Author byline as per print journal: Johanna Andrea García Cortes, MSc; Francisco Javier Sierra Esteban, MSc
|
Abstract: |
Submitted: 15 November 2017; Revised: 8 March 2018; Accepted: 8 March 2018; Published online first: 21 March 2018
Similar Biotherapeutic Products (SBPs or biosimilars) are a new class of biotherapeutic agent that are becoming increasingly developed and available worldwide. Since 2010, Colombia’s National Food and Drug Surveillance Institute (Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA) has been developing the regulatory framework for biotherapeutics approval and introduction. In January 2013, the Colombian Ministry of Health and Social Protection (Ministerio de Salud y Protección Social de Colombia) released a new law for biotherapeutics, including biosimilar products [1]. This was finalized in September 2014 when Decree 1782 was signed by the country’s Health Minister [2]. Colombia is now ready to implement this new legislation and increase the availability of biotherapeutics, including biosimilar products, which will improve the access to treatment across the country.
At the Second Colombian Scientific Meeting on Quality Assessment of Biosimilars/Similar Biotherapeutic Products, which took place on 15 August 2017 [3], Bogotá, Colombia, Ms Johanna Andrea Garcia Cortes, Professional Specialist of Medicines and Biological Products at INVIMA, outlined the current landscape for biological/biosimilar introduction in Colombia. This is summarized below.
In the old system, for a pharmaceutical product to be approved in Colombia it must first undergo a pharmacological assessment. Here, the product will be assessed with respect to safety and efficacy. Following this, the laboratory in charge of initial assessment can submit the product file for legal, quality and process assessment. After this process, INVIMA laboratories will oversee the quality control of products. There will be laboratory testing in order to enable batch-by-batch release of vaccine, haemoderivatives and antivenom that come to the market. In summary, under the previous legislation, approval process must be carried out sequentially and cannot be done simultaneously.
However, over the last seven years regulators and experts in Colombia have worked together to create a new process for the approval of biotherapeutics that will not compromise on safety and efficacy of products, but will improve access to therapy.
The new regulatory framework for biotherapeutics approval in Colombia will also start with a pharmacological assessment. This is divided into four assessment groups that simultaneously assess the case file for: safety and efficacy, quality and process, pharmacovigilance, and quality control. The groups will then meet to review and discuss the case file. Subsequently, there will be another official meeting with INVIMA’s specialized medications chamber and a decision will be made on the product. If approved, a market authorization application must be made before the product can reach the market.
This process is a more efficient way of connecting the pharmacological assessment groups and is the chosen method by which Decree 1782 (2014) will be implemented.
With regards to specific regulations set out by the decree, there are three pathways for the submission of biotherapeutics products: the complete dossier approach, the comparability approach, and the abbreviated comparability approach.
The three pathways have common elements including: manufacturing process and production place, expression system, biological identity, potency and purity, physiochemical properties, biological activities, and immunogenicity.
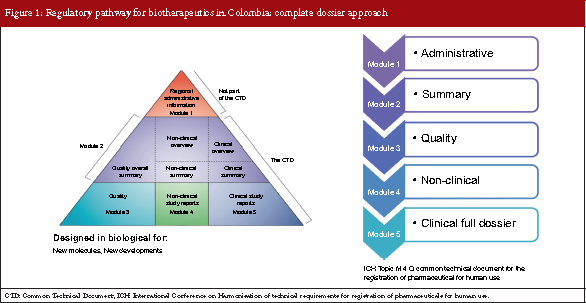
The complete dossier approach has been in place for over 20 years in Colombia and internationally, and is designed for the approval of new molecules and new process developments. There are five modules to the evaluation: 1. Administrative; 2. Summary; 3. Quality; 4. Non-clinical; and 5. Clinical full dossier, see Figure 1. This is called the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Topic M 4 Q, Common Technical Document for the Registration of Pharmaceutical for Human Use, and if all modules are carried out the complete dossier is achieved.
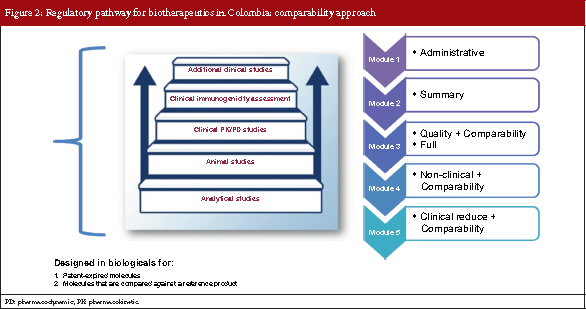
Comparability is based on analytical characterization/studies. It is a step-by-step process of clinical and non-clinical comparative studies designed for molecules with expired patents where molecules are compared against a reference product.
As for the comparability dossier approach, there are five modules. However, modules 3–5 include comparability exercises, take into account the clinical assessment required in module 5 is reduced, see Figure 2.
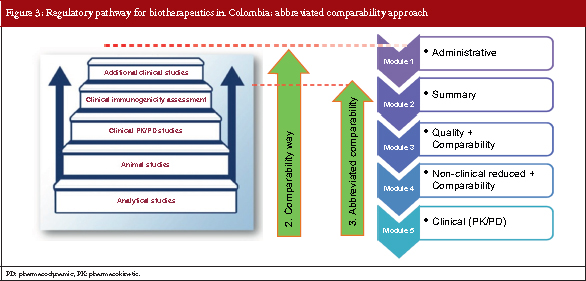
Based on the step-wise concept, the European Medicines Agency and the US Food and Drug Administration’s biosimilar guidelines recommend that clinical confirmatory studies are not always needed to demonstrate biosimilarity between reference and biosimilar products. Following a similar course, Colombia has developed a third pathway for the submission of biosimilar products which is known as the abbreviated comparability approach.
This pathway is followed if a molecule that will be used as a reference product is: sufficiently characterized, has well-defined and well-documented safety and efficacy profile, has strong clinical experience data and has strong pharmacovigilance evidence. So, for the abbreviated comparability approach, the module system resembles that of the comparability approach but for module 5, only pharmacokinetics (PKs) and pharmacodynamics (PDs) data may be required. see Figure 3.
In Colombia, INVIMA is to implement legislation for the approval of biotherapeutics. Three pathways for the submission of biotherapeutic products: the complete dossier approach, comparability approach, and abbreviated comparability approach, will be applied to pharmaceutical products. The pathway to be followed will depend on the biological/biosimilar molecule in question.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Francisco Javier Sierra Esteban, MSc
Director Técnico Dirección de Medicamentos y Productos Biológicos Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA)
Carrera 10, No. 64 – 28 piso 3, Bogotá DC, Colombia
References
1. GaBI Online – Generics and Biosimilars Initiative. Colombia issues draft decree for registration of biologicals [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2018 Mar 8]. Available from: www.gabionline.net/Guidelines/Colombia-issues-draft-decree-for-registration-of-biologicals
2. GaBI Online – Generics and Biosimilars Initiative. Colombian guidelines for productos bioterapéuticos similares [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2018 Mar 8]. Available from: www.gabionline.net/Guidelines/Colombian-guidelines-for-productos-bioterapeuticos-similares
3. Generics and Biosimilars Initiative. Second Colombian Scientific Meeting on Quality Assessment of Biosimilars/Similar Biotherapeutic Products 2017; 15 August 2017; Bogotá, Colombia. Available from: www.gabi-journal.net/second-colombian-scientific-meeting-on-quality-assessment-of-biosimilarssimilar-biotherapeutic-products-2017
|
Author for correspondence: Johanna Andrea García Cortes, MSc; Profesional Especializado, Coordinadora del Grupo de Registros Sanitarios de Medicamentos Biológicos, Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA), Carrera 10, No. 64 – 28 piso 3, Bogotá DC, Colombia |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/regulations-for-biotherapeutics-approval-in-colombia.html
Copyright ©2024 GaBI Journal unless otherwise noted.