|
Abstract: |
Submitted: 7 April 2015; Revised: 22 May 2015; Accepted: 24 May 2015; Published online first: 8 June 2015
The official pharmaceutical market for prescription products is worth around Euros 28 billion, and generic drugs represent Euros 6 billion, accounting for 22% of the market and around 70% of all substitution products registered to the Generics Repertory.
Within patent legislation, the term ‘generic drug’ refers to a copy of a listed brand-name drug, whose patent and patent term restoration have expired, making the drug publically usable by any drug manufacturer.
In France, the first legal definition of a generic drug was given by France’s National Office of Fair Trading (Commission de la Concurrence) on 21 May 1981:
‘a generic drug is defined as any copy of an original drug, whose production and marketing are made possible after the expiration of the original drug patent, hence becoming part of the public domain once the legal protection period comes to term. The term ‘generics’ includes drugs sold under a brand name or an invented name, as well as drugs sold under an internationally common or chemically-descriptive non-proprietary name, which must be accompanied by a brand or manufacturer name.’
It was subsequently necessary to define the content and quality of generic drugs more precisely. The order 96–345 of 24 April 1996 introduced the first technical legal definition of a generic drug [1], which corresponds to the European definition as cited in the minutes of the Executive Council of December 1986.
This definition was later used in 1998 in a judgment of the Court of Justice of the European Communities [2], highlighting the unique property of the qualitative and quantitative composition of active principles as well as of the bioequivalence between the original (brand-name) drug and the generic drug. This same definition was further modified in 2004, following a ruling of the Court of Justice of the European Communities [3], this time highlighting the bioequivalence criterion requirement.
In 2005, another legal ruling [4] permitted any drug containing the same active fraction as the brand-name drug (from a therapeutic standpoint) to benefit from a shortened registration procedure. The excipient can, on the other hand, be a different salt, isomer or ester. By focusing on therapeutic effect rather than on molecular structure, the notion of a generic drug became closer to the notion of therapeutic equivalence; that is, with the same qualitative and quantitative effects. The technical rationale behind generic drugs now focused on in vivo results for quality assessment rather than on galenic form for oral administration. On the basis of these legal rulings and the above logic, the Directive 2004/27/CE adopted the extensive definition of a ‘generic drug’, hence, replacing the vague term ‘similar drug’ [5].
This definition is due to be incorporated into the Public Health Code in the section L.5121–1,5°a) [6]. Within this framework, the sanitary authorities will require additional and sufficient proof of the safety and efficacy of a generic drug if its active principle is not identical in pharmaceutical or salt form to the brand-name drug. In France, a Repertory of Generic Groups [7] has been established to allow for easy identification of generics as substitutes to brand-name drugs.
The 1999 Health Insurance Funding Act introduced the notion of right of substitution for pharmacists, as long as the prescriber has not excluded this possibility. In 2008, the prescription of International Nonproprietary Name (INN) drugs became mandatory for all branded pharmaceutical products [8].
Compared with other high-income countries, e.g. Germany, the UK and the US, France is lagging behind in its introduction of generics to the pharmaceutical market. The first mention of the term ‘generic’ was in 1995 in the Prime Minister’s Health Plan to reduce public health spending:
‘The generic version of a reference branded product is defined as having the same qualitative and quantitative composition in terms of active principles, the same pharmaceutical form and whose bioequivalence with the reference product is demonstrated by relevant studies of bioavailability.’
At that time, pharmaceutical manufacturers considered generic drugs a potential threat to their products and doubted their economic benefit, given the high cost of drugs at the time; the medical profession, with lack of knowledge of health economics, were dismissive of pharmaceutical manufacturers, who they perceived to be rushing into the market to duplicate and exploit the discoveries of others.
Physicians believed that they were still in control of treatment, with the autonomy to choose the molecule, brand and manufacturer of a drug. Pharmacists were strongly opposed to the idea of cheaper drugs, because of the difficulty of stocking larger quantities of drugs, even if greater quantities should be sold, because they would be less profitable. The public, in the meantime, were completely unaware of developments in this area and what was at stake.
The French generics pharmaceutical company Laboratoire Français de Produits Génériques was established in the 1980s but was boycotted by pharmacist unions and by its parent pharmaceutical company Clin-Midy. This boycott of generics was condemned in July 1981 by the French Ministers of Finance, René Monory and after Jacques Delors.
In reality, generic drugs were available before 1995, and were copies of drugs whose patents had expired and which therefore benefited from a simplified registration procedure. Often, those were copies of the original molecule manufactured by the same brand as the original drug once the patent had expired, making them indistinguishable from their brand-name counterparts to both the public and health professionals. These generics were promoted among prescribers and sold under the same brand name by new pharmaceutical companies specially created to sell these generics (at a price lower, about 20%), and not well recognized by major companies at prices slightly lower than the brand-name drug prices. Biogalenique Laboratory was one of these new companies and was controlled secretly by Pierre Fabre Laboratories.
Generic drugs first gained real recognition with the announcement of the Retirement and Social Security plan by Prime Minister Alain Juppé on 15 November 1995.
In 1994, Mr Jean Marmot, magistrate at the audit office, became the first President of the Economic Committee for Healthcare Products, the interdepartmental government body in charge of regulating the prices of reimbursable drugs. A fervent supporter of generics, he inspired the provisions of Prime Minister Juppé’s plan before they were even set out, and drew up and signed agreements with pharmaceutical manufacturers to sell their products at 70% of the cost of brand-name drugs, using the INN in exchange for more favourable conditions for accessing the pharmaceutical market for their innovative products. This gave pharmaceutical manufacturers the freedom to fix the prices of their own new products.
This type of agreement ultimately benefitted the public accounts, but also became popular among innovative pharmaceutical manufacturers, without perturbing their brand image, who initiated acquisitions, began to develop a range of generic drugs, or both, often through affiliated companies dedicated to this task. In 1995, for example, Rhône-Poulenc Rorer acquired Biogalenique, later renamed ‘Rhône-Poulenc Génériques’. Sanofi also established a generics department, which subsequently became Ratiopharm. Although the price-agreement policy was instrumental to the emergence of generics, a fundamental element of the market was missing: demand.
The second act was in 1999, when the Chairman of the pharmaceutical unions federation of France (Fédération des Syndicats Pharmaceutiques de France), Mr Bernard Capdeville began campaigning for the ‘right of substitution’. Despite opposition from physicians, and, especially, general practitioners, he lobbied the medical profession and public authorities to support the principle of equal profit margins between generic and brand-name drugs.
Approval of this new measure ensured by all pharmacist unions reduced the potential economic gain of generics for public authorities. The authorities subsequently backed down admitting that they could not move forward without pharmacist support, and agreed to relax the system of ‘smoothed decreasing profit margin’ established in 1990.
In order to calm the strong medical opposition, public authorities restricted the ‘right of substitution’ to ‘generics groups’; that is, the ensemble composed of a brand-name drug and its generics registered by the Drug Agency. Then, all the generics groups were brought under the umbrella of the ‘Generics Repertory’, a unique reference directory used by western countries, which contains more than 1,000 generics groups relating to 370 molecules [9]. In France alone, a generic drug cannot be substituted unless it has been registered with the Generics Repertory. Therefore, even today, high-volume drugs such as paracetamol or aspirin whose generics have existed for a long time, are excluded from the substitution system under the pretext that a unique identifiable brand-name drug able to constitute a generic drug group no longer exists.
At this time, the strategic choice of betting on pharmacists paid off slowly but surely. The generics market took off but with rates of substitution well under the expected 35%. Physicians and patients still remained reluctant to switch to generics, and exercised their right to access original brand-name drugs, with no penalty.
The third act was in June 2002, when the newly constituted government, with Professor Jean-François Mattei as the new Minister of Health, gave in to the demands of the three main physicians unions (MG France, Syndicats des Médecins Libéraux (SML) and Confédération des Syndicats Médicaux de France (CSMF)) to increase the cost of a general practitioner’s medical consultation at Euros 20. Underpinning this was a focus on generics, with physicians requested to stop their active counter propaganda against generics. At the same time, the public authorities significantly increased the discounts agreed by manufacturers to pharmacists and further decreased the smoothed decreasing profit margin. Now it became more profitable for a pharmacist to sell a generic drug than a brand-name drug. The market finally took off and the expected substitution target of 35% was attained.
The fourth act was when the generic drug system was transferred from the government to the Health Insurance System in 2010 introducing innovative new changes in relation to generic drugs. First, an agreement was reached between the French National Health Insurance Agency and pharmacists to increase the substitution target to 80% for 20 of most prescribed molecules on the market, with advantages for pharmacists.
The second change, and one that had the biggest impact of all, was initially implemented by the Regional Health Insurance Agency of the ‘Alpes Maritimes’ region, during spring 2011. Only patients accepting substitution drugs (generics) were permitted to receive an advance on costs. The idea of a ‘third-party payer in exchange for generics’ was the first time that patients considering using generic drugs could benefit financially. This was also effective at the treasury level for accounting reasons, because if the patients wanted the brand-name drug they had to pay it and were reimbursed only three to four weeks after.
This initiative was quickly rolled out to other territories, covering all of France by June 2012: at this point, the plan included all pharmacists, all medical prescriptions, all medical products, and nearly all patients. The possibility of a ‘non-substitutable’ mention for the prescriber remained open but it had to be handwritten and was only applicable per product and not for the entire prescription.
The fifth act was between 2009 and 2012, when two important developments took place. First the French National Health Insurance Agency intervened to fix the depletion of the drug Repertory resulting from the expiration of drug patents for the most commonly used molecules, making them fall in the public domain.
A shift in prescribing was then observed to-wards products of the same therapeutic class as brand-name drugs but which were not generics. This move away from brand-name drugs, which blocked the substitution system, was a result of the weak promotion of generics in favour of competitors’ ‘non-repertory’ products still patented and actively promoted. This observation led to the decision to include prescribing in the scope of the Generics Repertory in the new performance-related pay system established in 2009 as part of the French general practitioner’s performance-related pay contract, and was presented to physicians willing to sign up.
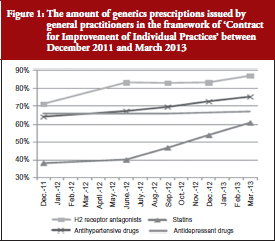
The French National Health Insurance Agency requested general practitioners to meet certain objectives (related to increased prescribing of substitution products for certain classes of commonly used drugs), which, if achieved, would be paid in the form of bonuses. This plan was a great success and was implemented with reinforced objectives, to be later added to the medical agreement under the name ‘Contract for Improvement of Individual Practices’. It was signed by 75,000 physicians and anticipated to make each physician earn up to Euros 5,000 more per year, see Figure 1.
A similar performance-related pay system was implemented for pharmacists through a conventional agreement on 4 May 2012. It included an explicit list of about 30 generic drug groups and set target substitution rates, which varied according to the molecule, and depended on their nature, time spent on the market, and mean age of the patient. Target rates varied between 42% for osteoporosis drugs and 95% for pravastatin. At this stage, the projected goal was a global rate of substitution of 85% by the end of 2012.
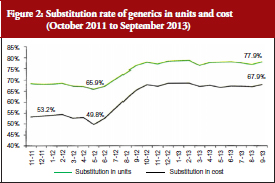
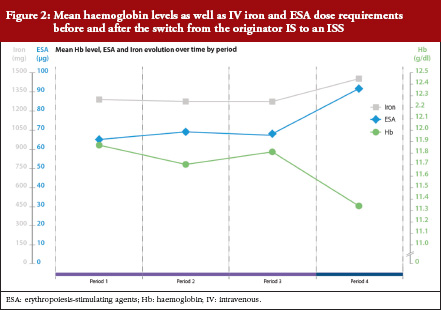
Coupled with the reinforcement of the ‘third-party payer in exchange for generics’ system mentioned above, this performance-related pay system was successful; reaching a 12% higher substitution from June 2012, see Figure 2. The rates stabilized at around 70% in cost and around 80% in units, with no observed decrease.
Finally, the sixth act was a ruling on the Bertrand Law (from 29 December 2011), which came into effect on 1 January 2015: the generalization of INN prescriptions recommended by the World Health Organization. This provision further promoted the substitution movement, as long as prescribers possessed the correct prescription softwares, of which 39 were certified by the French National Authority for Health.
The establishment and acceptance of generic drugs in France has been a protracted and cumbersome process compared with other countries, which have had a better take-up of generic drugs.
In countries with multiple insurance systems, either private (the US) or public (Germany and The Netherlands), the initiative to implement generics has always come from the insurers, with efforts primarily focused on convincing patients to accept prescribed generic drugs. The simplest system, known as the ‘reference price’ system, was introduced in Germany and The Netherlands in the late 1980s. This system took the price of generic drugs as the basis for reimbursing the cost of drugs, regardless of whether the patient chose a brand-name drug or the generic drug. A harsher version of this same system was implemented in the US by Managed Care organizations, and involved only reimbursing the cost of generic drugs listed in the formulary for reimbursement; a patient wishing to purchase a brand-name drug would be personally liable for the cost.
In one way or another, the policies that have succeeded in encouraging take-up of generic drugs by the patients are based on demand (prompted or forced), which have led to an immediate acceptance of a generics offer, later regulated by public authorities for clarification and organization purposes.
In France, with its centrally administered economy and mistrust of the market’s inner workings, a policy of ‘offer’ was initially devised, with a subsequent re-worked policy of ‘demand’ only when the former failed to produce results. The government had initially wanted to protect the patient, and, after the failure of the policy between the government and pharmaceutical manufacturers, the French Health Insurance Agency intervened to stimulate demand by offering financial incentives for health professionals and patients. This two-step process would have been fine, had it not taken 10 years to happen.
One of the reasons for this reverse logic is the persistence of an administrative system to fix the price of generics. Public authorities have precluded any system of competition, including between manufacturers, by fixing administrative prices for generic drugs (same prices for all generics belonging to the same group). In other countries, it is rather the competition between manufacturers that is responsible for price reductions, often greater than those obtained through the French public regulation system. This competition also leads to the emergence of global leaders and, in turn, to the delocalization of production sites in low cost locations.
Opening the generics market up to competition is still being debated today, and it has been loudly suggested that public authorities open the call for tender to entrust some pharmaceuticals with the entire market, e.g. statins, proton pump inhibitors, giving the best guarantees, as was the case in Germany and in The Netherlands, reducing prices drastically. The French Health Insurance fund prefers a system of well-advised buyers by making sure that public funds and pharmacists split the profits, a model similar to the UK system.
Ironically, the story of generics in itself demonstrates the paradox that financial incentives do produce results when the main health actors have little interest for health economic questions. Health professionals only agreed to the objective of efficacy because they were offered direct profits. Also patients, forced to switch to generics, responded positively to the ‘third-party payer in exchange for generics’ plan beyond expectations. Generics have therefore highlighted a contradiction in the French healthcare system, displaying an obvious aversion for economic issues while resorting to financial incentives to stimulate the generics market.
The poor handling of the generics issue, particularly in the context of drug dispensing, has been evident through Internet blogs and social networks, where the public has vented its frustration, often with a misinformed view of the subject, and with access to partial information. Such frustration is easily understandable in view of the obvious disregard for the patient’s best interests during implementation of the first generics policy. During that time, health authorities were focused on imposing generics on the physician community against their will via pharmacist arm-twisting, and patients were feeling left out in terms of financial benefits.
Health is priceless, but has its costs. The proclamation of experts that cheaper treatments are as efficient as costly treatments are not ringing true with French consumers, as they are used to intuitively factoring in price as a quality indicator. The questioning of pharmaceutical quality and similarity of generics has become even more widespread following the publication of a heated report from the Academy of Medicine [10]. Its high-level political, scientific and suspicious economic discourse has left the public with mixed feelings about generics.
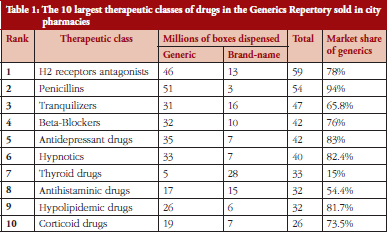
The future of generics in France and in developed countries is rather predictable. Generic drugs are about to become everyday drugs prescribed for almost all common diseases in all therapeutic domains. In most cases of common diseases, except for the thyroid disease, the current prescribed drugs are generics, see Table 1.
This situation is likely to last, at least for the next decade or two, until an important pharmaceutical innovation comes along to change the market share ranking of commonly sold molecules in these therapeutic areas. On the other hand, the flood of new generics is bound to dry up as a result of fewer innovations for commonly sold products in the past 20 years (duration of a patent). Therefore, the generics market seems stable for now and the model may spread to other countries where prices will yield under the pressure of payers either through administrative channels (as in western countries) or through competition.
Pharmaceutical research has long shifted from the big domains of traditional common diseases to the exploration of diseases such as cancers, autoimmune diseases and genetic diseases. It is here that true theoretical breakthroughs are needed, with new technological paradigms, e.g. targeted therapy, gene therapy, cell therapy, and that pharmaceutical manufacturers are not afraid to invest money in these. This active investment in innovative technologies is what shapes the market today. Among the top 20 products in the pharmaceutical market, two-thirds are recent high biotechnology products: monoclonal antibodies, antitumour necrosis factor, recombinant insulin, haematopoietic growth factors, granulocyte colony-stimulating factor, and erythropoietins, in addition to targeted anticancer therapies and, more recently, antiviruses.
The question is will this new costly innovation wave, which started in the 1980s and developed over a 20-year period, generate a new wave of generics when patents start to expire? No, it will not. These big molecules, copies of recombinant proteins with lost patents, are difficult to produce industrially from genetically modified live cells and cannot be compared with generics; either legally, medically or economically. Production conditions are such that, in 2005, the European Medicines Agency requested clinical efficacy trials to be conducted for ‘biosimilar’ drugs [11], contrary to generics, in which only a proof of pharmaceutical bioequivalence is necessary. European regulations state that, under certain circumstances, if the reference biological drug is prescribed in several indications, the biosimilar can be used for these same indications, even if it has been tested for only one of them. These products will create a new market for biosimilars, characterized by very different operational rules and by highly specialized high-tech pharmaceuticals. Biosimilars will not be generics, but their market may, in the coming years, play a role similar to the generics market.
Finally, the subject of small complex chemical molecules has been discussed internationally and also at the European level since 2009. These molecules are difficult to manufacture and, even though they may fit the definition of generic drugs, the generics market approach cannot be applied here. These molecules are commonly referred to as non-biological complex drugs [12]. As with biosimilar drugs, these few molecules are quite complex with iron sucrose (not registered in the Generics Repertory) being the main representative member of this class of active principles [13]. They are complex because the structure of these molecules depends partly on manufacturing conditions and is, therefore, specific to each manufacturer. This type of molecule could benefit from tailored registration regulations with appropriate risk-management plans.
With sustained financial means from society and from the French Health Insurance Agency, the savings generated by the use of generic drugs are ensuring better and cheaper access to costly therapeutic innovations for patients. The implementation of generics must hence be perceived as a sign of therapeutic progress and not as an obstacle. It is, therefore, paramount that health professionals unconditionally support all policies in favour of generics from now on, particularly because physicians are the main conduits of information for patients.
The authors wish to thank the English editing support provided by Ms Maysoon Delahunty, GaBI Journal Editor, for this manuscript.
Competing interest: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Jessica Nasica-Labouze, PhD, Laboratoire de Biochimie Théorique, IBPC, CNRS, Paris, France & International School for Advanced Studies (SISSA), Trieste, Italy
References
1. Journal Officiel de la République Française du 25 avril 1996 N° 98 page 6311-l’article 23 établit une définition des médicaments génériques et dispose que la publicité qui leur est relative mentionne leur nature de spécialités génériques.
2. CJCE, 3 décembre 1998, AFF, C368-396, Generics et al.
3. CJCE, 29 avril 2004, AFF, C-106/01, Novartis.
4. CJCE, 20 janvier 2005, AFF, C-74/03, Smithkline Beecham.
5. Médicaments génériques et droit de la concurrence, Evgéniya Petrova. Thèse de doctorat en droit des affaires- présentée et soutenue publiquement le 17 juillet 2009 – Université Jean Moulin Lyon 3 – école doctorale de droit. Available from: http://www.acadpharm.org/dos_public/RAPPORT_GEnEriques_VF_2012.12.21.pdf
6. Journal Officiel de la République Française du 27 février 2007 – Loi 2007-248 du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament.
7. Article L5121-10 et R.5121-8 du Code de la Santé Publique. Available from:
http://ansm.sante.fr/var/ansm_site/storage/original/application/52e5390a6f9e8580d73bca744a516503.pdf
8. Journal Officiel de la République Française du 26 décembre 2001- Loi n° 2001-1246 du 21 décembre 2001, art. 19-1 (article L5125-23 du Code de Santé Publique).
9. Agence nationale de sécurité du médicament et des produits de santé. Répertoire des Groupes Génériques. 10 avril 2013.
10. Menkes JC. Place des génériques dans la prescription. Académie nationale de Medicine. février 2012.
11. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005. EMEA. 2006 [homepage on the Internet]. [cited 2015 May 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
12. Schellekens H, Klinger E, Mühlebach S, Brin JF, Storm G, Crommelin DJ. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
13. Rottembourg J, Kadri A, Leonard E, Dansaert A, Lafuma A. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10):3262-7.
|
Author for correspondence: Professor Jacques Rottembourg, MD, Department of Nephrology, Hôpital de la Pitié, 83 Boulevard de l’Hôpital, FR-75013 Paris, France |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/the-implementation-of-generics-in-france.html
Author byline as per print journal: Professor Jacques Rottembourg, MD; Corinne Emery, MSc; Alessandra Moglia, PhD
|
Study objective: To describe and compare the haematological parameters and the anaemia medication use in haemodialysis patients who were switched from the originator iron sucrose (IS) to an iron sucrose similar (ISS). |
Submitted: 13 January 2014; Revised: 20 April 2014; Accepted: 21 April 2014; Published online first: 5 May 2014
Non-biological complex drugs (NBCDs) are a new category of molecule, distinct from complex biological molecules such as erythropoietin-stimulating agents and conventional drugs such as ibuprofen, tamoxifen. Difficult to fully characterize, NBCDs require an elaborate manufacturing process and, in contrast to small molecule drugs, cannot be considered therapeutically equivalent on the basis of pharmaceutical and pharmacokinetic criteria alone [1, 2].
Iron carbohydrate preparations are one important class of NBCDs. Such preparations are used to treat iron deficiency anaemia and iron deficiency in a variety of chronic clinical conditions [3–7], either as monotherapy or in combination with erythropoiesis-stimulating agents (ESAs) in order to reduce the required doses and mitigate the potential toxicity of ESA therapy [8, 9]. Since free ferric iron is inherently toxic, IV iron preparations take the form of iron carbohydrate complexes in which an iron-containing mineral core is surrounded by a carbohydrate ligand for stability [10]. Differences in the size and structure of the core and the carbohydrate shell profoundly influence the pharmacological and biological properties of the complex [10].
Iron sucrose (IS), first introduced in 1949 [11], is used widely to treat iron deficiency in a variety of medical conditions and results in prompt iron utilization by erythrocytes [12]. IS contains no dextran or dextran derivatives so cannot induce dextran-induced anaphylactic reactions [13]. It is generally considered to have a good safety profile, as documented in clinical studies and post-marketing analyses [14–16]. This can be attributed to the stable structure of IS, which consists of a ferric oxyhydroxide iron core complexed with sucrose in water, with a molecular weight of 34,000–60,000 Da [16]. The degree of stability of iron carbohydrate complexes is critical, since weakly bound iron may dissociate from the complex and catalyse the generation of reactive oxygen species [17, 19, 20]. Stability is highly dependent on the manufacturing process [17].
The fact that iron therapy is increasingly being recognized as an important component of treatment for iron deficiency and iron-deficiency anaemia in a variety of clinical conditions [18] has led to the development and introduction of new products (ferric carboxymaltose, ferumoxytol, isomaltoside 1,000) as well as a number of ‘iron sucrose similar’ (ISS) preparations.
ISS have been launched into the market under the assumption that they are identical to the originator IS. Generic drugs represent an important part of the drug armamentarium and the introduction of safe and effective generic formulation is welcome from an economic perspective and, occasionally, from a clinical standpoint if the generic drug shows a better safety and efficacy profile than the originator. However, replicating the physico-chemical identity of IS is challenging and achieving therapeutic interchangeability with the originator is even more difficult. Proof of pharmaceutical equivalence for ISS on the basis of comparable physico-chemical properties, as described in the United States Pharmacopeia (USP) [21], appears insufficient for NBCD drug classes since they cannot be fully characterized from a physic-chemical standpoint [1, 2]. Moreover, bioequivalence is difficult to demonstrate using IV preparations, a problem compounded with colloidal solutions such as iron carbohydrate.
It has been proposed that serum iron concentration can prove bioequivalence for ISS preparations versus IS. However, serum iron is uninformative since it is not related to haematopoietic efficacy [20] and more sophisticated monitoring of therapeutic effect is required. In the case of iron preparations, the true bioavailability of iron can only be assessed accurately by measuring iron incorporation into red blood cells, as undertaken previously for the originator IS [12], iron dextran [22], and ferric carboxymaltose [23], using ferrokinetic studies. An alternative approach could be to perform a thorough clinical evaluation to assess the therapeutic equivalence of ISS preparations. However, both ferrokinetic and clinical studies are almost entirely lacking for ISSs.
Even more alarming is the virtual absence of safety assessments, a cause of potentially serious concern in view of the risk of toxicity and the fragile clinical status of many patients requiring long-term IV iron therapy. A recent safety analysis showed the reporting rate of adverse events with ISSs to be nil, which is virtually impossible given the widespread, multinational use of ISS preparations [24] as well as the fact that several studies have demonstrated evidence for increased adverse events with the substitution of an ISS in clinical practice [25–27].
We have also previously reported short-term findings from an observational, single-cohort study demonstrating that switch from the originator IS to an ISS preparation led to destabilization of a well-controlled population of haemodialysis patients in terms of haemoglobin (Hb) levels and iron status [28]. However, a number of factors influence the haemoglobin levels in chronic kidney disease patients over time, including the modality of dialysis, iron status, co-morbidities and chronic inflammation [29] such that almost all haemodialysis patients exhibit endogenous fluctuation in Hb levels [30, 31].
We have therefore undertaken a two-year analysis to determine whether the Hb and iron status of the haemodialysis patients remained stable over the long term prior to the switch to ISS.
Period of the study
A long-term (108-week) observational, non-interventional, single-cohort study was undertaken at the Centre Suzanne Levy, Groupe Diaverum, Paris, France. The aim of the study was to compare anaemia-related haematological parameters and anaemia medication doses and costs in haemodialysis patients with iron deficiency anaemia before and after conversion from IS to an ISS as IV iron therapy at the centre.
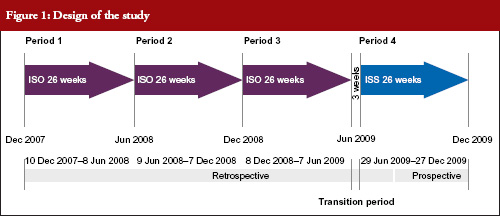
The study compared four time periods of 26 weeks each: Period 1, 19 November 2007 to 25 May 2008; Period 2, 26 May 2008 to 30 November 2008; Period 3, 1 December 2008 to 7 June 2009 and Period 4, 29 June 2009 to 3 January 2010. During Periods 1, 2 and 3, patients received IS. During Period 4, patients received an ISS, see Figure 1. Between 8 June 2009 to 28 June 2009 both IS and ISS preparations were simultaneously available at the centre so this period was excluded from analyses, as it was not possible to determine which preparation had been administered to each patient. Each of the above periods was six months in duration to avoid any differences that may have been due to seasonal practices or behaviours.
No other changes to medical management occurred during any of the study periods other than an adjustment of the Hb target level at the end of 2007 following publication of new evidence [32, 33], and international recommendations suggesting a lower target Hb [34].
Iron and erythropoiesis stimulating agent (ESA) administration
IS (Venofer®, Vifor (International) Ltd, St Gallen, Switzerland; 5 mL ampoules with 100 mg iron) or ISS (Fer Mylan®, ISS2, Mylan SAS, Saint Priest, France, manufactured by Help SA Pharmaceuticals, Athens, Greece; 5 mL ampoules with 100 mg iron) were injected IV once a week at a dose of 25–100 mg iron, adapted to iron parameters. Both IV iron preparations were diluted with saline solution (0.9%) up to 20 mL volume and infused over a one-hour period, between the second and the third hour of the dialysis session. The IV iron dose was titrated according to the most recent values of transferrin saturation (TSAT) and serum ferritin, targeting a TSAT level of 40–60% and a serum ferritin concentration of 500–800 μg/L [35].
The ESA darbepoetin (Aranesp®, Amgen, Neuilly-sur-Seine Cedex, France) was injected IV once every two weeks titrated according to the previous 3–4 Hb values and taking into account any surgical or clinical event [36].
The target Hb range for all patients at the centre was 11.5–12.0 g/dL. Medical management of the patients did not change during the study except for the switch from IS to ISS. Data to September 2009 were obtained retrospectively; subsequent data were collected prospectively.
Methods
The study population comprised all haemodialysis patients who had undergone at least 280 dialysis sessions in the unit during the study period and received at least one dose of IV iron. Measurement of Hb, serum calcium, serum phosphorus, haematology and urea (prior to dialysis) were obtained every two weeks. TSAT, serum ferritin, alkaline phosphatase, parathyroid hormone (PTH), 25-OH vitamin D, albumin, urea (before and after the haemodialysis session), adequacy of dialysis (Kt/V), C-reactive protein (CRP), fibrinogen, liver enzymes (asparamate aminotransferase [AST], alanine aminotransferase [ALT], gamma-glutamyl transpeptidase [γ-GT] and total bilirubin) were measured every three months.
Routine data collection included demographics, primary cause of end-stage renal disease, number of dialysis sessions, and consumption of IV iron and darbepoeitin. All adverse events were reported according to applicable regulations and adverse events resulting in hospitalization were recorded.
Statistical methods
Mean values for Hb, TSAT and serum ferritin, and consumption of IV iron and darbepoetin, were compared using analysis of variance (ANOVA): (a) across the four treatment periods, and (b) during Period 3 (IS) versus Period 4 (IS). Paired data were also analysed using the Wilcoxon test for continuous data, and the McNemar test (Χ2) for qualitative data.
The cost analysis took the approach of an anaemia drug budget impact for a third-party healthcare provider (French Sickness Funds). The costs of IV iron medications and darbepoetin were compared during IS administration and ISS administration. The unit cost of IV iron (public price) was Euros 12.98/ampoule (100 mg iron) for IS and Euros 10.20/ ampoule (100 mg iron) for ISS (Fer Mylan®). The cost of darbepoetin-α at the centre was Euros 1.638/μg. Recently, drug costs changed to Euros 10.00/ampoule (100 mg iron) for IS, Euros 8.00/ampoule (100 mg iron) for ISS (Fer Mylan®) and Euros 1.36/μg for darbepoetin-α. As a sensitivity analysis the cost assessments were repeated using the new values.
All statistical analyses except the mixed-effects model used SAS V9.2 software (SAS Software Inc, Cary, NC, USA).
Sixty-six patients were eligible for inclusion in the analysis and 45 (68.2%) were male. The mean age was 60 ± 15 years and the mean duration of dialysis at the start of the analysis was 62 ± 39 months. The primary causes of end-stage renal failure were diabetes (n = 22, 33.3%), glomerulonephritis (n = 15, 22.7%), hypertension (n = 16, 24.2%) and other nephropathies (n = 13, 19.7%). The mean number of dialysis sessions was 73 ± 9 sessions per patient in Periods 1, 2, 3 and 4. The mean number of Hb values recorded per patient was 50.7 (range 39–55), with a total number of 813, 828, 888 and 818 values obtained during Periods 1, 2, 3 and 4, respectively.
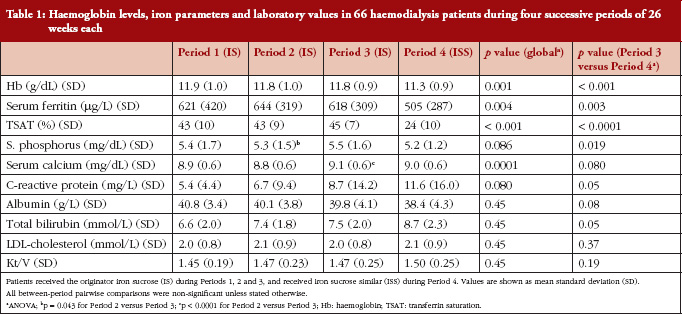
Mean Hb concentration during Periods 1, 2 and 3, when IS was administered, was 11.9 ± 1.1, 11.7 ± 0.9 and 11.8 ± 0.9 g/dL, respectively. This decreased to 11.3 ± 0.9 g/dL during Period 4 (ISS treatment, p < 0.0001 versus Period 3 [ANOVA]), see Table 1 and Figure 2). The mixed-effects model showed that Hb remained relatively stable during Periods 1 to 3, with only minor variations that coincided with introduction of new ESA dosing guidelines at the end of 2007. During the ISS period, the model showed a mean decrease in Hb values of 0.45 g/dL (95%CI -0.66 to -0.24, p < 0.0001).
Levels of serum ferritin and TSAT were stable during Periods 1 to 3, see Table 1 and Figure 2). During Periods 3 and 4, the mean concentration of serum ferritin was 618 ± 308 μg/L and 504 ± 286 μg/L, respectively (p = 0.003); corresponding values for TSAT were 45 ± 7% and 24 ± 10% (p < 0.0001). There were no significant differences between Periods 1, 2 and 3 in terms of Hb concentration, serum ferritin or TSAT.
Serum concentrations of phosphorus and calcium varied across the four study periods, see Table 1. No significant differences across Periods 1 to 4, or between Period 3 and Period 4 were observed for PTH, albumin, fibrinogen, γ-GT, alkaline phosphatase, ALT, AST, leucocytes, platelets, neutrophils or Kt/V. There was a significant increase between P1–P3 to P4 on 2 values, CRP and total bilirubin, indicating both the role of the oxidative stress due to the change from IS to ISS, see Table1.
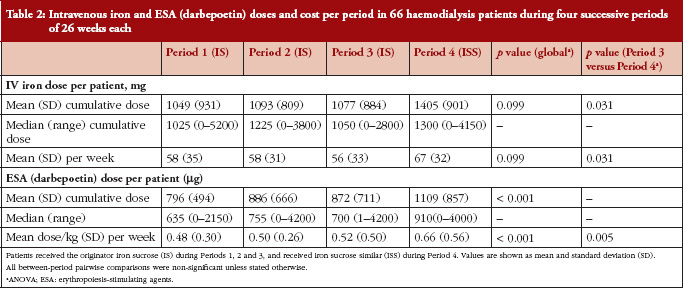
Intravenous iron therapy was received by all patients during each of the four treatment periods other than seven, four, ten and two patients in Periods 1, 2, 3 and 4, respectively. Figure 2 and Table 2 summarize the mean doses of IV iron and ESA administered during the four treatment periods. Doses of both therapies were stable during IS treatment, with no significant differences between Periods 1, 2 and 3, see Table 2. Values for mean IV iron dose per patient increased significantly from Period 3 (56 ± 33 mg/week) to Period 4 (67 ± 32 mg/week), an increase of 21.1% (p = 0.031). The mean ESA dose per patient also increased from Period 3 to Period 4 (0.52 ± 0.50 μg/kg/week to 0.66 ± 0.56 μg/kg/week) by 26.9% (p = 0.005), see Table 2. This increase in anaemia medication use led to a 24.7% increase in the mean cost of medication per patient per period from Period 3 (Euros 1,422) to Period 4 (Euros 1,773). Using updated prices, the sensitivity analysis showed a similar increase in anaemia medication cost from Period 3 to Period 4 (27.3%).
During the four treatment periods there were no adverse events considered by the investigators to be related to the study drugs. There were no differences in the frequency or duration of hospitalization either across all four study periods or between Periods 3 and 4.
Anaemia is a common co-morbidity in chronic kidney disease [37] resulting from reduced erythropoietin production by the impaired kidney and iron deficiency secondary to blood loss and uremic status. Virtually all patients on dialysis require iron supplementation, and administration of an effective iron preparation is essential. Given the therapeutic importance of effectively controlling iron-deficiency anaemia [19] and the risk of oxidative stress and hypersensitivity reactions [38] the IV iron therapy must be selected carefully, particularly since it is typically injected into patients with severe chronic disease over the long term.
There is evidence from these clinical studies that ISS preparations are not equivalent to IS in either setting [25–27]. In other areas of medicine, concerns have already been expressed about the risks associated with switching to non-originator compounds in the absence of adequate clinical testing [39, 40]. Moreover, in the current analysis, conversion to ISS resulted in a substantial increase in the total cost of anaemia medication (+ 27.3%) due to requirement for higher doses of both IV iron (+ 30.3%) and ESA therapy (+ 27.1%), thus negating the rationale for switch.
In this population of stable haemodialysis patients, switch to an ISS, was associated with a significant reduction in Hb level and reduced iron indices. This deterioration was observed despite an increase in both IV iron and ESA dosing. Variations in complex structure and stability are likely to have accounted for the differences in Hb control, since the kinetics of iron dissociation influence the pattern of iron release, distribution and storage [18]. In this population of iron-deficient individuals, TSAT values decreased dramatically after the switch, indicating that less iron was available for erythropoiesis. This may signify that iron released from the ISS had been sequestered by other compartments of the body such as the liver, consistent with the more extensive iron deposits observed in liver tissues within the ISS groups of the experimental studies, published by Toblli [41–43]. The increased levels of liver enzymes recorded in the animal model were not mirrored by evidence of hepatotoxicity in the dialysis population because doses were far lower in the clinical study, but such an effect cannot be ruled out during long-term ISS therapy in dialysis patients. It has previously been observed that the administration of IV iron carbohydrate complexes with low stability, such as sodium ferric gluconate, can result in severe and extended parenchymal liver necrosis secondary to iron-induced lipid peroxidation in non-clinical models [18] and the high level of total bilirubin and low TSAT seen in the current clinical trial are consistent with some degree of hepatic toxicity and less stable molecular structures in the ISS preparations [41]. The significant increase in serum iron and TSAT described in the experimental study from Toblli [42, 43], coupled with greater iron deposition, indicates more rapid release of iron compared to IS due to overloading of serum transport proteins.
Findings from the animal model described by Toblli [42, 43] were consistent with the observed clinical outcomes and may also sign post long-term toxicity risks. The examination of haemodynamic, biochemical and oxidative/nitrosative stress parameters in the non-clinical model demonstrated significant differences between the four European ISS complexes versus both the control group and the originator IS. IS, in contrast, exhibited a toxicity profile comparable to controls. Furthermore, significant differences were observed between the ISS preparations for most biochemical, oxidative stress and nitrosative stress parameters, with ISS1 and ISS2 showing higher levels of toxicity than ISS3 or ISS4. Under normal circumstances, iron in the body remains protein-bound until required for metabolic processes, to avoid tissues becoming exposed to the free cation. Free iron in the cytosol can react with oxidatively susceptible biomolecules, catalysing the formation of reactive oxygen species [44] with potentially damaging effects [45–47]. These variations may at least partly explain the differences observed in the current clinical and non-clinical studies.
In conclusion, these clinical studies point to important variations between ISS complexes and IS and raise doubts about the interchangeability of IS and ISS preparations. Moreover, the requirement for higher dose of both IV iron and ESA not only reversed the expected cost benefit for conversion from IS but also can raise concerns about the need for higher doses of medication. In both cases, this is worrying given the current emphasis on minimizing ESA dosing as well as the preclinical data suggesting damage to liver, heart and kidney with the use of an ISS. Hence, clinical evidence on safety and therapeutic benefit should be established before use (or approval) of iron similar compounds.
Data were recorded by Hemodial (PHP development). The authors wish to thank Dr Antoine Lafuma for contributing to the statistical analysis of the observational study.
Disclosure of financial and competing interests: Vifor (International) Ltd financially supported the independent statistical analysis of the long-term observational study by CEMKA/EVAL. Vifor Pharma Ltd did not contribute to the study design. Professor Jacques Rottembourg has received a consultancy fee from Vifor Pharma Ltd following the conclusion of this study.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor Jacques Rottembourg1, MD
Corinne Emery2, MSc
Alessandra Moglia3, PhD
1Hemodialysis Unit, Groupe Diaverum, Paris, France
2CEMKA/EVAL, Bourg-la-Reine, France
3Vifor Pharma Ltd, Glattbrugg, Switzerland
References
1. Schellekens H, Klinger E, Mühlebach S, et al. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
2. Schellekens H, Stegemann S, Weinstein V, de Vlieger JS, Flühmann B, et al. How to regulate nonbiological complex drugs (NBCD) and their follow-on version: points to consider. The AAPS Journal. 2014;16:15-21.
3. Fishbane S, Pollack S, Feldman HI, Joffe MM. Iron indices in chronic kidney disease in the National Health and Nutritional Examination Survey 1988-2004. Clin J Am Soc Nephrol. 2009;4(1):57-61.
4. Valderrábano F, Hörl WH, Macdougall IC, et al. PRE-dialysis survey on anaemia management. Nephrol Dial Transplant. 2003;18(1):89-100.
5. Jankowska EA, Rozentryt P, Witkowska A, et al. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J. 2010;31(15):1872-80.
6. Wilson A, Reyes E, Ofman J. Prevalence and outcomes of anemia in inflammatory bowel disease: a systematic review of the literature. Am J Med. 2004;116(Suppl 7A): 44S-49S.
7. Knight K, Wade S, Balducci L. Prevalence and outcomes of anemia in cancer: a systematic review of the literature. Am J Med. 2004;116 Suppl 7A:11S-26S.
8. Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med. 2010;363(12):1146-55.
9. Pfeffer MA, Burdmann EA, Chen C-Y, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361(21):2019-32.
10. Danielson BG. Structure, chemistry, and pharmacokinetics of intravenous iron agents. J Am Soc Nephrol. 2004;15 Suppl 2:S93-8.
11. Paschen HW. Efficient anaemia treatment with large intravenous iron doses. Geburtshilfe Fauenheilkunde. 1949;9:604-16.
12. Beshara S, Lundqvist H, Sundin J et al. Pharmacokinetics and red cell utilization of iron(III) hydroxide-sucrose complex in anaemic patients: a study using positron emission tomography. Br J Haematol. 1999;104(2):296-302.
13. Gasche C, Berstad A, Befrits R, et al. Guidelines on the diagnosis and management of iron deficiency and anemia in inflammatory bowel diseases. Inflamm Bowel Dis. 2007;13(12):1545-53.
14. Locatelli F, Aljama P, Bárány P, et al. Best Practice Guidelines Working Group. European Best Practice Guidelines (EBPG). Nephrol Dial Transplant. 2004;19 Suppl 2:ii1-47.
15. Bailie GR, Clark JA, Lane CE, Lane PL. Hypersensitivity reactions and deaths associated with intravenous iron preparations. Nephrol Dial Transplant. 2005;20(7):1443-9.
16. Yee J, Besarab A. Iron sucrose: the oldest iron therapy becomes new. Am J Kidney Dis. 2002;40(6):1111-21.
17. Geisser P, Baer M, Schaub E. Structure/histotoxicity relationship of parenteral iron preparations. Arzneimittelforschung. 1992;42(12):1439-52.
18. Stewart T, Freeman J, Stewart J, et al. Anaemia in heart failure: a prospective evaluation of clinical outcome in a community population. Heart Lung Circ. 2010;19:703-5.
19. Qunibi WY, et al. The efficacy and safety of current intravenous iron preparations for the management of iron-deficiency anaemia: a review. Arzneimittelforschung. 2010;60(6a):399-412.
20. Geisser P, Burckhardt S. The pharmacokinetics and pharmacodynamics of iron preparations. Pharmaceutics. 2011;3(1):12-33.
21. U.S. Pharmacopeial Convention [homepage on the Internet]. [cited 2014 Apr 20]. Available from: http://www.usp.org
22. Speyer BE, Doney VJ, Fielding J. Transfer of iron from Ferastral and other organic complexes to transferrin as measured by reticulocyte uptake. Scand J Haematol Suppl. 1977;32:215-21.
23. Beshara S, Sörensenj, Lubberink M, et al. Pharmacokinetics and red cell utilization of 52/Fe/59/Fe-labelled iron polymaltose in anaemic patients using positron emission tomography. Br J Haematol. 2003;120(5):853-9.
24. Bailie GR, Hörl WH, Verhoef JJ. Differences in spontaneously reported hypersensitivity and serious adverse events for intravenous iron preparations: comparison of Europe and North America. Arzneimittelforschung. 2011;61(5):265-75.
25. Stein J, Dignass A, Chow AK. Clinical case reports raise doubts about the therapeutic equivalence of an iron sucrose similar preparation compared with iron sucrose originator. Curr Med Res Opin. 2012;28(2):241-3.
26. Martin-Malo A, Merino A, Carracedo J, Alvarez-Lara MA, Ojeda R, Soriano S, et al. Effect of intravenous iron on mononuclear cells during haemodialysis session. Nephrol Dial Transplant. 2012;27(6):2465-71.
27. Lee ES, Park BR, Kim JS, Choi GY, Lee JJ, Lee IS. Comparison of adverse event profile of intravenous iron sucrose and iron sucrose similar in postpartum and gynaecologic operative patients. Curr Med Res Opin. 2013;29(2):141-7.
28. Rottembourg J, Kadri A, Leonard E, et al. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10):3262-7.
29. Richardson D. Clinical factors influencing sensitivity and response to epoetin. Nephrol Dial Transplant. 2002;17 Suppl 1:53-9.
30. Ebben JP, Gilbertson DT, Roley RN, Collins AJ. Hemoglobin level variability: associations with comorbidity, intercurrent events, and hospitalizations. Clin J Am Soc Nephrol. 2006;1(6):1205-10.
31. Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int. 2005;68(3):1337-43.
32. Drueke TB, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006;335(20):2071-84.
33. Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355(20):2085-98.
34. KDOQI. KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50(3):471-530.
35. Gaweda AE, Bhat P, Maglinte GA, Chang CL, Hill J, et al. TSAT is a better predictor than ferritin of hemoglobinresponse to Epoetin alfa in US dialysis patients. Hemodial Int. 2014;18(1):38-46.
36. Rottembourg JB, Dansaert A. Feasibility strategy of darbepoetin alfa administration every other week: 2005-2007 experience in a dialysis unit. Nephrol Ther. 2011;7(7):549-57. [French].
37. McClellan W, Aronoff SL, Bolton WK, et al. The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin. 2004;20(9):1501-10.
38. Bailie GR. Breaking new ground in intravenous iron therapy. Eur Haematol Touch Briefings. 2008;2(1):58-60.
39. Pitt B, Julius S. Easy money? Health cost savings resulting from the switch from a branded drug to a low-cost generic drug in the same class. Int J Clin Pract. 2011;65(3):242-4.
40. Johnston A. Challenges of therapeutic substitution of drugs for economic reasons: focus on CVD prevention. Curr Med Res Opin. 2010;26(4):871-8.
41. Toblli JE, Cao G, Oliveri L, Angerosa M. Comparison of the renal, cardiovascular and hepatic toxicity data of original intravenous iron compounds. Nephrol Dial Transplant. 2010;25(11):3631-40.
42. Toblli JE, Cao G, Oliveri L, Angerosa M. Differences between original intravenous iron sucrose and iron sucrose similar preparations. Arzneimittelforschung. 2009;59(4):176-90.
43. Toblli JE, Cao G, Oliveri L, Angerosa M. Differences between the original iron sucrose complex Venofer® and the iron sucrose similar Generis®, and potential implications. Port J Nephrol Hypert. 2009;23:53-63.
44. Evans RW, Rafique R, Zarea A, et al. Nature of non-transferrin-bound iron: studies on iron citrate complexes and the thalassemic sera. J Biol Inorg Chem. 2008;13(1):57-74.
45. Geisser P. Safety and Efficacy of Iron(III)-hydroxyde polymaltose complex/ a review of over 25 years experience. Drug Res. 2007;57:439-52.
46. Devaki PB, Chandra RK, Geisser P. Effect of oral supplementation with iron(III)-hydroxide polymaltose complex on the immunological profile of adolescents with varying iron status. Arzneimittelforschung. 2007;57(6A):417-25.
47. Crichton RR, Danielson BG, Geisser P. Iron therapy with special emphasis on intravenous administration. 4th ed. Bremen, Germany: UNI-MED Verlag AG; 2008.
|
Author for correspondence: Professor Jacques Rottembourg, MD, Department of Nephrology, Groupe Hospitalier Pitié-Salpêtrière, 83 Boulevard de l’Hôpital, FR-75013 Paris, France |
Copyright © 2014 Pro Pharma Communications InternationalDisclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/retrospective-chart-review-disrupted-anaemia-control-in-haemodialysis-patients-following-the-switch-to-an-iron-sucrose-similar-iss-after-long-term-treatment-with-the-originator-iron-sucrose-is.html
Copyright ©2024 GaBI Journal unless otherwise noted.