|
Abstract: |
Submitted: 23 June 2015; Revised: Not applicable; Accepted: 1 July 2015; Published online first: 14 July 2015
The South African guideline for the development and registration of biosimilar medicines [1] defines a biosimilar as a biological medicine that is similar, but not necessarily identical, in terms of quality, safety and efficacy to an already registered reference biological medicine. The reference biological medicine is the innovator medicine that is used as comparator in head-to-head comparative quality, non-clinical and clinical studies with the biosimilar product to demonstrate similarity. A requirement for the reference medicine is that it must be registered in South Africa on the basis of efficacy and safety data; however, samples of the reference product used in comparability studies do not need to be procured from the South African market but can be sourced from a country with which the Medicines Control Council (MCC) is aligned. These include countries generally recognized to have stringent regulatory systems such as those forming part of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) regions as well as Australia, Canada and Switzerland.
The guideline for biosimilars applies only to those biologicals composed of well-characterized recombinant DNA-derived therapeutic proteins. It excludes vaccines, even if these are derived from recombinant DNA technology. Guidance for the development and registration of biosimilar monoclonal antibody (mAb) products is set out in an annex to the main guideline document [1].
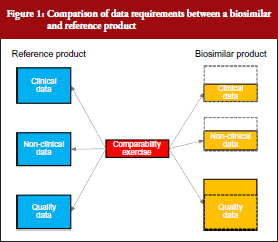
The South African biosimilars guideline is essentially based on the corresponding guidelines of the European Medicines Agency (EMA) [2] and World Health Organization (WHO) [3]. All three guidelines stipulate that biosimilars, being biological molecules with complex structures, would require proof of efficacy and safety in humans prior to registration. The generic medicines registration pathway used for small well-characterized molecules and which require only the demonstration of pharmaceutical equivalence (comparative dissolution in three pH media) and bioequivalence with the innovator (in the case of most solid oral dosage forms) would not be appropriate. Although the extent of clinical and non-clinical studies required for the registration of biosimilars would be less than for innovator medicines, it will to a large degree be dependent on how well the active ingredient has been characterized and its similarity to that of the reference drug substance. Complete physico-chemical characterization of the active ingredient coupled with a high degree of similarity with the reference substance, using state-of-the-art analytical methods, would require substantially reduced clinical and non-clinical data. This is illustrated in Figure 1, which shows that whereas the amount of quality data are greater than for an innovator product (since it includes a comparability study with the reference besides full structural characterization to the same extent as for the reference), the non-clinical and clinical data packages are significantly less. Specific requirements for quality, safety and efficacy are briefly reviewed below.
A full quality data package detailing the structural characteristics, chemical and physical properties of the biosimilar active pharmaceutical ingredient (API), its validated manufacturing and control processes and stability profile must be submitted. In addition, the formulation, manufacturing, filling and packaging procedures as well as the stability profile of the final product must also be included. These data for the biosimilar are equivalent in amount and extent to that required for the registration of the innovator. However, apart from the quality data on the biosimilar API and final product, the manufacturer of a biosimilar must also submit data on side-by-side comparability studies that show that the biosimilar and reference products are indeed similar in terms of their structures, chemistry and physical properties. This means that for a biological to be considered a biosimilar, it must have an identical primary structure, i.e. amino acid sequence and disulphide bonding, to the innovator reference substance as the primary structure determines the biological function(s) of the substance [4]. Furthermore, it must be shown to be highly similar in terms of post-translational modifications, e.g. glycosylation, three-dimensional structure, e.g. α-helix, β-sheet, and even impurity profiles, although a product with fewer impurities would be preferred [1, 2]. The comparability studies must be carried out in line with the principles of ICH Q6B [5] as well as ICH Q5E [6].
Non-clinical studies should be conducted before initiation of clinical investigations. The non-clinical studies should be comparative, and be designed to detect differences between the biosimilar and reference medicine. Ideally, a number of endpoints should be monitored such as pharmacodynamic effects as well as toxic effects as determined in at least one repeat dose study. Toxicokinetic measurements should be included and must involve analysis of immunogenicity as the latter may be of value in demonstrating similarity of immune responses to both reference and biosimilar products. Animal immunogenicity studies cannot, however, serve as alternative to immunogenicity studies in humans. In general, other routine toxicological studies such as safety pharmacology, reproduction pharmacology, mutagenicity and carcinogenicity studies are not required for biosimilar medicines.
The trend nowadays is to move away from animal studies; hence, if the minimum required data can be obtained with in vitro biological assays, then these should be used or considered. Justification will, however, be required.
Clinical comparability studies should only proceed after acceptable biosimilarity has been established at a physicochemical level. Non-clinical (or in vitro bioassay) comparability data should support the conclusion of biosimilarity based on physicochemical results. The clinical comparability exercise is a stepwise procedure that should begin with pharmacokinetic (PK) and pharmacodynamic (PD) studies followed by clinical efficacy and safety trials. In general, an equivalence trial design is preferred although other designs could be acceptable if properly motivated and justified. Since the clinical data package for a biosimilar is not as extensive as for an innovator medicine, a patient population that is highly sensitive should be selected and the patient population should be large enough to detect meaningful differences in safety, efficacy and immunogenicity.
Clinical studies should be provided for each indication for which the reference product is authorized. However, in cases where the clinical effects have the same underlying mechanism of action, data from a clinical trial of the biosimilar in one indication may be used to support approval of the biosimilar for other indications for which the reference medicine is approved.
Demonstration of comparable efficacy alone does not constitute clinical biosimilarity; equivalent safety and particularly immunogenicity with the reference medicine are also important. Preregistration safety data, derived from the efficacy trials, should be obtained to address the adverse effect profiles of the biosimilar and reference medicines. Preclinical safety data are, however, not sufficient to identify all potential differences and, hence, post-approval pharmacovigilance must be continued accompanied by the submission of regular periodic safety update reports (PSURs) to MCC. This, in fact, is a condition of the registration of all new biologicals, including biosimilars. A suitable Risk Management Plan (RMP) for monitoring immunogenicity, inherent safety concerns and unknown safety signals that could result from the impurity profile and other properties of the biosimilar, should be submitted at the time of application for registration of the biosimilar product.
For most applications for the registration of a biosimilar, comparative clinical trials will be required. However, in certain instances well-designed comparative PK/PD studies may be sufficient to demonstrate clinical comparability where sufficient justification is provided and certain conditions are met. A critical condition is that at least one PD marker is accepted as a surrogate marker for efficacy and that the relationship between dose/exposure to the biosimilar or reference product and this surrogate marker is validated and well known. Examples include absolute neutrophil count to assess the effect of granulocyte colony-stimulating factor and early viral load reduction in chronic hepatitis C to assess the effect of alpha interferon [1].
Finally, since biosimilars, unlike generics, are not considered identical to their innovator reference products, they are neither interchangeable nor substitutable with their reference products or other medicines of the same class. This is in line with similar approaches of other regulatory authorities, such as EMA [2] and Health Canada [7]. Even the WHO guideline stipulates that biosimilars are not interchangeable with their reference products. Interestingly, a few national regulatory authorities in Europe such as The Netherlands [8] and Finland [9] do allow substitution of biosimilars for their reference medicines, albeit under special conditions. In the case of The Netherlands, substitution is allowed but only with adequate clinical monitoring and after the patient has been informed [8]. In Finland, it is permitted but must take place under supervision of a healthcare professional. In France, provisions in the law that allow the substitution of originators by generics have been adapted to extend to biosimilars; however, it has still to be enacted and consequently has not been implemented yet [10].
As of July 2015, no biosimilars have been registered in South Africa. This is despite several biosimilar applications for products containing erythropoietin, filgrastim and insulin having been received by MCC over the past 10 years. None of those which have been reviewed have complied with the registration requirements for a biosimilar medicine. The major deficiencies identified in these applications included the following:
It was clear that many of the biosimilars submitted and which were registered in their countries of origin obtained registration in those countries via a non-biosimilars pathway, i.e. by means of the generics route. Although MCC published its final version of the biosimilars guideline only in 2014 [1], it applied the criteria of EMA and WHO biosimilars guidelines to biosimilar registration applications received prior to 2014. An exception was an application received for the registration of a low molecular weight heparin, enoxaparin, of which the innovator product, namely Clexane® was registered as a pharmaceutical medicine in South Africa. The new enoxaparin application was consequently assessed as a generic drug and was approved as such. No clinical or non-clinical data were submitted with the application. The owner of the Clexane® brand subsequently appealed the MCC’s decision on the grounds that enoxaparin was a biological and that all subsequent enoxaparin applications should be reviewed according to criteria for the registration of biosimilars as opposed to those applicable to generics. The appeal was successful and in August 2009, MCC through the Registrar of Medicines, issued a letter to the industry stating that enoxaparin was regarded as a biological and that clinical data will be required for the registration of all enoxaparin products [11]. Interestingly, the US Food and Drug Administration came to the exact opposite decision and rejected the appeal of the innovator company, which suggests that they may have registered their application of enoxaparin made by Sandoz as a generic drug [12].
The biosimilar guideline of MCC follows the same principles as those of EMA, Health Canada and WHO with respect to quality, safety and efficacy. Although several countries in the European Union now allow substitution of innovator biologicals with their biosimilars, it is unlikely that MCC will revise their guideline to allow substitution in the near future since the local population in South Africa has thus far had no therapeutic exposure to biosimilars.
The views expressed in this paper are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Medicines Control Council of South Africa or one of its committees or working groups.
Competing interest: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Henry MJ Leng, PhD
School of Public Health, University of the Western Cape, Private Bag X17, Bellville 7130, South Africa
Khamusi Mutoti, BSc, BPharm Sci
Medicines Control Council, National Department of Health, Private Bag X828, Pretoria 0001, South Africa
Professor Nontombe Mbelle, MBChB, MMed, FC Path (CMSA)
Department of Medical Microbiology, University of Pretoria, Private Bag X20, Hatfield 0028, South Africa
References
1. Medicines Control Council. Biosimilar medicines: quality, non-clinical and clinical requirements. August 2014. Ver. 3 [homepage on the Internet]. [cited 2015 Jun 23]. Available from: http://mccza.com/genericdocuments/2.30_Biosimilars_Aug14_v3.pdf
2. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (EMEA/CHMP/BWP/49348/2005). 22 February 2006 [homepage on the Internet]. 2006 Mar 8 [cited 2015 Jun 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003953.pdf
3. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs) [homepage on the Internet]. 2010 Jun 6 [cited 2015 Jun 23]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
4. Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A, et al. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34(Web Server issue):W645-50.
5. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Specifications: test procedures and acceptance criteria for biotechnological/biological products. Q6B. Current step 4 version. 10 March 1999 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Jun 23]. Available from: http://www.ich.org/fileadmin/public_web_site/ich_products/guidelines/quality/q6b/step4/q6b_guideline.pdf
6. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Comparability of biotechnological/biological products subject to changes in their manufacturing process. Q5E. Current step 4 version. 18 November 2004 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Jun 23]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf
7. Health Canada. Health Products and Food Branch. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs) [homepage on the Internet]. 2010 Mar 8 [cited 2015 Jun 23]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf
8. GaBI Online – Generics and Biosimilars Initiative. Dutch medicines agency says biosimilars ‘have no relevant differences’ to originators [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Biosimilars/General/Dutch-medicines-agency-says-biosimilars-have-no-relevant-differences-to-originators
9. GaBI Online – Generics and Biosimilars Initiative. Finnish drug regulator recommends interchangeability of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Policies-Legislation/Finnish-drug-regulator-recommends-interchangeability-of-biosimilars
10. GaBI Online – Generics and Biosimilars Initiative. France to allow biosimilars substitution [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Policies-Legislation/France-to-allow-biosimilars-substitution
11. Medicines Control Council. Applications for registration of medicines containing enoxaparin [homepage on the Internet]. [cited 2015 Jun 23]. Available from: http://mccza.com/dynamism/default_dynamic.asp?Grpid=25&doc=dynamic_generated_page.asp&categid=169&groupid=25
12. GaBI Online – Generics and Biosimilars Initiative. Good news for biosimilar enoxaparin sodium [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Jun 23]. Available from: www.gabionline.net/Biosimilars/News/Good-news-for-biosimilar-enoxaparin-sodium
|
Author for correspondence: Henry MJ Leng, PhD, School of Public Health, University of the Western Cape, Private Bag X17, Bellville 7130, South Africa |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/regulatory-requirements-for-the-development-and-registration-of-biosimilars-in-south-africa.html
Copyright ©2024 GaBI Journal unless otherwise noted.