these applications included the following:
- Poor characterization of the active ingredient. Data presented in the quality part of dossiers did not convincingly prove that the primary structure, including carbohydrate chains in the case of glycosylated proteins, was fully elucidated. Isoform profiles were either not determined or poorly characterized. Similarly, impurities were often not sufficiently described and determined.
- Head-to-head comparability studies between the innovator and biosimilar at the quality level were either absent or not comprehensive enough to demonstrate similarity at this level.
- Non-clinical studies were in many cases absent without justification.
- Clinical studies were either not performed or in cases where such studies were conducted, the data were often not comparative. Where comparative data were submitted, the studies did not provide sufficient evidence to support claims of similarity or absence of differences.
It was clear that many of the biosimilars submitted and which were registered in their countries of origin obtained registration in those countries via a non-biosimilars pathway, i.e. by means of the generics route. Although MCC published its final version of the biosimilars guideline only in 2014 [1], it applied the criteria of EMA and WHO biosimilars guidelines to biosimilar registration applications received prior to 2014. An exception was an application received for the registration of a low molecular weight heparin, enoxaparin, of which the innovator product, namely Clexane® was registered as a pharmaceutical medicine in South Africa. The new enoxaparin application was consequently assessed as a generic drug and was approved as such. No clinical or non-clinical data were submitted with the application. The owner of the Clexane® brand subsequently appealed the MCC’s decision on the grounds that enoxaparin was a biological and that all subsequent enoxaparin applications should be reviewed according to criteria for the registration of biosimilars as opposed to those applicable to generics. The appeal was successful and in August 2009, MCC through the Registrar of Medicines, issued a letter to the industry stating that enoxaparin was regarded as a biological and that clinical data will be required for the registration of all enoxaparin products [11]. Interestingly, the US Food and Drug Administration came to the exact opposite decision and rejected the appeal of the innovator company, which suggests that they may have registered their application of enoxaparin made by Sandoz as a generic drug [12].
Conclusion
The biosimilar guideline of MCC follows the same principles as those of EMA, Health Canada and WHO with respect to quality, safety and efficacy. Although several countries in the European Union now allow substitution of innovator biologicals with their biosimilars, it is unlikely that MCC will revise their guideline to allow substitution in the near future since the local population in South Africa has thus far had no therapeutic exposure to biosimilars.
Disclaimer
The views expressed in this paper are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Medicines Control Council of South Africa or one of its committees or working groups.
Competing interest: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Henry MJ Leng, PhD
School of Public Health, University of the Western Cape, Private Bag X17, Bellville 7130, South Africa
Khamusi Mutoti, BSc, BPharm Sci
Medicines Control Council, National Department of Health, Private Bag X828, Pretoria 0001, South Africa
Professor Nontombe Mbelle, MBChB, MMed, FC Path (CMSA)
Department of Medical Microbiology, University of Pretoria, Private Bag X20, Hatfield 0028, South Africa
References
1. Medicines Control Council. Biosimilar medicines: quality, non-clinical and clinical requirements. August 2014. Ver. 3 [homepage on the Internet]. [cited 2015 Jun 23]. Available from: http://mccza.com/genericdocuments/2.30_Biosimilars_Aug14_v3.pdf
2. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (EMEA/CHMP/BWP/49348/2005). 22 February 2006 [homepage on the Internet]. 2006 Mar 8 [cited 2015 Jun 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003953.pdf
3. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs) [homepage on the Internet]. 2010 Jun 6 [cited 2015 Jun 23]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
4. Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A, et al. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34(Web Server issue):W645-50.
5. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Specifications: test procedures and acceptance criteria for biotechnological/biological products. Q6B. Current step 4 version. 10 March 1999 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Jun 23]. Available from: http://www.ich.org/fileadmin/public_web_site/ich_products/guidelines/quality/q6b/step4/q6b_guideline.pdf
6. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Comparability of biotechnological/biological products subject to changes in their manufacturing process. Q5E. Current step 4 version. 18 November 2004 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Jun 23]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf
7. Health Canada. Health Products and Food Branch. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs) [homepage on the Internet]. 2010 Mar 8 [cited 2015 Jun 23]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf
8. GaBI Online – Generics and Biosimilars Initiative. Dutch medicines agency says biosimilars ‘have no relevant differences’ to originators [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Biosimilars/General/Dutch-medicines-agency-says-biosimilars-have-no-relevant-differences-to-originators
9. GaBI Online – Generics and Biosimilars Initiative. Finnish drug regulator recommends interchangeability of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Policies-Legislation/Finnish-drug-regulator-recommends-interchangeability-of-biosimilars
10. GaBI Online – Generics and Biosimilars Initiative. France to allow biosimilars substitution [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Policies-Legislation/France-to-allow-biosimilars-substitution
11. Medicines Control Council. Applications for registration of medicines containing enoxaparin [homepage on the Internet]. [cited 2015 Jun 23]. Available from: http://mccza.com/dynamism/default_dynamic.asp?Grpid=25&doc=dynamic_generated_page.asp&categid=169&groupid=25
12. GaBI Online – Generics and Biosimilars Initiative. Good news for biosimilar enoxaparin sodium [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Biosimilars/News/Good-news-for-biosimilar-enoxaparin-sodium
|
Author for correspondence: Henry MJ Leng, PhD, School of Public Health, University of the Western Cape, Private Bag X17, Bellville 7130, South Africa
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/regulatory-requirements-for-the-development-and-registration-of-biosimilars-in-south-africa.html
Pro-generics policies and the backlog in medicines registration in South Africa: implications for access to essential and affordable medicines
|
Abstract:
The backlog in medicines registration in South Africa is a result of the implementation of pro-generics policies without strengthening the regulator to handle the substantial increase in registration applications that followed. Despite the backlog, more than enough generics are registered to promote price competition and ensure access to affordable medicines.
|
Submitted: 25 February 2015; Revised: 10 April 2015; Accepted: 11 April 2015; Published online first: 24 April 2015
Background
The Medicines Control Council (MCC) of South Africa has been under considerable pressure to increase the rate of medicines registration and has been accused of delaying patients’ access to affordable and essential medicines. A study commissioned by the Minister of Health in 2006 to investigate the slow pace at which medicines were being registered ascribed it to a lack of skilled human resources, poor infrastructure and inefficient regulatory processes. It was thought that the MCC processes did not keep pace with developments in the pharmaceutical industry. Although these factors certainly contributed to the backlog in medicines registration, which developed at the MCC, it seemed unlikely that they could have been the cause since the MCC, prior to 2005, was viewed as a highly efficient organization and did not have a backlog. In this review we followed the history of medicines registration and application submissions from 2000 to 2012 to determine whether the development of the backlog was a gradual process, which could have been due to factors mentioned above, or a sudden occurrence that may have been precipitated by one or more critical events, such as a change in policy or relaxation of standards for registration. We have found that the backlog originated with the implementation of policies to promote the availability and access to generics but without anticipating, and providing for, the impact this would have on the resources of the MCC. The new policies caused a flood of application submissions to the MCC, which was not equipped in terms of manpower or administrative processes to handle the substantial increase in submissions. In spite of the backlog, the claim that it compromised access to affordable generic medicines appears unfounded since our analysis for a group of tracer medicines showed that only 54% of registered medicines were being marketed and that a maximum of only five brands account for 80% or more of the market for a particular medicine.
Introduction
In 2006, the South African Minister of Health appointed a ministerial task team (MTT) to review the MCC of South Africa and to make recommendations for the establishment of a new medicines regulatory authority. This was prompted by complaints from pharmaceutical companies [1, 2], private clinical research organizations [3, 4] academic clinical research groups [5] and civil society organizations [6, 7] that delays and the backlog in medicines registration were harming patients’ access to affordable medicines. The MTT report was published in 2008 and recommended that the MCC be replaced by a new regulatory agency to be funded on a 50% cost recovery basis from industry [8]. The proposed new agency that will be known as the South African Health Products Regulatory Authority (SAHPRA) was scheduled for implementation in April 2013. However, the Act has since been further amended to include provisions for SAHPRA to be a public entity with an independent board chaired by a Chief Executive Officer (CEO) and a stronger governance structure than in the previous draft. The latest version of the draft amendments to the Bill (Bill 6 of 2014) was tabled in parliament in February 2015. No date has since been specified for when SAHPRA will be established [9, 10].
Among the shortcomings in the MCC and its secretariat, the Medicines Regulatory Affairs (MRA), identified in the MTT report, were: a shortage of in-house skilled human resource capacity and its dependency on external reviewers employed at academic and research institutions (in-house staff turnover is high as many migrate to industry); lack of an efficient electronic document management system to track applications as they progress through the regulatory system; and a shortage of funds to improve infrastructure and attract and retain competent staff.
Since the MTT report did not examine how or when the backlog originated [8], we undertook a review of the data on the number of applications submitted for registration and the number of registration certificates (marketing authorization certificates) issued by the MCC between 2000 and 2012. The premise was that if the two remained closely correlated over this time period it would suggest that there was no real crisis at the MCC and that it could cope with its regulatory workload. A gradual divergence in correlation over time would imply that the regulatory processes, resources and infrastructure were not keeping pace with the developments in the local pharmaceutical industry. If a sudden break or divergence occurred at a specific time during the review period it would signal the occurrence of a critical event such as a change in policy.
In order to determine whether the current rate of registration by the MCC could potentially impede access to medicines we compared the availability of the number of branded generics of eight tracer medicines in the total South African market, i.e. the combined private and public sector markets, with the number of brands registered for each of the eight tracer medicines.
Selection criteria for tracer medicines
The criteria used to select the tracer medicines included: a) diseases that are prevalent in South Africa [11]; b) treatment established according to standard treatment guidelines; c) inclusion on the Essential Medicines List (EML); and d) the Millennium Development Goals (MDGs) of the United Nations, which deal specifically with health (MDG 4: to reduce child mortality; MDG 5: to improve maternal health; and MDG 6: to combat HIV/AIDS, malaria and other diseases). One medicine to treat each of the following conditions was selected: bacterial infections (ciprofloxacin); tuberculosis (rifampicin); human immunodeficiency virus (HIV) infection (lamivudine); diabetes (metformin); hypertension (amlodipine); atherosclerosis (simvastatin); maternal health (oxytocin); and depression (fluoxetine). It is important to note that although many of the medicines selected may be widely used in the treatment of the conditions for which they are registered, that they are not necessarily representative of the entire pharmaceutical market in South Africa. Consequently, generalizing the findings to other medicines or pharmacological classes requires caution.
The backlog in medicine registrations
By 2010 the backlog in registrations was estimated to be in excess of 3,000 applications
[12, 13]. Prior to 2005, the number of applications received and registration certificates issued were in equilibrium. From 2005 the number of applications submitted more than doubled whereas the number of certificates issued remained approximately the same, see Figure 1.
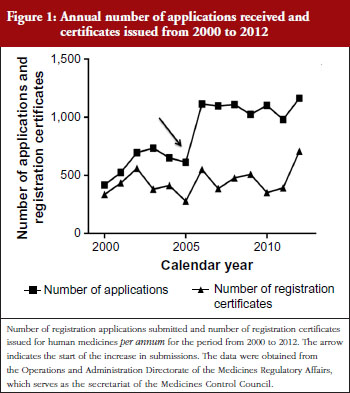
The relatively constant rate at which certificates were being issued from 2000 to 2011 was because additional staff were not appointed to meet the high demand for registration of new products. This shortcoming was also identified by the MTT in its report [8]. The increase in certificates issued after 2011 was due to the recruitment of temporary technical and administrative staff in 2009, with funding from the UK Department for International Development (DFID). It takes around 24 to 36 months from the start of a dossier review until a registration certificate is issued; hence, the increase in product approvals was only seen in 2012.
Pro-generics policy and the backlog in approvals
The large increase in submissions since 2005 is due to the introduction of pro-generics policies, which includes the mandatory generics substitution policy and fast track registration policy. The increase includes applications submitted via routine and fast track pathways.
In 1997 the Medicines Act was amended (Act 90 of 1997) with the aim of making medicines more affordable and accessible. The amendments included provisions for, amongst others, a transparent pricing system, parallel importation of innovator medicines from countries where they are sold for less than in South Africa, and generics substitution to promote the use of generic medicines, i.e. the progenerics policy. The legislation was challenged in early 1998 by 41 pharmaceutical companies [14] resulting in a delay in its implementation until 2003 when the legal challenge was withdrawn [15]. Not all of the policies were introduced; the generics substitution policy was implemented in 2003 whereas policies on pricing such as the single exit price (SEP) was introduced in 2004. Other amendments to the Act (pharmacists dispensing fee) were implemented in 2014.
Apart from the generics substitution provision made in 2003, the Department of Health (DoH) also introduced a fast track registration policy at that time, not only for new chemical entities (NCEs) considered essential for national health and which may not be on the EML, but also for all medicines on the EML [16], the majority of which are generics [17]. This contrasts with the fast track policy of the Food and Drug Administration (FDA) in the US, which is restricted to investigational new drugs designed to fill an unmet medical need [18].
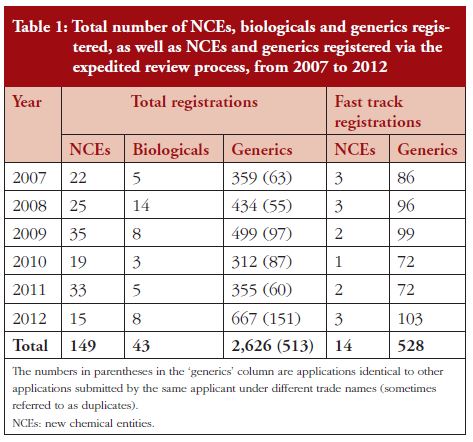
Table 1 shows the number of registrations per year by product type, i.e. NCEs, biological and generic medicines and the number of fast track and duplicate registrations. The data were obtained from the Operations and Administration Directorate of the MRA. Between 2007 and 2012, registration of generic medicines outnumbered that of NCEs (more than 17-fold) and biological medicines (more than 60-fold). More generic medicines were registered through expedited review (fast track) than NCEs. Some generics manufacturers submit two or more copies of the same dossier under different tradenames. Each of these applications is for a separate registration certificate for a product and each product is considered independent. The numbers in parentheses indicate the number of such duplicate or multiplicative applications.
The practice of certain companies to register the same product under different trade names reflects both the very low cost to the company of registering medicines in South Africa (less than Euros 3,000 for a generic medicine [19]) and the commercial advantage they stand to gain when they licence or sell one of the duplicates (if they registered the product under two different names) to another company. This practice, known as ‘dossier farming’, is well known in the industry but is under researched and seldom reported in the literature.
The backlog in medicines registration may, thus, have been caused by the large number of generics submissions which followed soon after the implementation of mandatory generics substitution (pharmacists are required by law to dispense the generic drug, unless the patient, or the patient’s doctor, expressly refuses the substitution, or the price of the generic drug is higher than that of the branded product). This was compounded by legislative requirements for the expedited review of medicines on the EML (most of which are generics) and the lack of control over the number of generics applications for the same innovator medicine that can be submitted by a single company. The question that arises is whether the uncontrolled registration of generic medicines can be justified in terms of promoting access to affordable medicines. If a large number of generics in the market continually drive down the cost of medicines through competition, then the MCC should endeavour to acquire the necessary resources to increase its efficiency and shorten review times. However, if there is no price advantage to the public after a certain number of generics of an innovator product becomes available, then it may be necessary to review the current policies governing the registration of generic medicines.
Does the backlog impede the accessibility and availability of affordable medicines?
The pro-generics policy is intended to promote competition; the theory being that as more competitors enter a market, prices of products will fall [20]. Thus, registering a large number of generic brands of a particular medicine should drive down the price of that medicine. The question is whether the current backlog prevents both important medicines from coming onto the market and reduces competition and, hence, availability of affordable medicines.
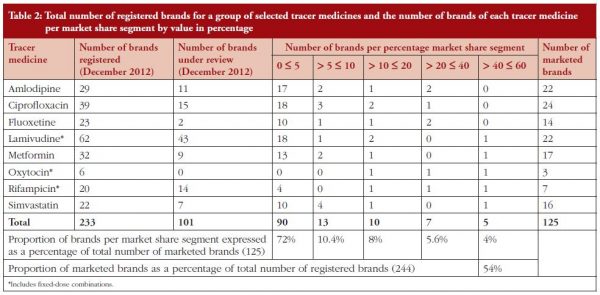
Using IMS data for each of the tracer medicines we analysed the percentage market share (as measured by annual sales value in both private and public markets) for each branded product (generic and innovator) from 5% or less to more than 40%. The highest market share for a brand for most tracer medicines was between 40% and 50%. The number of registered brands and brands under review for each medicine was also included in the analysis to show the relationship between current (number of registered brands) and future (number of registered brands plus brands under review) product availability for each medicine, see Table 2.
The data show that two to five brands for each medicine account for 80% or more of the market value. Approximately 70% of marketed generics have a market share of less than 5% and only 54% of all registered generic brands are actually being marketed. This suggests that for most of the tracer medicines, with the exception of oxytocin, more than enough branded generics have been registered to ensure robust competition in their markets. Their markets have in fact become oversaturated since more than 40% of the registered products are not being marketed. Exceptions would be medicines, such as lamivudine and rifampicin where changes in treatment guidelines for HIV and tuberculosis (TB) may render some products obsolete. For example, in South Africa the intention is to transition all HIV patients to fixed-dose combinations (FDCs) containing tenofovir, emtricitabine and efavirenz or tenofovir, lamivudine and efavirenz [21]. Other antiretrovirals (ARVs), such as lamivudine, whether available in FDCs with other ARVs or as single agent products, may eventually be phased out. Oxytocin represents a special case since there are only three brands registered with none in the registration pipeline awaiting registration. The local market for oxytocin products is very small, just over US$2.8 million in 2012, and the product is only used in the hospital setting in South Africa. This, coupled with the need for cold chain storage, probably discourages local companies from including oxytocin formulations in their product portfolio. This contention is supported by more than 50 oxytocin formulations for human use that are available in the world market [22]. The limited availability of oxytocin products is thus more due to market failure than competition. The MCC should therefore prioritize registration of oxytocin products, should it receive applications, to ensure greater access.
Rifampicin is another case where the data do not follow that for the other tracer medicines. Changes in the treatment guidelines for TB may have made some FDC products containing rifampicin obsolete, which would account for them not being marketed. This would be further impacted by the development of resistant TB, an issue which is particularly pertinent in South Africa with its high incidence of the disease [11]. New FDC formulation with rifampicin should, thus, also be prioritized for registration by the MCC.
In the US, Reiffen and Ward (2005) used regression analysis to estimate the effect of the number of generics entries on the pre-expiry price of the original branded product for 31 drugs [23]. The authors also used price data obtained from IMS Inc. They found that a negative relationship existed between price and the number of generics entries and that the negative effect of increased competition on prices continues at least until the fifth firm enters the market but is not likely to be important after the eighth firm has entered. In other words, after eight generics, prices are unlikely to decline further since the average generics price will now approach the long-run marginal cost of production. South Africa had a population of 48.8 million in 2012 compared with the US population of 316.4 million [24]. Our market is thus smaller than that of the US and consequently one could expect that five generics would ensure sufficient competition in a market for prices to decline significantly from that of the innovator prior to patent expiry. Our findings appear to support this, since five generics competitors dominated the market for most of our tracer medicines.
The large percentage of generic drug products for some of our tracer medicines, which are currently not being marketed, could be due to market saturation. Thus, for some medicines (those for which there are already many generics in the market) it is likely that the backlog does not impede effective competition or, by extension, access to affordable medicines.
Although there is currently no formal mutual recognition agreement between the MCC in South Africa and the regulatory authorities of countries in the Southern African Development Community (SADC), companies may seek to register their products with the MCC in South Africa in order to obtain faster registration in neighbouring countries that have minimal regulatory capacity. For example, Namibia has, since 2010, granted abbreviated reviews for registration of essential medicines that are registered by Medicines Regulatory Authorities with which the Namibia Medicines Regulatory Council (NMRC) aligns itself, such as the MCC of South Africa, provided that the pharmaceutical
manufacturing site complies with current Good Manufacturing Practices (cGMP) and a complete dossier in terms of the applicable registration application format is submitted. (Gaeseb J, Registrar of Medicines, Namibia Medicines Regulatory Council, Ministry of Health and Social Services, Namibia. Personal communication. 6 November 2014).
The proliferation of pharmaceutical products, particularly in developing country markets, either through importation or local manufacture, places a tremendous burden on small and poorly resourced regulatory agencies. There is, thus, a need to review the legislation underpinning the registration process to include a medical needs clause, so that medicine registration can be prioritized on that basis. Provision should also be made to ensure that there is enough competition in the market to make medicines affordable.
Revisit pro-generics policy in the context of medical need
When suffi cient competition for an offpatent medicine exists in the market, regulatory backlog for registering further generics of that product will not have an appreciable effect on market competition, price or availability, and therefore not on access. It is only when competition is limited or non-existent, that the backlog may have those effects. Thus, the claims of industry that the backlog in registration applications of generic medicines impedes access to affordable medicines are generally not supported by our data. Our findings suggest that the backlog could be the result of government implementing progenerics policies without first providing the MCC and MRA with additional resources to handle the substantial increase in generic medicine registration applications that followed. It is important that these policies be reviewed so that further registration applications for products that are already widely available do not prevent generics of critical medicines from gaining speedy access to the registration system, while ensuring robust competition. It may be necessary to consider introducing a policy that will limit the number of generics for a specific medicine to prevent oversaturation of its market. The finding that a large proportion of generics for certain medicines are not marketed suggests that, in these cases, further generics may not lead to greater access.
The South African Government has committed itself to the establishment of a new medicines regulatory authority. The new authority, SAHPRA, is intended to be better resourced in terms of staff and infrastructure and is, therefore, likely to cost the public purse more to operate than the MCC, even if the fees to the industry will be significantly higher than the fees currently levied by the MCC. The scope of the new authority is to be expanded to include regulation of medical devices and complementary medicines. However, there is concern that if the primary motivation for SAHPRA is to register medicines regardless of need, it will serve the interests of the pharmaceutical industry more than that of the public. The industry will receive the benefi ts of shorter timelines while the burden in the form of tax payers’ funds to support the larger agency will be borne by the public. Another concern is that SAHPRA could expose itself to industry capture should it become dependent on funds sourced from industry to pay for its operational costs. When the funding formula for SAHPRA is thus determined it is essential that the regulator never be placed in a situation in which it might lose its independence, i.e. not to industry or to government [25, 26].
Conclusion
The findings from our analysis of the backlog in registration applications and market dynamics of generic drug products, although limited to only eight medicines, allow us to make a few tentative recommendations. Firstly, the National DoH should review its policy on expedited review, which currently applies to all generic medicines on the EML, and consider developing a medical needs clause to restrict this registration pathway to NCEs, new biologicals or new formulations for critical medicines, e.g. FDCs of ARVs. Fast track reviews for generics should be limited to the first five to eight applications received from different companies prior to expiry of the patent on the original. These should be reviewedconcurrently, if possible, so that they can enter the market more or less at the same time to effect significant price reductions through competition, thereby, making the medicine more affordable. This will link the expedited review pathway for registration not only to medical, but to economic need as well. Clone applications, i.e. a copy or exact duplicate of the originator and marketed by the manufacturer or applicant of the originator medicine but with only the labelling (including the brand name) being different, should be excluded from this provision. This is because the innovator applicant has the advantage of submitting such applications at any time before the patent on its original brand expires and can also market the clone before the expiry date of the originator brand in anticipation of imminent generics entry. Secondly, MCC should consider cancelling the registration of products after two years if they have not been marketed (sunset clause), unless the company can provide adequate justification for retaining registration beyond this period.
This recommendation stems from the observation that a high percentage of generics for certain medicines are not marketed after registration. The finding suggests that companies do not conduct adequate market research before submitting applications for registration to assess the viability of their products. One of the reasons could be the low cost for registering a generic drug in South Africa (US$2,260) compared with countries, such as Canada (US$38,006) and Australia (US$73,900). The resources spent on evaluating an application for a product that is eventually not marketed could have been applied to other regulatory activities, such as the regulation of medical devices, which are currently not controlled by the MCC because of resource constraints. Thirdly, generic medicines companies should be discouraged from submitting several applications for the same medicine under different trade names unless this can be justified from a public health perspective. This is because companies may be trading in registration certificates (dossier farming) at a significant premium to what they spent on obtaining registration with the MCC. Finally, we recommend that the new regulatory agency, SAHPRA, not be placed in a position in which it will become overly reliant on industry fees to fund its operations as this may compromise its independence and lead to industry capture.
Acknowledgements
The authors are grateful to IMS Health Inc for generously providing the market share data and the Medicines Regulatory Affairs for providing data on product registrations and registration applications. The authors would also like to thank Dr Petra Sevcikova, Ms Karen Maigetter, Professor Roger Jeffery and Professor Richard Laing for their valuable comments.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Medicines Control Council of South Africa or one of its committees or working groups.
Funding sources
This paper results from research funded by the European Union Seventh Framework Programme Theme: Health-2009-4.3.2-2 (Grant no. 242262) under the title ‘Accessing Medicines in Africa and South Asia [AMASA]’ (http://ec.europa.eu/research/health/public-health/public-health-andhealth-systems/projects/amasa_en.html). The project team includes partners at the University of Edinburgh (UK), Foundation for Research in Community Health (India), University of Ghent (Belgium), Mbarara University of Science and Technology (Uganda), Makerere University (Uganda), Queen Mary University London (UK), Swiss Tropical and Public Health Institute at the University of Basel (Switzerland) and the University of the Western Cape (South Africa).
Competing interest: The authors declare that they have no competing interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Henry MJ Leng1, PhD
David Sanders1, MRCP
Professor Allyson M Pollock2, MBChB, FFPH, MRCP (Ed), MRCGP
1School of Public Health, University of the Western Cape, Private Bag X17, Bellville, 7535, South Africa
2Centre for Primary Care and Public Health, Queen Mary University of London, London E1 2AB, UK
References
1. Kahn T. Cipla slumps on MCC delays. Business Day Live. 2012 Oct 18 [cited 2015 Apr 10]. Available from: http://www.businessday.co.za/articles/Content.aspx?id=167676
2. Kahn T. Litha forced to shut down cardiac unit. Business Day Live. 2012 Aug 8 [cited 2015 Apr 10]. Available from: http://www.bdlive.co.za/articles/2012/03/20/litha-forced-to-shut-downcardiac-unit
3. Buthelezi L. Drug listing delays laid at door of MCC. Accessed 3 August 2012. Available: http://www.mm3admin.co.za/documents/docmanager/2D5ED792-878C-4371-9575-8281A96BBB26/00031267.pdf
4. Kahn T. Blow to SA drugs sector as red tape snares trials. Business Day Live. 2012 Aug 8 [cited 2015 Apr 10]. Available from: http://www.businessday.co.za/articles/Content.aspx?id=174618
5. Thom A. MCC blocking access to lifesaving meds – HIV. Health-E News. 2010 Mar 4 [cited 2015 Apr 10]. Available from: http://www.healthe.org.za/news/article.php?uid = 20032666
6. Geffen N. Medicines Control Council needs new leaders. Quackdown. 2012 Jul 17 [cited 2015 Apr 10]. Available from: http://www.quackdown.info/article/medicines-control-council-needs-new-leaders/
7. Thom A. Change at the MCC – too little, too late? Health-E News. [cited 2015 Apr 10]. Available from: http://www.health-e.org.za/2010/05/28/changeat-the-mcc-too-little-too-late/
8. Report of the Ministerial Task Team on the restructuring of the Medicines Regulatory Affairs and Medicines Control Council and recommendations for the new Regulatory Authority for Health roducts
of South Africa (2008). Available from: oldgov.gcis.gov.za/documents/download.php?f=81967
9. Gray AL. Our medicines regulatory authority: plans for reform in South Africa. NSP Review. 2012;3. [cited 2015 Apr 10]. Available from: http://www.nspreview.org/2012/10/03/our-medicines- regulatory-authorityplans-for-reform-in-south-africa/
10. Kahn T. SA step closer to new medicines regulator. Business Day Live. 2014 Mar 3 [cited 2015 Apr 10]. Available from: http://www.bdlive.co.za/national/health/2014/03/03/sa-step-closerto-new-medicines-regulator
11. Coovadia H, Jewkes R, Barron P, Sanders D, McIntyre D. The health and health system of South Africa: historical roots of current public health challenges. Lancet. 2009;374(9692):817-34.
12. Thom A. The MCC mess. Health 24. 2010 May 28 [cited 2015 Apr 10]. Available from: http://www.health24.com/Medical/Meds-and-you/Using-medicines/The-MCC-mess-20120721
13. Project Conclusion Report. MCC/MRA backlog task team. 2010 Oct [cited 2015 Apr 10]. Available from: http://www.sarrahsouthafrica.org/LinkClick.aspx?fi leticket=AW7KP7zEHKM%3d&tabid=2339.
14. Department of Health. Defending the Medicines Control Amendment Act. 2 March 2001. Pretoria: Department of Health
15. Berger J. Negotiating the new medicines regulatory framework: some basic facts and observations. The South African Journal of HIV Medicine. 2004;5(2):38-40.
16. Republic of South Africa. Health Department. Medicines Control Council. General information guideline. 2008 [homepage on the Internet]. [cited 2015 Apr 10]. Available from: www.mccza.co.za.
17. Republic of South Africa. Department: Health. National EML [homepage on the Internet]. [cited 2015 Apr 10]. Available from: http://www.health.gov.za/index.php/component/phocadownload/
18. U.S. Food and Drug Administration. Fast track, accelerated approval and priority review. Accelerating availability of new drugs for patients with serious diseases [homepage on the Internet]. cited 2015 Apr 10]. Available from: http://www.fda.gov/For-Patients/Approvals/Fast/default.htm
19. Medicines and Related Substances Act, 1965. Schedules. Fees payable in terms of the Act. Government Gazette. 2012;569(35857).
20. Bagwell K, Lee GM. Number of fi rms and price competition. [cited 2015 Apr 10]. Available from: http://www.stanford.edu/~kbagwell/papers/Bagwell%20 Lee%20s%20021214.pdf
21. Davies NECG. Advice document. Fixed-dose combination for adults accessing antiretroviral therapy. S Afr J HIV Med. 2013;14(1 Suppl):41-3.
22. Drugs.com. Oxytocin [homepage on the Internet] [cited 2015 Apr 10]. Available from: http://www.drugs.com/international/oxytocin.html
23. Reiffen D, Ward MR. Generic drug industry dynamics. The Rev Econ Stats. 2005;87(1):37-49.
24. The world in fi gures: countries. The World in 2012. The Economist: 2012;97-105.
25. Stigler GJ. The theory of economic regulation. The Bell Journal of Economic and Management Science. 1971;2(1):3-21.
26. Abraham J. The pharmaceutical industry as a political player. Lancet. 2002;360(9344):1498-502.
|
Author for correspondence: Henry MJ Leng, PhD, School of Public Health, University of the Western Cape, Private Bag X17, Bellville, 7130, South Africa
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/pro-generics-policies-and-the-backlog-in-medicines-registration-in-south-africa-implications-for-access-to-essential-and-affordable-medicines.html
Pro-generics policies and the backlog in medicines registration in South Africa: implications for access to essential and affordable medicines
Abstract:
The backlog in medicines registration in South Africa is a result of the implementation of pro-generics policies without strengthening the regulator to handle the substantial increase in registration applications that followed. Despite the backlog, more than enough generics are registered to promote price competition and ensure access to affordable medicines.
Submitted: 25 February 2015; Revised: 10 April 2015; Accepted: 11 April 2015; Published online first: 24 April 2015
Background
The Medicines Control Council (MCC) of South Africa has been under considerable pressure to increase the rate of medicines registration and has been accused of delaying patients’ access to affordable and essential medicines. A study commissioned by the Minister of Health in 2006 to investigate the slow pace at which medicines were being registered ascribed it to a lack of skilled human resources, poor infrastructure and inefficient regulatory processes. It was thought that the MCC processes did not keep pace with developments in the pharmaceutical industry. Although these factors certainly contributed to the backlog in medicines registration, which developed at the MCC, it seemed unlikely that they could have been the cause since the MCC, prior to 2005, was viewed as a highly efficient organization and did not have a backlog. In this review we followed the history of medicines registration and application submissions from 2000 to 2012 to determine whether the development of the backlog was a gradual process, which could have been due to factors mentioned above, or a sudden occurrence that may have been precipitated by one or more critical events, such as a change in policy or relaxation of standards for registration. We have found that the backlog originated with the implementation of policies to promote the availability and access to generics but without anticipating, and providing for, the impact this would have on the resources of the MCC. The new policies caused a flood of application submissions to the MCC, which was not equipped in terms of manpower or administrative processes to handle the substantial increase in submissions. In spite of the backlog, the claim that it compromised access to affordable generic medicines appears unfounded since our analysis for a group of tracer medicines showed that only 54% of registered medicines were being marketed and that a maximum of only five brands account for 80% or more of the market for a particular medicine.
Introduction
In 2006, the South African Minister of Health appointed a ministerial task team (MTT) to review the MCC of South Africa and to make recommendations for the establishment of a new medicines regulatory authority. This was prompted by complaints from pharmaceutical companies [1, 2], private clinical research organizations [3, 4] academic clinical research groups [5] and civil society organizations [6, 7] that delays and the backlog in medicines registration were harming patients’ access to affordable medicines. The MTT report was published in 2008 and recommended that the MCC be replaced by a new regulatory agency to be funded on a 50% cost recovery basis from industry [8]. The proposed new agency that will be known as the South African Health Products Regulatory Authority (SAHPRA) was scheduled for implementation in April 2013. However, the Act has since been further amended to include provisions for SAHPRA to be a public entity with an independent board chaired by a Chief Executive Officer (CEO) and a stronger governance structure than in the previous draft. The latest version of the draft amendments to the Bill (Bill 6 of 2014) was tabled in parliament in February 2015. No date has since been specified for when SAHPRA will be established [9, 10].
Among the shortcomings in the MCC and its secretariat, the Medicines Regulatory Affairs (MRA), identified in the MTT report, were: a shortage of in-house skilled human resource capacity and its dependency on external reviewers employed at academic and research institutions (in-house staff turnover is high as many migrate to industry); lack of an efficient electronic document management system to track applications as they progress through the regulatory system; and a shortage of funds to improve infrastructure and attract and retain competent staff.
Since the MTT report did not examine how or when the backlog originated [8], we undertook a review of the data on the number of applications submitted for registration and the number of registration certificates (marketing authorization certificates) issued by the MCC between 2000 and 2012. The premise was that if the two remained closely correlated over this time period it would suggest that there was no real crisis at the MCC and that it could cope with its regulatory workload. A gradual divergence in correlation over time would imply that the regulatory processes, resources and infrastructure were not keeping pace with the developments in the local pharmaceutical industry. If a sudden break or divergence occurred at a specific time during the review period it would signal the occurrence of a critical event such as a change in policy.
In order to determine whether the current rate of registration by the MCC could potentially impede access to medicines we compared the availability of the number of branded generics of eight tracer medicines in the total South African market, i.e. the combined private and public sector markets, with the number of brands registered for each of the eight tracer medicines.
Selection criteria for tracer medicines
The criteria used to select the tracer medicines included: a) diseases that are prevalent in South Africa [11]; b) treatment established according to standard treatment guidelines; c) inclusion on the Essential Medicines List (EML); and d) the Millennium Development Goals (MDGs) of the United Nations, which deal specifically with health (MDG 4: to reduce child mortality; MDG 5: to improve maternal health; and MDG 6: to combat HIV/AIDS, malaria and other diseases). One medicine to treat each of the following conditions was selected: bacterial infections (ciprofloxacin); tuberculosis (rifampicin); human immunodeficiency virus (HIV) infection (lamivudine); diabetes (metformin); hypertension (amlodipine); atherosclerosis (simvastatin); maternal health (oxytocin); and depression (fluoxetine). It is important to note that although many of the medicines selected may be widely used in the treatment of the conditions for which they are registered, that they are not necessarily representative of the entire pharmaceutical market in South Africa. Consequently, generalizing the findings to other medicines or pharmacological classes requires caution.
The backlog in medicine registrations
By 2010 the backlog in registrations was estimated to be in excess of 3,000 applications
[12, 13]. Prior to 2005, the number of applications received and registration certificates issued were in equilibrium. From 2005 the number of applications submitted more than doubled whereas the number of certificates issued remained approximately the same, see Figure 1.
The relatively constant rate at which certificates were being issued from 2000 to 2011 was because additional staff were not appointed to meet the high demand for registration of new products. This shortcoming was also identified by the MTT in its report [8]. The increase in certificates issued after 2011 was due to the recruitment of temporary technical and administrative staff in 2009, with funding from the UK Department for International Development (DFID). It takes around 24 to 36 months from the start of a dossier review until a registration certificate is issued; hence, the increase in product approvals was only seen in 2012.
Pro-generics policy and the backlog in approvals
The large increase in submissions since 2005 is due to the introduction of pro-generics policies, which includes the mandatory generics substitution policy and fast track registration policy. The increase includes applications submitted via routine and fast track pathways.
In 1997 the Medicines Act was amended (Act 90 of 1997) with the aim of making medicines more affordable and accessible. The amendments included provisions for, amongst others, a transparent pricing system, parallel importation of innovator medicines from countries where they are sold for less than in South Africa, and generics substitution to promote the use of generic medicines, i.e. the progenerics policy. The legislation was challenged in early 1998 by 41 pharmaceutical companies [14] resulting in a delay in its implementation until 2003 when the legal challenge was withdrawn [15]. Not all of the policies were introduced; the generics substitution policy was implemented in 2003 whereas policies on pricing such as the single exit price (SEP) was introduced in 2004. Other amendments to the Act (pharmacists dispensing fee) were implemented in 2014.
Apart from the generics substitution provision made in 2003, the Department of Health (DoH) also introduced a fast track registration policy at that time, not only for new chemical entities (NCEs) considered essential for national health and which may not be on the EML, but also for all medicines on the EML [16], the majority of which are generics [17]. This contrasts with the fast track policy of the Food and Drug Administration (FDA) in the US, which is restricted to investigational new drugs designed to fill an unmet medical need [18].
Table 1 shows the number of registrations per year by product type, i.e. NCEs, biological and generic medicines and the number of fast track and duplicate registrations. The data were obtained from the Operations and Administration Directorate of the MRA. Between 2007 and 2012, registration of generic medicines outnumbered that of NCEs (more than 17-fold) and biological medicines (more than 60-fold). More generic medicines were registered through expedited review (fast track) than NCEs. Some generics manufacturers submit two or more copies of the same dossier under different tradenames. Each of these applications is for a separate registration certificate for a product and each product is considered independent. The numbers in parentheses indicate the number of such duplicate or multiplicative applications.
The practice of certain companies to register the same product under different trade names reflects both the very low cost to the company of registering medicines in South Africa (less than Euros 3,000 for a generic medicine [19]) and the commercial advantage they stand to gain when they licence or sell one of the duplicates (if they registered the product under two different names) to another company. This practice, known as ‘dossier farming’, is well known in the industry but is under researched and seldom reported in the literature.
The backlog in medicines registration may, thus, have been caused by the large number of generics submissions which followed soon after the implementation of mandatory generics substitution (pharmacists are required by law to dispense the generic drug, unless the patient, or the patient’s doctor, expressly refuses the substitution, or the price of the generic drug is higher than that of the branded product). This was compounded by legislative requirements for the expedited review of medicines on the EML (most of which are generics) and the lack of control over the number of generics applications for the same innovator medicine that can be submitted by a single company. The question that arises is whether the uncontrolled registration of generic medicines can be justified in terms of promoting access to affordable medicines. If a large number of generics in the market continually drive down the cost of medicines through competition, then the MCC should endeavour to acquire the necessary resources to increase its efficiency and shorten review times. However, if there is no price advantage to the public after a certain number of generics of an innovator product becomes available, then it may be necessary to review the current policies governing the registration of generic medicines.
Does the backlog impede the accessibility and availability of affordable medicines?
The pro-generics policy is intended to promote competition; the theory being that as more competitors enter a market, prices of products will fall [20]. Thus, registering a large number of generic brands of a particular medicine should drive down the price of that medicine. The question is whether the current backlog prevents both important medicines from coming onto the market and reduces competition and, hence, availability of affordable medicines.
Using IMS data for each of the tracer medicines we analysed the percentage market share (as measured by annual sales value in both private and public markets) for each branded product (generic and innovator) from 5% or less to more than 40%. The highest market share for a brand for most tracer medicines was between 40% and 50%. The number of registered brands and brands under review for each medicine was also included in the analysis to show the relationship between current (number of registered brands) and future (number of registered brands plus brands under review) product availability for each medicine, see Table 2.
The data show that two to five brands for each medicine account for 80% or more of the market value. Approximately 70% of marketed generics have a market share of less than 5% and only 54% of all registered generic brands are actually being marketed. This suggests that for most of the tracer medicines, with the exception of oxytocin, more than enough branded generics have been registered to ensure robust competition in their markets. Their markets have in fact become oversaturated since more than 40% of the registered products are not being marketed. Exceptions would be medicines, such as lamivudine and rifampicin where changes in treatment guidelines for HIV and tuberculosis (TB) may render some products obsolete. For example, in South Africa the intention is to transition all HIV patients to fixed-dose combinations (FDCs) containing tenofovir, emtricitabine and efavirenz or tenofovir, lamivudine and efavirenz [21]. Other antiretrovirals (ARVs), such as lamivudine, whether available in FDCs with other ARVs or as single agent products, may eventually be phased out. Oxytocin represents a special case since there are only three brands registered with none in the registration pipeline awaiting registration. The local market for oxytocin products is very small, just over US$2.8 million in 2012, and the product is only used in the hospital setting in South Africa. This, coupled with the need for cold chain storage, probably discourages local companies from including oxytocin formulations in their product portfolio. This contention is supported by more than 50 oxytocin formulations for human use that are available in the world market [22]. The limited availability of oxytocin products is thus more due to market failure than competition. The MCC should therefore prioritize registration of oxytocin products, should it receive applications, to ensure greater access.
Rifampicin is another case where the data do not follow that for the other tracer medicines. Changes in the treatment guidelines for TB may have made some FDC products containing rifampicin obsolete, which would account for them not being marketed. This would be further impacted by the development of resistant TB, an issue which is particularly pertinent in South Africa with its high incidence of the disease [11]. New FDC formulation with rifampicin should, thus, also be prioritized for registration by the MCC.
In the US, Reiffen and Ward (2005) used regression analysis to estimate the effect of the number of generics entries on the pre-expiry price of the original branded product for 31 drugs [23]. The authors also used price data obtained from IMS Inc. They found that a negative relationship existed between price and the number of generics entries and that the negative effect of increased competition on prices continues at least until the fifth firm enters the market but is not likely to be important after the eighth firm has entered. In other words, after eight generics, prices are unlikely to decline further since the average generics price will now approach the long-run marginal cost of production. South Africa had a population of 48.8 million in 2012 compared with the US population of 316.4 million [24]. Our market is thus smaller than that of the US and consequently one could expect that five generics would ensure sufficient competition in a market for prices to decline significantly from that of the innovator prior to patent expiry. Our findings appear to support this, since five generics competitors dominated the market for most of our tracer medicines.
The large percentage of generic drug products for some of our tracer medicines, which are currently not being marketed, could be due to market saturation. Thus, for some medicines (those for which there are already many generics in the market) it is likely that the backlog does not impede effective competition or, by extension, access to affordable medicines.
Although there is currently no formal mutual recognition agreement between the MCC in South Africa and the regulatory authorities of countries in the Southern African Development Community (SADC), companies may seek to register their products with the MCC in South Africa in order to obtain faster registration in neighbouring countries that have minimal regulatory capacity. For example, Namibia has, since 2010, granted abbreviated reviews for registration of essential medicines that are registered by Medicines Regulatory Authorities with which the Namibia Medicines Regulatory Council (NMRC) aligns itself, such as the MCC of South Africa, provided that the pharmaceutical
manufacturing site complies with current Good Manufacturing Practices (cGMP) and a complete dossier in terms of the applicable registration application format is submitted. (Gaeseb J, Registrar of Medicines, Namibia Medicines Regulatory Council, Ministry of Health and Social Services, Namibia. Personal communication. 6 November 2014).
The proliferation of pharmaceutical products, particularly in developing country markets, either through importation or local manufacture, places a tremendous burden on small and poorly resourced regulatory agencies. There is, thus, a need to review the legislation underpinning the registration process to include a medical needs clause, so that medicine registration can be prioritized on that basis. Provision should also be made to ensure that there is enough competition in the market to make medicines affordable.
Revisit pro-generics policy in the context of medical need
When suffi cient competition for an offpatent medicine exists in the market, regulatory backlog for registering further generics of that product will not have an appreciable effect on market competition, price or availability, and therefore not on access. It is only when competition is limited or non-existent, that the backlog may have those effects. Thus, the claims of industry that the backlog in registration applications of generic medicines impedes access to affordable medicines are generally not supported by our data. Our findings suggest that the backlog could be the result of government implementing progenerics policies without first providing the MCC and MRA with additional resources to handle the substantial increase in generic medicine registration applications that followed. It is important that these policies be reviewed so that further registration applications for products that are already widely available do not prevent generics of critical medicines from gaining speedy access to the registration system, while ensuring robust competition. It may be necessary to consider introducing a policy that will limit the number of generics for a specific medicine to prevent oversaturation of its market. The finding that a large proportion of generics for certain medicines are not marketed suggests that, in these cases, further generics may not lead to greater access.
The South African Government has committed itself to the establishment of a new medicines regulatory authority. The new authority, SAHPRA, is intended to be better resourced in terms of staff and infrastructure and is, therefore, likely to cost the public purse more to operate than the MCC, even if the fees to the industry will be significantly higher than the fees currently levied by the MCC. The scope of the new authority is to be expanded to include regulation of medical devices and complementary medicines. However, there is concern that if the primary motivation for SAHPRA is to register medicines regardless of need, it will serve the interests of the pharmaceutical industry more than that of the public. The industry will receive the benefi ts of shorter timelines while the burden in the form of tax payers’ funds to support the larger agency will be borne by the public. Another concern is that SAHPRA could expose itself to industry capture should it become dependent on funds sourced from industry to pay for its operational costs. When the funding formula for SAHPRA is thus determined it is essential that the regulator never be placed in a situation in which it might lose its independence, i.e. not to industry or to government [25, 26].
Conclusion
The findings from our analysis of the backlog in registration applications and market dynamics of generic drug products, although limited to only eight medicines, allow us to make a few tentative recommendations. Firstly, the National DoH should review its policy on expedited review, which currently applies to all generic medicines on the EML, and consider developing a medical needs clause to restrict this registration pathway to NCEs, new biologicals or new formulations for critical medicines, e.g. FDCs of ARVs. Fast track reviews for generics should be limited to the first five to eight applications received from different companies prior to expiry of the patent on the original. These should be reviewedconcurrently, if possible, so that they can enter the market more or less at the same time to effect significant price reductions through competition, thereby, making the medicine more affordable. This will link the expedited review pathway for registration not only to medical, but to economic need as well. Clone applications, i.e. a copy or exact duplicate of the originator and marketed by the manufacturer or applicant of the originator medicine but with only the labelling (including the brand name) being different, should be excluded from this provision. This is because the innovator applicant has the advantage of submitting such applications at any time before the patent on its original brand expires and can also market the clone before the expiry date of the originator brand in anticipation of imminent generics entry. Secondly, MCC should consider cancelling the registration of products after two years if they have not been marketed (sunset clause), unless the company can provide adequate justification for retaining registration beyond this period.
This recommendation stems from the observation that a high percentage of generics for certain medicines are not marketed after registration. The finding suggests that companies do not conduct adequate market research before submitting applications for registration to assess the viability of their products. One of the reasons could be the low cost for registering a generic drug in South Africa (US$2,260) compared with countries, such as Canada (US$38,006) and Australia (US$73,900). The resources spent on evaluating an application for a product that is eventually not marketed could have been applied to other regulatory activities, such as the regulation of medical devices, which are currently not controlled by the MCC because of resource constraints. Thirdly, generic medicines companies should be discouraged from submitting several applications for the same medicine under different trade names unless this can be justified from a public health perspective. This is because companies may be trading in registration certificates (dossier farming) at a significant premium to what they spent on obtaining registration with the MCC. Finally, we recommend that the new regulatory agency, SAHPRA, not be placed in a position in which it will become overly reliant on industry fees to fund its operations as this may compromise its independence and lead to industry capture.
Acknowledgements
The authors are grateful to IMS Health Inc for generously providing the market share data and the Medicines Regulatory Affairs for providing data on product registrations and registration applications. The authors would also like to thank Dr Petra Sevcikova, Ms Karen Maigetter, Professor Roger Jeffery and Professor Richard Laing for their valuable comments.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Medicines Control Council of South Africa or one of its committees or working groups.
Funding sources
This paper results from research funded by the European Union Seventh Framework Programme Theme: Health-2009-4.3.2-2 (Grant no. 242262) under the title ‘Accessing Medicines in Africa and South Asia [AMASA]’ (http://ec.europa.eu/research/health/public-health/public-health-andhealth-systems/projects/amasa_en.html). The project team includes partners at the University of Edinburgh (UK), Foundation for Research in Community Health (India), University of Ghent (Belgium), Mbarara University of Science and Technology (Uganda), Makerere University (Uganda), Queen Mary University London (UK), Swiss Tropical and Public Health Institute at the University of Basel (Switzerland) and the University of the Western Cape (South Africa).
Competing interest: The authors declare that they have no competing interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Henry MJ Leng1, PhD
David Sanders1, MRCP
Professor Allyson M Pollock2, MBChB, FFPH, MRCP (Ed), MRCGP
1School of Public Health, University of the Western Cape, Private Bag X17, Bellville, 7535, South Africa
2Centre for Primary Care and Public Health, Queen Mary University of London, London E1 2AB, UK
References
1. Kahn T. Cipla slumps on MCC delays. Business Day Live. 2012 Oct 18 [cited 2015 Apr 10]. Available from: http://www.businessday.co.za/articles/Content.aspx?id=167676
2. Kahn T. Litha forced to shut down cardiac unit. Business Day Live. 2012 Aug 8 [cited 2015 Apr 10]. Available from: http://www.bdlive.co.za/articles/2012/03/20/litha-forced-to-shut-downcardiac-unit
3. Buthelezi L. Drug listing delays laid at door of MCC. Accessed 3 August 2012. Available: http://www.mm3admin.co.za/documents/docmanager/2D5ED792-878C-4371-9575-8281A96BBB26/00031267.pdf
4. Kahn T. Blow to SA drugs sector as red tape snares trials. Business Day Live. 2012 Aug 8 [cited 2015 Apr 10]. Available from: http://www.businessday.co.za/articles/Content.aspx?id=174618
5. Thom A. MCC blocking access to lifesaving meds – HIV. Health-E News. 2010 Mar 4 [cited 2015 Apr 10]. Available from: http://www.healthe.org.za/news/article.php?uid = 20032666
6. Geffen N. Medicines Control Council needs new leaders. Quackdown. 2012 Jul 17 [cited 2015 Apr 10]. Available from: http://www.quackdown.info/article/medicines-control-council-needs-new-leaders/
7. Thom A. Change at the MCC – too little, too late? Health-E News. [cited 2015 Apr 10]. Available from: http://www.health-e.org.za/2010/05/28/changeat-the-mcc-too-little-too-late/
8. Report of the Ministerial Task Team on the restructuring of the Medicines Regulatory Affairs and Medicines Control Council and recommendations for the new Regulatory Authority for Health roducts
of South Africa (2008). Available from: oldgov.gcis.gov.za/documents/download.php?f=81967
9. Gray AL. Our medicines regulatory authority: plans for reform in South Africa. NSP Review. 2012;3. [cited 2015 Apr 10]. Available from: http://www.nspreview.org/2012/10/03/our-medicines- regulatory-authorityplans-for-reform-in-south-africa/
10. Kahn T. SA step closer to new medicines regulator. Business Day Live. 2014 Mar 3 [cited 2015 Apr 10]. Available from: http://www.bdlive.co.za/national/health/2014/03/03/sa-step-closerto-new-medicines-regulator
11. Coovadia H, Jewkes R, Barron P, Sanders D, McIntyre D. The health and health system of South Africa: historical roots of current public health challenges. Lancet. 2009;374(9692):817-34.
12. Thom A. The MCC mess. Health 24. 2010 May 28 [cited 2015 Apr 10]. Available from: http://www.health24.com/Medical/Meds-and-you/Using-medicines/The-MCC-mess-20120721
13. Project Conclusion Report. MCC/MRA backlog task team. 2010 Oct [cited 2015 Apr 10]. Available from: http://www.sarrahsouthafrica.org/LinkClick.aspx?fi leticket=AW7KP7zEHKM%3d&tabid=2339.
14. Department of Health. Defending the Medicines Control Amendment Act. 2 March 2001. Pretoria: Department of Health
15. Berger J. Negotiating the new medicines regulatory framework: some basic facts and observations. The South African Journal of HIV Medicine. 2004;5(2):38-40.
16. Republic of South Africa. Health Department. Medicines Control Council. General information guideline. 2008 [homepage on the Internet]. [cited 2015 Apr 10]. Available from: www.mccza.co.za.
17. Republic of South Africa. Department: Health. National EML [homepage on the Internet]. [cited 2015 Apr 10]. Available from: http://www.health.gov.za/index.php/component/phocadownload/
18. U.S. Food and Drug Administration. Fast track, accelerated approval and priority review. Accelerating availability of new drugs for patients with serious diseases [homepage on the Internet]. cited 2015 Apr 10]. Available from: http://www.fda.gov/For-Patients/Approvals/Fast/default.htm
19. Medicines and Related Substances Act, 1965. Schedules. Fees payable in terms of the Act. Government Gazette. 2012;569(35857).
20. Bagwell K, Lee GM. Number of fi rms and price competition. [cited 2015 Apr 10]. Available from: http://www.stanford.edu/~kbagwell/papers/Bagwell%20 Lee%20s%20021214.pdf
21. Davies NECG. Advice document. Fixed-dose combination for adults accessing antiretroviral therapy. S Afr J HIV Med. 2013;14(1 Suppl):41-3.
22. Drugs.com. Oxytocin [homepage on the Internet] [cited 2015 Apr 10]. Available from: http://www.drugs.com/international/oxytocin.html
23. Reiffen D, Ward MR. Generic drug industry dynamics. The Rev Econ Stats. 2005;87(1):37-49.
24. The world in fi gures: countries. The World in 2012. The Economist: 2012;97-105.
25. Stigler GJ. The theory of economic regulation. The Bell Journal of Economic and Management Science. 1971;2(1):3-21.
26. Abraham J. The pharmaceutical industry as a political player. Lancet. 2002;360(9344):1498-502.
Author for correspondence: Henry MJ Leng, PhD, School of Public Health, University of the Western Cape, Private Bag X17, Bellville, 7130, South Africa
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/pro-generics-policies-and-the-backlog-in-medicines-registration-in-south-africa-implications-for-access-to-essential-and-affordable-medicines.html