A white paper: US biosimilars market on pace with Europe
|
Abstract:
In the US, 28 biosimilars have been approved, with 10 in the last two years. The US is keeping pace with the EU who pioneered biosimilars approvals a decade earlier. Herein, current FDA regulations and hurdles encountered for US biosimilar approval and uptake are discussed.
|
Submitted: 12 October 2020; Revised: 28 October 2020; Accepted: 29 October 2020; Published online first: 9 November 2020
While many sources claim that Europe is winning the race when it comes to biosimilars, a broader assessment of the landscape reveals a more encouraging story for the US. Although the European Medicines Agency (EMA) pioneered the framework for biosimilar regulation, the US Food and Drug Administration (FDA) is moving at approximately the same pace as EMA based on the number of approvals at the same time after implementation of its regulatory pathway [1].
The biosimilar regulatory framework in Europe was implemented in 2004. Within this framework, the Committee for Medicinal Products for Human Use (CHMP) provides initial assessments for marketing authorization of new medicines that are ultimately approved centrally by EMA [2]. In the US, the Biologics Price Competition and Innovation Act of 2009 (BPCIA Act) created an abbreviated licensure pathway for biosimilars and granted FDA the authority to approve these near-identical biologicals [3]. This legislation was implemented in 2010 as part of the Patient Protection and Affordable Care Act signed by President Barack Obama.
As of September 2020, approximately 10 years after implementation of the biosimilar approval pathway, 28 biosimilars have been approved by FDA, with 18 of those approvals granted in the last two years [4]. In the 10-year time period following the creation of Europe’s biosimilar regulatory pathway, EMA approved 13 biosimilar products (some of which were marketed under several different brands) [5]. From this perspective, the US appears to be on a faster pace than the EU in terms of biosimilar approvals. Currently, there are 45 biosimilars approved in Europe; however, these estimates fall to 35 when products approved in the US as follow-on biologicals via the 505(b)(2) pathway, e.g. somatropin, insulin, teriparatide, or abbreviated new drug application (ANDA) are excluded, see Table 1. Furthermore, Europe’s filgrastim biosimilar Tevagrastim®/Ratiograstim® was approved as Granix® (tbo-filgrastim) in the US via a Biologic License Application (BLA) prior to the implementation of a biosimilar approval pathway and is not included in the US biosimilar count.
Biosimilars have been developed in various therapeutic areas, including oncology and rheumatology, and are based on nine reference products. For some reference products, numerous biosimilars have been developed; for example, five biosimilars of Genentech’s Herceptin® (trastuzumab) and Amgen’s Neulasta® (pegfilgrastim) have been approved in the US and Europe. A summary of the biosimilars landscape in the US and EU is presented in Table 1.
There are currently nine biosimilar applications under CHMP evaluation, including biosimilar candidates for adalimumab (2), bevacizumab (5), pegfilgrastim (1), and trastuzumab (1) [6]. Unlike the transparency on biosimilar filings in Europe, FDA does not provide information on biosimilar candidates under review until they are approved.
Since the creation of a regulatory approval pathway, numerous guidance documents have been developed by both EMA and FDA to guide manufacturers in their development of biosimilar candidates. At least 10 documents have been released by FDA that provide guidance on scientific and quality considerations in the demonstration of biosimilarity and interchangeability [7]. EMA has published three overarching guidelines and nine product-specific guidelines for biosimilars manufacturers that address both non-clinical and clinical issues as well as quality-related issues [8].
Because EMA pioneered biosimilar regulations, the question has commonly been raised: To what extent does the European biosimilar experience translate to the US? Although the US and the EU are on a similar trajectory in terms of the number of biosimilar products approved, these markets differ in several respects. A critical point of divergence between the US and EU biosimilars terrain is the concept of interchangeability. Unique to the US, FDA may designate a product ‘interchangeable’ if it meets additional requirements beyond being biosimilar, which translates to more clinical development that includes switching studies and increased cost from a manufacturer’s perspective. To date, no biosimilar products have interchangeability status. Having a separate designation of interchangeability for an approved biological has been said to give the impression that interchangeable biosimilars are superior in quality to non-interchangeable biologicals ‒ which is not the case. A major focus area for the Association for Accessible Medicines’ Biosimilars Council has been educating physicians and payers on interchangeability designations as they are ‘waiting for interchangeability to really get on board with biosimilars’ [9].
Perceptions of prescribing physicians about the safety of biosimilars is an ongoing issue. There is a need for increased confidence, particularly around switching from a reference product to a biosimilar. Concerns are particularly heightened in the oncology therapeutic area, where treatments can be curative and clinicians fear the loss of efficacy and increased immunogenicity [10]. One of the key concerns around switching to a biosimilar is the negative counterpart of the placebo effect: the nocebo effect, a phenomenon in which negative expectations lead to worsening of symptoms [11]. Some physicians continue to have doubts regarding the rigorous approval process for biosimilars and switching studies that have been performed thus far and transfer these negative concerns to patients through body language or tone of voice when discussing treatment options. Raising awareness about the nocebo effect by educating healthcare professionals on this phenomenon may mitigate its effect in patients receiving biosimilar products [12, 13].

The biosimilars marketplace
While the European biosimilars market has been credited with higher uptake compared to the US market, rates of uptake differ from country to country in Europe and can vary significantly by product class. A report by KPMG commissioned by Medicines for Europe to analyse the procurement of medicines in hospitals in eight European countries highlighted the variability in biosimilar sales against originator in these different Member States [14]. An average of hospital biosimilar volume in March 2019 showed that Denmark achieved 63% overall biosimilar volume, with the UK coming in second at 45%. Germany had 40% biosimilar volume, France had 34%, and Belgium tied with Switzerland for last place among the countries studied at 14%. In a recent assessment of the impact of biosimilar competition in Europe, 16 European countries were reported to have achieved > 90% biosimilar utilization for filgrastim and pegfilgrastim in 2018, while utilization in Ireland was just 27%. Among antitumour necrosis factor biosimilars (adalimumab, etanercept and infliximab), Norway and Denmark had 81% and 96% biosimilar uptake, respectively, while every other country’s utilization was less than 50% [15]. Variations in adoption rates among individual European countries as well as across therapeutic areas are influenced by government involvement, reimbursement structures and tender procurement policies.
In the US, biosimilars have gained significant share in the majority of therapeutic areas in which they have been introduced, ranging on average from 20% to 25% within the first year of launch, with some projected to reach greater than 50% within the first two years [16, 17]. As expected, first-to-market biosimilars tend to capture a greater portion of the segment compared to later entrants. Filgrastim biosimilars have been on the market the longest at five years and have achieved a 72% share, while bevacizumab and trastuzumab biosimilars have approximately 40% share. Rituximab and infliximab have had the most limited adoption, with approximately 20% market share [16].
Biosimilar competition has a significant potential to impact overall drug spending, with the amount of savings per country based on the volume and list price of products prior to biosimilar entry [15, 17]. Manufacturers are reducing healthcare costs by launching biosimilars at a wholesale acquisition cost (WAC) that is generally 15% to 37% lower than the reference product WAC and 3% to 40% below the reference product average sales price (ASP). Manufacturers of originator products have had to adapt their strategies in order to stay competitive, whether it be launching a second-generation product, or a new formulation aimed at reducing side effects. Biosimilar competition and originator manufacturer responses are translating into significant savings. The annualized savings reached US$6.5 billion in the second quarter of 2020, where reference molecules sold US$20.8 billion (annualized) pre-biosimilar [18]. Savings over the next five years as a result of biosimilar alternatives are projected to exceed US$100 billion [17].
Biosimilars include both self-administered drugs obtained at retail pharmacies as well as provider-administrated drugs in inpatient and outpatient settings. In the US, the balance of cost savings to healthcare payers, providers and patients is different for each of these due to differences in payment and cost-sharing arrangements [19]. Most self-administered pharmacy-dispensed biologicals are paid for on a fee-for-service basis and the final amount paid by insurers reflects several transactions, including a confidential rebate payment. On the other hand, the costs associated with provider-administered biologicals are purchased directly from manufacturers and wholesalers or through group purchasing organizations either incorporated into prospective, bundled payments or under fee-for-service arrangements. Access to these biologicals (reference or biosimilar) is determined by formularies that may reflect the highest price concession rather than the lowest list price.
To date, there have been no self-administered biosimilars launched in the US. This is in stark contrast to Europe, where biosimilars administered via self-injection at home are available, including a recently approved subcutaneous form of infliximab [20]. It remains to be seen how self-administered biosimilars will fare in the US system where higher list price drugs may be ‘preferred’ on the formulary based on their profitability for those constructing the formulary.
Overcoming the hurdles
Despite having approved 28 biosimilars in 10 years, the US faces one of its biggest biosimilar hurdles when it comes to launching these products. So far, 18 of 28 biosimilars have been made available to patients, while the others have not been commercialized due to patent-related issues. In an effort to protect their originator products, brand manufacturers block competition by filing numerous follow-on patents. The expense of challenging these patents can serve as a deterrent to potential biosimilar competitors [21]. Brand manufacturers also disincentivize biosimilar utilization by leveraging a ‘rebate trap’ wherein brand manufacturers offer significant rebates for their products that disable competition from biosimilars coming on the market [22].
While there is work to be done to accelerate commercialization of biosimilars, FDA has taken active steps to mitigate some of these barriers to biosimilar utilization. In July 2018, Health and Human Services Secretary Alex Azar announced a Biosimilars Action Plan (BAP) to advance policies that facilitate the development of the biosimilars market and increase competition for biological drugs [23].
Key strategies of FDA’s BAP of 2018:
- Improve the efficiency of the biosimilar and interchangeable product development and approval process
- Maximize scientific and regulatory clarity for the biosimilar product development community
- Develop effective communications to improve understanding of biosimilars among patients, healthcare providers and payers
- FDA said these actions are meant to prevent companies from blocking new biosimilars from entering the market and to stop manufacturers of reference products from manipulating the exclusivity provision to fend off biosimilar entry to the marketplace. Each of the four key elements of the BAP was associated with a number of priority deliverables, and since then FDA has been taking steps to meet those.
FDA’s most recent actions addressed biosimilar market competition in a joint statement issued by FDA and the Federal Trade Commission (FTC) that identified four goals to help combat anti-competitive practices [24]. In particular, the agencies noted their concern with false or misleading statements comparing biological reference products and biosimilars, which may be hampering biosimilar uptake by creating negative misperceptions about the safety and efficacy of biosimilar therapies. To further clarify how data and information about biosimilars should be presented in a truthful and non-misleading manner in regulated promotional materials, FDA announced the release of the ‘Draft guidance for industry: promotional labeling and advertising considerations for prescription biological reference and biosimilar products ‒ questions and answers’ and invited comment by stakeholders in the docket. As part of their efforts to promote greater competition, FDA and FTC held a public workshop in March 2020 to discuss their ‘collaborative efforts to support appropriate adoption of biosimilars, discourage false or misleading communications about biosimilars, and deter anticompetitive behaviors in the biological product marketplace’.
Targeting faster reviews, FDA issued new draft guidance in February 2020 detailing how it will speed its review of biosimilar or interchangeable application supplements, which can be used to update the initial biosimilar approval when it is for fewer than all the reference product’s licensed conditions of use [25]. At the time of submission, applicants may decide not to seek licensure of a proposed biosimilar for conditions of use that are protected by patent for the innovator. FDA had previously committed to reviewing and acting on original 351(k) BLA supplements with clinical data within 10 months of receipt; however, the agency now says such supplements will be reviewed and acted upon in a 6-month timeframe.
The US Centers for Medicare and Medicaid Services (CMS) have also revised a number of their payment policies in an effort to promote biosimilar competition. Three policy changes made under Medicare Part B to incentivize biosimilar uptake were: 1) BPCIA required Medicare Part B to pay for biosimilars at the ASP of the biosimilar plus 6% of the innovator product’s ASP [26]; 2) unique payment codes were provided for each biosimilar (reversing policy that grouped drugs under single codes) [27]; and 3) biosimilars were allowed ‘pass-through’ status under the 340B Drug Pricing Program, which meant they could be paid at ASP plus 6% rather than being heavily discounted (–22.5%) [28]. Furthermore, under Medicare Part D, the Bipartisan Budget Act of 2018 changed the treatment of biosimilars in the coverage gap (doughnut hole) discount programme, requiring manufacturers to give discounts for biosimilars [29]. Furthermore, in 2019, there was a lowering of the maximum copay amount on biosimilars to rates commensurate with generic copay amounts for lower income beneficiaries.
In parallel with updated regulatory policies that encourage development of the biosimilars market, a growing body of evidence has amassed to support the clinical safety of biosimilars. In a systematic review of primary data from 90 studies that enrolled 14,225 unique individuals, the nature and intensity of safety signals reported after switching from reference medicines to biosimilars were the same as those already known from continued use of the reference biological [30]. Similar results were observed in a systematic review of 57 switching studies evaluating the efficacy, safety and immunogenicity risk of transitioning between an originator biological and a biosimilar [31]. These results should increase confidence of patients, healthcare professionals and the public in biosimilars, leading to increased acceptance of these safe and effective medicines.
Conclusion
Although it has been suggested that biosimilars are not living up to their promise in the US, the current landscape suggests otherwise. FDA has approved 18 biosimilars in the past two years alone and a total of 28 since the implementation of a regulatory pathway just 10 years ago. Although getting these drugs into the hands of patients has hit some stumbling blocks due to anti-competitive behaviours from brand manufacturers, FDA has been proactive in addressing these ‘shenanigans’ and has created a BAP as well as additional guidances to mitigate such barriers to uptake and ensure the biosimilars market is poised for success. Clinical studies on the impact of switching between reference medicines and biosimilars have underscored the safety of biosimilars and impart confidence in their utilization for both physicians and patients.
In short, the overall outlook on biosimilars is positive. Pharmaceutical Research and Manufacturers of America (PhRMA), an organization representing biopharmaceutical manufacturers, believes that biosimilars are helping to bring down the cost of drugs in the US market and are positioned to achieve further savings. Katie Verb, Director of Policy and Research for PhRMA, says that federal policies are having a positive effect. These sentiments were echoed by Dr Leah Christl, Amgen’s Executive Director for Global Regulatory and Research and Development Policy, who believes the US biosimilars market is showing healthy vital signs.
Funding sources
This paper is funded by the Alliance for Safe Biologic Medicines (ASBM).
The ASBM is an organization composed of diverse healthcare groups and individuals ‒ from patients to physicians, innovative medical biotechnology companies and others ‒ who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Dr Madelaine Feldman is the Chairperson of the Alliance for Safe Biologic Medicines. She has participated in advisory boards for Gilead, Lilly, Pfizer and Samsung. Mr Michael S Reilly, Esq is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002 to 2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Madelaine Feldman, MD, FACR
Michael S Reilly, Esq
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Mehr SR, Brook RA. Biosimilars in the USA: will new efforts to spur approvals and access spur uptake and cost savings? Pharmaceut Med. 2019;33(1):1-8.
2. European Commission. Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending 2001/83/EC on the Community code relating to medicinal products for human use [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2004_27/dir_2004_27_en.pdf
3. Carver KH, Elikan J, Lietzan E. An unofficial legislative history of the Biologics Price Competition and Innovation Act of 2009. Food Drug Law J. 2010;65(4):671-818, ii.
4. U.S. Food and Drug Administration. Biosimilar product information. FDA-approved biosimilar products [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
5. European medicines Agency. Centrally authorized biosimilar medicines [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_bio-similar.
6. European Medicines Agency. EMA/535780/2020. Applications for new human medicines under evaluation by the Committee for Medicinal Products for Human Use. 8 October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/documents/report/applications-new-human-medicines-under-evaluation-chmp-october-2020_en.pdf
7. U.S. Food and Drug Administration. Biosimilar guidances. Current as of 21 June 2019 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances.
8. European Medicines Agency. Scientific guidelines on biosimilar medicinal products. 2020 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/ multidisciplinary/multidisciplinary-biosimilar
9. The Center for Biosimilars. AAM’s Christine Simmon on Biosimilars Council Initiatives and educating around interchangeability. 16 February 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.centerforbiosimilars.com/news/aams-christine-simmon-on-biosimilars-council-initiatives-and-educating-around-interchangeability
10. Konstantinidou S, Papaspiliou A, Kokkotou E. Current and future roles of biosimilars in oncology practice. Oncol Lett. 2020;19(1):45-51.
11. Planès S, Villier C, Mallaret M. The nocebo effect of drugs. Pharmacol Res Perspect. 2016;4(2):e00208.
12. Ebbers HC, Schellekens H. Are we ready to close the discussion on the interchangeability of biosimilars? Drug Discov Today. 2019;24(10):1963-7.
13. Rezk MF, Pieper B. To see or NOsee: the debate on the nocebo effect and optimizing the use of biosimilars. Adv Ther. 2018;35(6):749-53.
14. KPMG. Improving healthcare delivery in hospitals by optimized utilization of medicines: A study into 8 European countries. Commissioned by Medicines for Europe. 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2019/10/20190903_Hospital-Reform-Study_final.pdf
15. IQVIA. White Paper. The impact of biosimilar competition in Europe. Oct 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://ec.europa.eu/docsroom/documents/ 31642/attachments/1/translations/en/renditions/native
16. Amgen. 2020 Biosimilar trends report [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.amgenbiosimilars.com/-/media/Themes/Amgen/amgenbiosimilars-com/Amgenbiosimilars-com/spandf/USA-CBU-80723-2020-Amgen-Biosimilar-Trends-Report.pdf
17. IQVIA. Biosimilars in the United States 2020-2024. Competition, savings and sustainability. October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.iqvia.com/-/media/iqvia/spandfs/institute-reports/iqvia-institute-biosimilars-in-the-united-states.pdf
18. Fein AJ. Drug Channels Institute®; The Booming biosimilar market of 2020. 6 October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.drugchannels.net/2020/10/the-booming-biosimilar-market-of-2020.html
19. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3.
20. GaBI Online – Generics and Biosimilars Initiative. Celltrion launches infliximab biosimilar Remsima SC in Europe. 20 March 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Oct 28]. Available from: www.gabionline.net/Biosimilars/News/Celltrion-launches-infliximab-biosimilar-Remsima-SC-in-Europe
21. Chen BK, Yang YT, Bennett CL. Why biologics and biosimilars remain so expensive: despite two wins for biosimilars, the Supreme Court’s recent rulings do not solve fundamental barriers to competition. Drugs. 2018;78(17):1777-81.
22. U.S. Food and Drug Administration. Speech by Scott Gottlieb: Capturing the benefits of competition for patients.7 March 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/news-events/speeches-fda-officials/capturing-benefits-competition-patients-03072018
23. U.S. Food and Drug Administration. Biosimilars action plan: balancing innovation and competition. July 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/114574/download
24. U.S. Food and Drug Administration. Joint statement of the Food & Drug Administration and the Federal Trade Commission regarding a collaboration to advance competition in the biologic marketplace. 3 February 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/134864/download
25. U.S. Food and Drug Administration. Draft guidance for industry. Biosimilars and interchangeable biosimilars: licensure for fewer than all conditions of use for which the reference product has been licensed. February 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/134932/download
26. Centers for Medicare & Medicaid Services. MLN Matters® Number SE1509. 2014 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/Downloads/SE1509.pdf
27. Centers for Medicare & Medicaid Services. Part B Biosimilar biological product payment and required modifiers. Last modified 2 February 2018 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice/Part-B-Biosimilar-Biological-Product-Payment
28. Centers for Medicare & Medicaid Services. MLN Matters® Number MM11099 Revised. Update of the Hospital Outpatient Prospective Payment System (OPPS). Jan 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM11099.pdf
29. Congress.Gov. 115th Congress. H.R.1892 ‒ Bipartisan Budget Act of 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.congress.gov/bill/115th-congress/house-bill/1892
30. Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463-78.
31. McKinnon RA, Cook M, Liauw W, Marabani M, Marschner IC, Packer NH, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32(1):27-52.
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-white-paper-us-biosimilars-market-on-pace-with-europe.html
European prescribers’ attitudes and beliefs on biologicals prescribing and automatic substitution
|
Introduction: The European Union (EU) and the European Medicines Agency (EMA) have led the development of a regulatory framework for biosimilars since 2004. By end of December 2019, 64 biosimilars of 15 originator biological medicines have a marketing authorization in Europe. Now, for the second time, the Alliance for Safe Biologic Medicines (ASBM) asked European prescribers for their views on the prescribing, adverse drug reaction reporting, automatic substitution and switching of biologicals and biosimilars.
Methods: In March 2019, the ASBM surveyed 579 prescribers in France, Germany, Italy, Spain, Switzerland and the UK. Prescribers were asked for their views on authority over prescribing and dispensing of biologicals/biosimilars, reporting biological/biosimilar use and adverse drug reactions (ADR) and switching. There were also questions related to their familiarity with, knowledge of, attitudes to, and beliefs in, biosimilars.
Results: Since the previous European prescriber study conducted in 2013, the percentage of respondents considering themselves highly familiar with biosimilar medicines has increased from 76% to 90%. Four out of five prescribers said they are legally required to report ADR that are brought to their attention and they file detailed ADR reports taking 10–20 minutes. Four out of five prescribers feel very strongly about having control over what is prescribed and dispensed to their patients. While highly comfortable prescribing biosimilars to naïve patients, physician comfort level decreased when switching a stable patient to a biosimilar. Comfort level decreased further when prescribers were asked about switching a patient to a biosimilar for non-medical reasons, e.g. cost, and further still if the switch is initiated by a third party.
Conclusion: European physicians have increased their familiarity with biosimilars since the 2013 survey. Physicians increasingly believe they should always have control of treatment decisions including the decision to switch to a biosimilar. It was also highlighted that governments should make multiple therapeutic options available through tenders.
|
Submitted: 9 July 2020; Revised: 9 August 2020; Accepted: 19 August 2020; Published online first: 26 August 2020
Introduction
Healthcare systems across the globe face resource and budget constraints. Biosimilar drug products offer less expensive alternatives to brand-name originator drug products and can thus offer some relief to healthcare costs. Biosimilars are highly similar and have no clinical meaningful differences; but are not identical to originator biologicals. As countries seek to control health costs and expand access to biological therapies, building physician confidence in biosimilars is critical to promoting their use and reaping the cost benefits.
The European Union (EU) and the European Medicines Agency (EMA) have led the development of a regulatory framework for biosimilars. In 2005, EMA established the first biosimilars approval pathway that was distinct from generics approval [1]. Since then, EMA has developed and refined a comprehensive set of regulatory guidelines on which biosimilar applications are reviewed and approved or rejected. By the end of 2019, 58 biosimilars of 15 originator biological medicines have a marketing authorization in Europe [2]. The European biosimilars market is currently the largest in the world, representing approximately 60% of the global biosimilar market and growing consistently year on year [3].
At present, once authorized, EMA applies a ‘same-label’ (generic) approach to biosimilar product labels [4]. However, there are concerns over whether this is sufficient to ensure appropriate drug switching and product traceability. There is ongoing debate about what information is appropriate in the naming and labelling of biosimilars. In the US, FDA released its requirements for the non-proprietary naming of biological products in January 2017 [4]. Prior to this, the Alliance for Safe Biologic Medicines (ASBM) carried out surveys of Australia and EU prescribers and US pharmacist perspectives on the naming of these products. Overall, both groups believed that naming should make biosimilars distinguishable from originator products [5–7]. The ASBM is an organization composed of diverse healthcare groups including patients, physicians and medical innovators. It is funded by its many member partners that are made up of international organizations and companies [8].
The interchangeability of biosimilars is viewed differently in countries across the world. This is particularly marked by the approaches to interchangeability and substitution in the US and Europe [9]. ‘In the US, insurance mandates can result in formulary changes requiring patients to be switched from a reference product to a biosimilar strictly for cost reasons’ . In Europe, automatic substitution of originator biologicals with biosimilars is rare as this practice excludes physicians from decisions regarding the treatment of patients. There have been a number of surveys and workshops carried out across the world (Australia [5], Europe [4, 6], South America [10] and the US [11]) that have asked for prescriber opinions on prescribing practices, naming and labelling of biologicals. In terms of naming, prescribers in Australia, Europe and the US, overall, agreed that there is a need for distinguishable non-proprietary names to be given to all medications. In South America, knowledge about biosimilars varied in different countries surveyed (Argentina, Brazil, Colombia and Mexico) and revealed gaps in understanding and in the use of distinguishable names for biologicals.
In 2019, the ASBM commissioned 15-minute web-based surveys to be carried out by biological prescribers in six Western European countries (France, Germany, Italy, Spain, Switzerland and the UK) to document their perspectives on biological substitution. This survey mirrors their previous European prescriber survey conducted in 2013 [6] (both survey reports can be found at www.safebiologics.org/surveys).
Overall, the 2019 survey showed that awareness of biosimilars in the countries had increased since the 2013 survey. Specifically, more physicians (90%) rated themselves as being ‘Familiar’ or ‘Very familiar’ with biosimilars than did in 2013 (76%). A strong majority of respondents (82%) felt that it is either ‘Very important’ or ‘Critical’ for them to decide which biological medicine is dispensed to their patients, representing a 10% increase over the results of the 2013 survey. Again, a strong majority of respondents (84%) considered authority to prevent a substitution either ‘Very important’ or ‘Critical’ , another 10% increase over the 2013 findings. In 2019, physicians remained uncomfortable with switching a stable patient to a biosimilar for non-medical reasons. Since the 2013 survey, there has also been a sharp increase in physicians who are highly uncomfortable with a non-medical substitution performed by a third party.
It is hoped that the findings of this study may serve as a resource for other countries in developing biosimilar policies that can build physician confidence in biosimilars. Confidence that will increase biosimilar uptake and reduce government expenditures on biological products.
Sample characteristics and methodology
In March 2019, 579 prescribers practising in six specified European countries (France, Germany, Italy, Spain, Switzerland and the UK) completed the 15-minute web-based survey that was administered in the respondents’ native language – French, German, Italian, Spanish, or English. The survey was commissioned by the ASBM and was a refreshed version of that carried out in 2013 [6]. The questionnaires were developed as a collaboration among ASBM management, ASBM membership and Industry Standard Research (ISR) management. No ‘validation’ was conducted as the instruments did not measure higher level ‘constructs’ . They are purely direct measures of opinion and attitude.
Potential respondents were identified in – and recruited from – a large, global, commercial database/panel of healthcare professionals. The response rate was high because people in this database/panel have already indicated a willingness to participate in market research. In addition, their specialties were known prior to recruitment, which decreased the rate of disqualification, as if someone was identified as representing a specialty that did not qualify for the study, they were not invited.
Respondents were paid a stipend for their participation. Stipends ranged from US$37.00 to US$48.00, depending on the specialty.
Prescriber eligibility criteria
- Must prescribe biological medicines in their practice
- Must practice in France, Germany, Italy, Spain, Switzerland, or UK
- Must specialize in one of 10 practice areas: Dermatology, Endocrinology, Gastroenterology, Haematology-Oncology, Immunology, Nephrology, Neurology, Oncology, Ophthalmology, Rheumatology
- Must have been in practice for one year or more
Online survey
The surveys were administered by ISR. In summary, prescribers were asked to rate:
- The importance of retaining sole authority to decide the most suitable biological for their patients.
- The importance of retaining the authority to deny/prevent a substitution by indicating “Do Not Substitute” or similar language when prescribing.
- Their comfort level with: a) prescribing a biosimilar to a new (treatment-naïve) patient; and b) switching a stable patient from an originator biological to a biosimilar.
- Their comfort level with a biosimilar switch for non-medical reasons, e.g. cost, coverage, a) when performed by the physician; and b) when performed by a third party.
- The importance of awarding government tenders on originator biologicals and biosimilars to multiple suppliers.
- The importance of national tender offers including factors besides price.
ISR provided statistical significance tests by country and practice area for most questions. The information from these tests made it possible to determine which answers were most significant amongst prescribers from different countries and working in different practice areas.
Information on survey participants
Participants were sourced from six countries and across 10 therapeutic areas. The detailed breakdown of this information is as follows. Table 1 provides details of the survey sample disposition.

A total of 579 responses were received:
- France: 97 (17%)
- Germany: 97 (17%)
- Italy: 97 (17%)
- Spain: 96 (17%)
- Switzerland: 95 (17%)
- United Kingdom: 97 (17%)
The breakdown of the practitioner’ s primary therapeutic areas is as follows:
Neurology (14%), Rheumatology (14%), Gastroenterology (13%), Ophthalmology (12%), Nephrology (12%), Endocrinology (11%), Dermatology (10%), Oncology (6%), Immunology (5%) and Haematology-Oncology (3%).
The largest group of prescribers (47%) practice in a hospital setting, with the remainder in academic medical centres (23%), private/family practice (18%), multi-specialty clinics (8%), community settings (3%), and other settings (1%).
Respondents’ mean experience level was 15.5 years in practice. Forty per cent of responders had been practising medicine for 11–20 years, 24% for more than 21 years, 23% for 6–10 years, and 13% for 1–5 years.
Seventy-nine per cent of responders said they commonly treat patients who are using biological medicines prescribed by another healthcare provider.
Respondents use different sources to learn about the details of a medicine for prescribing and monitoring, see figure 1.
All data refer only to those who completed the survey. All data were analysed in MS Excel and checked manually.

Results
Familiarity with biosimilars
Familiarity with biologicals versus biosimilars
When asked about familiarity with biological medicines, 58% of prescribers said they are ‘Very familiar’ , and have a complete understanding of them, compared to 41% who said the same about biosimilars. Thirty-seven per cent of prescribers said they are “Familiar”, with a basic understanding of biologicals, compared to 49% who said the same about biosimilars. And 4% had heard of biologicals but could not define them, compared to 8% who said the same about biosimilars. All prescribers had heard of biologicals whereas 2% of prescribers have not heard of biosimilars.
Since the 2013 European prescribers study, familiarity with biosimilar medicines increased from 76% to 90%; and a prescriber’ s awareness that a biosimilar may be approved for several or all indications of the reference product on the basis of clinical trials in only one of those indications increased from 63% (2013) to 83% (2019.) Strongest familiarity with biosimilars was among prescribers in Italy, Spain and Germany (48%, 47% and 44% are very familiar/have complete understanding). Prescribers in Switzerland had the lowest familiarity with only 31% stating they are very familiar/have complete understanding of biosimilars; 19% of Swiss prescribers either could not define biosimilars or have never heard of biosimilars.
Strongest familiarity with biological medicines was among Rheumatology and Gastrointestinal prescribers (96% and 88% are very familiar/have complete understanding) when compared to the other practice areas. Strongest familiarity of biosimilars was also among Rheumatology, Gastrointestinal and Endocrinology prescribers (70%, 61% and 60% are very familiar/have complete understanding).
Preferred route to familiarity
Of the respondents (n = 517) that said they were very familiar/familiar with biosimilar medicines. The top five sources of information were: 1) scientific publications (70%); 2) national medical conferences/symposia (70%); 3) international medical conferences/symposia (61%); 4) self-study (42%); and 5) CME/IME (40%).
The sources varied among the countries. For example, prescribers in the UK became more familiar with biosimilars through self-study (66%) and scientific publications (56%), while in Spain scientific publications (73%) and CME/IME (66%) were the most utilized.
The top five sources to learn about biosimilars among the respondents (n = 62) who had never heard of, nor could define biosimilar medicines were: 1) scientific publications (68%); 2) international medical conferences/symposia (61%); 3) national medical conferences/symposia (55%); 4) CME/IME (37%); and 5) reference product company sponsored education (35%).
There were no significant differences in the preferred method for becoming familiar with biosimilars among practitioners in different countries.
Biosimilar approval awareness
Prescribers in Italy (94%) had significantly higher biosimilar approval awareness compared to the rest of the countries. The specialties with the highest biosimilar approval awareness were Rheumatology (96%), Endocrinology (95%), Oncology (94%) and Gastrointestinal (92%) prescribers. All had significantly higher awareness than other specialties.
Adverse drug reaction reporting: mechanism, recording, information required, barriers
The survey showed that four out of five prescribers are legally required to report adverse drug reactions (ADRs) that are brought to their attention.
Italian prescribers rated the highest percentage for being required to report ADRs (96%), and French prescribers the lowest (69%). The practice areas in which the highest number of prescribers are required to report ADRs were Oncology (91%) and Immunology (90%).
More than half (54%) of prescribers said they are most likely to report an ADR to the National Competent Authority (NCA). The UK is significantly more likely to report to a combination of the NCA, the Marketing Authorization Holder (MAH), i.e. the manufacturer, and EMA (54%) as opposed to the NCA alone (29%).
ADR report mechanisms, time spent and follow-up
Email was utilized by almost half (49%) of prescribers (n = 550) to report ADRs to the NCA or MAH. However, when looking at specific countries, prescribers in Germany (58%) and the UK (52%) had a majority preference for paper. Prescribers in France (57%) and Italy (60%) had a preference for email, while Spain (53%) preferred a web-based tool/app.
Two-thirds (65%) of prescriber respondents said that the amount of time spent on filing a report ranged from 10 to 20 minutes, with 25% requiring less than 10 minutes and 10% requiring more than 20 minutes (average 36 minutes) While prescribers do file detailed reports, the time varies among the specialties. Dermatology (38%) prescribers need less time to file compared to other practice areas, whilst Neurology (19%), Immunology (17%) and Nephrology (14%) prescribers need more than 20 minutes to file the ADR report.
In terms of follow up from the NCA or MAH, 24% of prescribers responded they always receive follow-up, 21% very often, 30% sometimes, 19% rarely and 6% never. Prescribers in Switzerland have one of the highest rates of follow up from reporting entities (Always, 35%) compared to several other countries.
Information included in the ADR reports
When ADR reports are filed for a biological medication, 92% of practitioners responded that information about the ADR experienced by the patient are included, 84% include brand name of the biological suspected to have caused the incident, 80% include date and time of report, 72% include the non-proprietary name of the biological suspected to have caused the incident, 69% include batch number of the biological suspected to have caused the incident, including the manufacturer of the product suspected to have been associated with the reaction.
Prescribers in Italy (79%) are better about including batch number in the ADR report; Germany (74%) prescribers are better about including the manufacturer of the product; prescribers in the UK (90%) are better about including date and time.
When asked about how frequently the NCA or MAH follow-up to request the brand name or manufacturer of the product, 55% of prescribers responded either always or very often, 28% said sometimes, while 18% said rarely or never.
Fifty-five per cent of practitioners said that the level of detail required in ADR reports deters them from reporting minor events. When looking at the country specific data, prescribers in France are significantly more deterred from reporting minor events, while those in Italy are significantly less deterred.
Barriers to reporting ADRs
Fifty-five per cent of the prescribers responded that the amount of information necessary to report an adverse drug reaction deters them from reporting minor events. France (74%) is significantly more deterred from reporting minor events, while Italy (38%) is significantly less deterred.
More than half (56%) of prescribers responded that reporting infrastructure, e.g. the mechanism of reporting ADRs, was the biggest barrier to accurate reporting; another 20% responded no barriers exist. When looking at the country specific data, prescribers in Spain identified reporting infrastructure (70%) and lack of integration of electronic health records (55%) as barriers to accurate reporting more so than most countries.
Nearly all prescribers responded that they were somewhat confident (62%) or highly confident (36%) in the European pharmacovigilance system’ s ability to accurately identify the specific product at the brand-name level that might be responsible for the ADR. However, prescribers in the UK were less confident in the European pharmacovigilance system than the other countries surveyed, with only 24% reporting they were ‘highly confident’ the system would be able to accurately identify the product responsible.
Frequency of including batch number when reporting adverse events was mixed; 37% always, 27% very often, 20% sometimes, 17 % rarely/never. The survey showed that prescribers in Italy (55%) were best about including batch number (always) when compared to most of other countries. Of the prescribers who said they only included batch number sometimes, rarely, or never, more than half (53%) of prescribers responded that the reason for this was due to not having it available at time of reporting.
Automatic substitution, switching and physician choice
A high majority of prescribers (82%) feel very strongly about having control over what is prescribed and dispensed to their patients.
Opinion on sole authority for prescribers
Most prescribers agreed that it is either critical or very important (82%) that they had the sole authority, together with their patients, to decide on the most suitable biological medicine for their disease. When looking at each country, it is significantly more critical to have sole authority in deciding medicine for prescribers in Italy (94%), Switzerland (91%) and Germany (84%). When looking at specific fields, it was most important/critical to have sole authority in deciding biological medicine for Immunology (86%), Dermatology (86%) and Ophthalmology (86%) prescribers. It was least important/critical for Haematology-Oncology prescribers, 20% of whom considered it slightly/not important, compared to an average of 2% across all specialties which thought this, see figure 2.

It is significantly more critical to have sole authority in deciding medicine for Immunology, Rheumatology, Dermatology and Endocrinology.
Government tenders
Most prescribers stated that they believe it is very important or critical (63%) that government tenders for biosimilars are awarded to multiple suppliers. Prescribers in Spain and the UK, while considering this very important, do not think it is as critical for government tenders to be awarded when compared to the other countries surveyed. Only 7% and 9% considered this ‘critical’ compared to an 18% average across all other respondents.
Most prescribers agreed that it is either critical or very important (83%) that factors besides price to be taken into account in national tender offers, e.g. reliability of supply, patient support services, manufacturer reputation.
Prescriber authority to deny substitution
Most prescribers agreed that it is either critical or very important (84%) that, in a situation where substitution by a pharmacist was an option in their country, they have the authority to designate a biological medicine as ‘DISPENSE AS WRITTEN’ or ‘DO NOT SUBSTITUTE’ . It was significantly more critical for those in Switzerland (94%) to have authority to deny substitution for a biological medicine, and least so for those in the UK (73%), compared to those in the other countries. It was significantly less important for Haematology-Oncology prescribers to be able to deny substitution when compared to almost all other practice areas, see figure 3.

Identifying medicines
Eighty-five per cent of prescribers said that, when prescribing medicine including biologicals, they identify the medicine in the patient record by brand name. When looking at country and practice area responses, UK (68%) and Oncology (56%) identify medicine in a patient’ s record by brand name significantly less than those in other countries and practice areas, see figure 4.

Forty-three per cent of prescribers responded they rarely or never prescribe biological products by non-proprietary name only. When compared to the other countries, prescribers in Switzerland (40%) are most likely never to use the non-proprietary name of a product. When compared to those in other practice areas, Dermatology (32%) and Rheumatology (28%) prescribers are more likely never to use the non-proprietary name of a product, see figure 5.

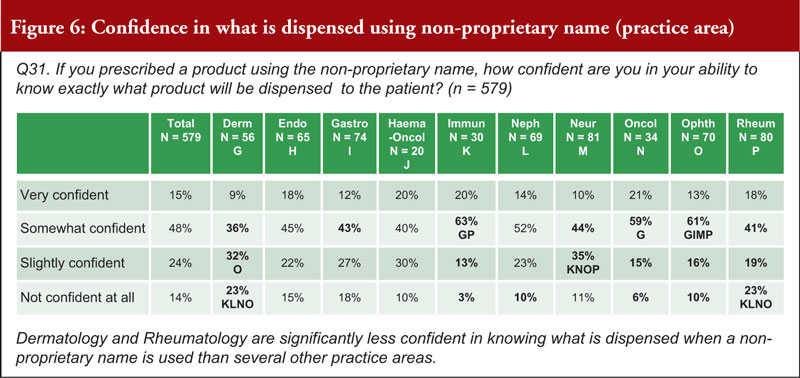
When asked about how confident a prescriber can be in their ability to know exactly what product is dispensed to a patient when using a non-proprietary name, 63% were very or somewhat confident, while 38% were slightly confident or not confident at all. Prescribers in Switzerland (26% are not confident at all) noted that they are significantly less confident in knowing what is dispensed when a non-proprietary name is used than those in Italy, Spain and the UK. Prescribers in the fields of Dermatology (55%) and Rheumatology (42%) are significantly less confident in knowing what is dispensed when a non-proprietary name is used compared to those in several other practice areas, see figure 6.
Dispensing in pharmacies
When asked about biological products dispensed directly to patients in a pharmacy, 61% of prescribers said that they were either very confident or somewhat confident that, if the pharmacy dispenses a drug that is different from the one that is prescribed (whether it is biosimilar 1, 2, or 3 or even the reference product), they have the ability to identify exactly what drug was dispensed to the patient. Thirty-nine per cent were either slightly confident or not confident at all. Prescribers in the UK (73%) said they are significantly more confident in knowing what is dispensed by pharmacy than those in Germany (49%) and Spain (60%); and those in Switzerland (24% not confident at all) are significantly less confident than several countries. It was shown that Oncology (82%) prescribers are significantly more confident in knowing what is dispensed than those in almost all of the other practice areas, see figure 7.

Eighty-three per cent of prescribers said it was critical or very important to be notified by the pharmacist if a patient has received a biological other than the one prescribed, if the patient was receiving chronic (repeated) treatment. It was shown to be significantly more critical for prescribers in Switzerland (80%) to be notified that a different biological was prescribed than for those in all other surveyed countries. It was also shown that it is significantly more critical for Rheumatology (84%) prescribers to be notified that a different biological was prescribed than those in several other practice areas; and it is significantly less important for Haematology-Oncology prescribers to be notified.
Only 5% of prescribers thought it was totally acceptable for a pharmacist to determine which biological (reference product or biosimilar) to dispense to a patient at the initiation of treatment. Fifty-eight per cent thought this was acceptable if the pharmacist’ s ability to determine the product was agreed to by clinicians in advance, and 37% thought it not acceptable. It was shown to be significantly not acceptable for a pharmacist to make the decision for prescribers in Spain (52%) and Switzerland (51%) when compared to the other countries surveyed. It was shown to be significantly not acceptable for a pharmacist to make decision more so for Rheumatology (60%) and Dermatology (52%) prescribers compared to those in other practice areas, see figure 8.

Prescribing biosimilars and switching
Seventy-four per cent of prescribers agreed that the correct definition for a ‘naïve’ patient is: a patient that has never received any biological treatment from this class of medicines. Eighty-four per cent of prescribers said they were very comfortable or somewhat comfortable in prescribing biosimilars to treat naïve patients. Prescribers in France, Germany, Italy, Switzerland and the UK are significantly more comfortable (very) than those in Spain (18%) in prescribing a biosimilar to a naïve patient. Rheumatology (60%) prescribers are more comfortable (very) than those of many other practice areas in prescribing a biosimilar to a naïve patient; Ophthalmology (10%) prescribers are the least comfortable.
Comfort level decreases when asked about switching a stable patient to a biosimilar versus to a naïve patient. While 17% are uncomfortable in prescribing a biosimilar to a naïve patient, see figure 9; twice as many (40%) are uncomfortable with switching a stable patient from an originator to a biosimilar. Spain (54%) prescribers are the least comfortable with switching a stable patient to a biosimilar. Haematology-Oncology prescribers are more comfortable switching a stable patient from an originator to a biosimilar than those in several other practice areas; Ophthalmology and Rheumatology prescribers are less comfortable, see figure 10.


Comfort level decreases further when asked about switching a patient to a biosimilar for non-medical reasons. More than half of prescribers (58%) said they are uncomfortable with switching their patients to a biosimilar for non-medical reasons. Prescribers in France are significantly more comfortable (very) switching a patient to a biosimilar for non-medical reasons than several other countries; prescribers in Italy and Spain are the least comfortable. Haematology-Oncology prescribers are significantly more comfortable (very) switching a patient to a biosimilar for non-medical reasons than those in most other practice areas, see figure 11.

Even more prescribers are uncomfortable (73%) when asked about a third party initiating such a switch. In the UK and France, prescribers were shown to be most comfortable with switching their patients (40% and 35% comfortable, respectively), while in Spain, prescribers are the least comfortable with having a third party make the switch (14%). Haematology-Oncology prescribers were shown to be significantly more comfortable with a third party switching a patient to a biosimilar for non-medical reasons (60% versus an average of 27%) than those in several other practice areas, see figure 12.

Conclusion
In summary, the survey reveals that European physicians have increased their familiarity with biosimilars since last surveyed in 2013. After 13 years of experience with biosimilars in Europe, physicians:
- Increasingly consider maintaining physician control of treatment decisions to be highly important
- Are more than twice as uncomfortable switching a stable patient to a biosimilar than they are prescribing a biosimilar to a treatment-naïve patient
- Remain uncomfortable with switching a patient to a biosimilar for non-medical reasons
- Are highly uncomfortable with a non-medical substitution performed by a third party. This figure has increased sharply since the 2013 survey
- Consider it highly important for governments to make multiple therapeutic choices available in tenders; and believe these tenders should take into account factors besides price.
Key points of the 2019 European prescribers survey on biosimilar
- More than half of prescribers are most likely to report an ADR to the National Competent Authority
- Two-thirds of prescribers said amount of time spent on filing a report is 10 to 20 minutes
- Prescribers do file detailed reports; this level of detail in turn deters 55% from reporting minor events
- More than half of prescribers said reporting infrastructure was the biggest barrier to accurate reporting; another 20% said no barriers exist
- Frequency of including batch number is mixed; not having the number available at time of reporting was selected by more than half of prescribers who said sometimes, rarely, or never
- Control over prescribing and dispensing – four out of five prescribers feel very strongly about having control over what is prescribed AND dispensed to their patients. Italy prescribers expressed the highest importance in having sole authority to decide the medicine, while France prescribers expressed the least. Switzerland prescribers expressed the highest importance in having the ability to deny a pharmacist’ s substitution, while UK prescribers expressed the least. Having this level of control was most important to Immunology, Rheumatology, Endocrinology and Dermatology prescribers.
- Product Name and Pharmacist Control
- More than 40% of prescribers said they rarely or never prescribe biological products by non-proprietary name only
- More than one-third said confidence would be lacking in knowing exactly what was dispensed to patient if they prescribed a product using non-proprietary name
- Four out of five prescribers said it would be critical or very important to be notified by pharmacist that patient received a biological medication other than one they prescribed
- Fifty-eight per cent of prescribers said it would be acceptable for a pharmacist to determine which biological to dispense on initiation of treatment, but would require clinician agreement in advance
- Prescribe Biosimilar versus Switch to Biosimilar – comfort level decreases when asked about switching a stable patient to a biosimilar versus prescribing a biosimilar to a naïve patient. About 20% are uncomfortable in prescribing a biosimilar to a naïve patient; twice as many (40%) are uncomfortable with switching a stable patient from an originator to a biosimilar. France, Switzerland and UK prescribers are most comfortable with prescribing a biosimilar to a naïve patient, while Spain prescribers are the least comfortable with switching a stable patient to a biosimilar.
- Prescriber Switch versus Third-Party Switch – Comfort level decreases when asked about switching a patient to a biosimilar for non-medical reasons. More than half of prescribers (58%) are uncomfortable with switching their patients to a biosimilar for non-medical reasons; this percentage increases to 73% when asked about a third party initiating such a switch. UK and France prescribers are most comfortable with switching their patients, while Spain prescribers are the least comfortable with having a third party make the switch.
|
Funding sources
The survey study was sponsored by Alliance for Safe Biologic Medicines (ASBM) and administered by Industry Standard Research, LLC.
This paper is funded by the ASBM.
The ASBM is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’ s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Dr Madelaine Feldman is the Chairperson of the Alliance for Safe Biologic Medicines. She has participated in advisory boards for Gilead, Lilly, Pfizer and Samsung. Mr Michael S Reilly, Esq, is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Madelaine Feldman, MD, FACR
Michael S Reilly, Esq
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Aug 9]. Available from: www.gabionline.net/Guidelines/EU-guidelines-for-biosimilars
2. Bird E. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(1):37-44. doi:10.5639/gabij.2020.0901.007
3. Schneider PJ, Reilly MS. Policy recommendations for a sustainable biosimilars market: lessons from Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):76-83. doi:10.5639/gabij.2020.0902.013
4. Jensen AR. Biosimilar product labels in Europe: what information should they contain? Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):38-40. doi:10.5639/gabij.2017.0601.008
5. Murby SP, Reilly MS. A survey of Australian prescribers’ views on the naming and substitution of biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):107-13. doi:10.5639/gabij.2017.0603.022
6. Dolinar RO, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018
7. Schneider PJ, Reilly MS. Naming and labelling of biologicals – the perspective of hospital and retail pharmacists. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(4):151-5. doi:10.5639/gabij.2016.0504.040
8. Safe Biologics. Members partners [homepage on the Internet]. [cited 2020 Aug 9]. Available from: www.safebiologics.org/member-partners/
9. Derbyshire M. USA and Europe differ in interchangeability of biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):183-4. doi:10.5639/gabij.2017.0604.039
10. Reilly MS, Gewanter HL. Prescribing practices for biosimilars: questionnaire survey findings from physicians in Argentina, Brazil, Colombia and Mexico. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):161-6. doi:10.5639/gabij.2015.0404.036
11. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/european-prescribers-attitudes-and-beliefs-on-biologicals-prescribing-and-automatic-substitution.html
Clear naming, traceability of biological medicines will protect patients
Author byline as per print journal: Adjunct Professor Valderilio Feijó Azevedo, MD, PhD, MBA; Associate Professor Brian Bressler, MD, MS, FRCPC; Madelaine A Feldman, MD, FACR; Professor Alejandro Mercedes, MD
Abstract:
As more biological medicines and biosimilars become increasingly available worldwide, clear product identification is critical for accurate pharmacovigilance. |
Submitted: 25 September 2017; Revised: 25 September 2017; Accepted: 27 September 2017; Published online first: 2 October 2017
When a patient experiences an adverse event from a medication or the medicine stops working, it is important for both the patient and their physician to know which medicine produced these effects. Yet, with a fairly new and effective class of medicines called biologicals, it is becoming more difficult for physicians worldwide to accurately track patient’s response and monitor potential problems. Luckily, the World Health Organization (WHO) – which assigns the scientific or ‘non-proprietary’ names for medicines – has developed a solution that will help physicians and regulators keep patients safe in whatever country they seek treatment.
Biological medicines – effective therapies grown in living cells – are used to treat patients suffering from serious conditions like rheumatoid arthritis, Crohn’s disease and cancer. Biosimilars are highly similar (but not exact) copies of these medicines and are becoming increasingly available worldwide. Biosimilars offer patients more treatment options at a lower healthcare cost. The complexity of biologicals and how they are produced in living cells mean that no two biologicals – whether originator or biosimilar – will ever be identical. This complexity can make these medications vulnerable to unintended properties that could result in adverse events or reduced efficacy. Clear product identification is particularly important with biologicals due to the risk of unwanted immune reactions in patients, and the sensitive and complex nature of these medicines.
For these reasons, WHO has proposed a modification to their naming system to ensure precise identification of biologicals. Biological naming is currently governed by a patchwork of policies that vary widely by country.
For example in Europe, five different biological medications manufactured by seven different companies, all sharing the non-proprietary name ‘filgrastim’ are currently being sold. While each is marketed by its trade name in Europe, many physicians around the world, including in Europe, prescribe using the non-proprietary name alone. This ambiguity leaves it unclear to the dispenser which biological medicine was intended, and unclear to the prescriber which was actually received by the patient. This makes it difficult to accurately assess which drug the patient is responding to or having a side effect from.
In addition, adverse events could be pooled or potentially misattributed to the wrong medication, making tracking the problem difficult. This leaves untraceable patients at risk for the adverse event. In Thailand, more than 15 biological medicines shared the same non-proprietary name. When a significant increase in adverse events occurred, the regulator could not easily determine which product (or products) was responsible.
Distinct naming, however, ensures accurate tracking to the exact medicine responsible, if any problems should arise. This also promotes greater manufacturer accountability for their products.
WHO, which assigns International Nonproprietary Names (INNs) to medicines, has proposed a solution that will make a uniform option available for national regulators to adopt. Their plan consists of a four-letter code called the Biological Qualifier (BQ), which will be attached to the INN name and will indicate where and by whom the biological was manufactured. When fully implemented, the BQ will extend the protections of distinct naming to patients worldwide, regardless of which country they seek treatment. This is especially helpful in the many countries without a robust pharmacovigilance system in place. For those countries which do, the BQ serves as an additional safeguard.
We commend WHO for their leadership in proposing a global solution to this problem, and await its availability for implementation. National regulatory authorities should follow suit and volunteer to be among the first to implement the WHO’s BQ proposal, bringing the many benefits of distinguishable biological naming to their patients.
Competing interests: This paper is funded by Alliance for Safe Biologic Medicines.
Adjunct Professor Valderílio Feijó Azevedo is a speaker for AbbVie, AstraZeneca, BMS, Celltrion, Janssen, Novartis, Pfizer, Roche, Sanofi, UCB; and had produced graphic material to AbbVie, Janssen, BMS, Pfizer. He is also a member of the advisory board of AbbVie, AstraZeneca, BMS, Janssen, Merck Serono, Pfizer.
Associate Professor Brian Bressler is an advisor and speaker for AbbVie, Actavis, Ferring, Genentech, Janssen, Merck, Pfizer, Shire, Takeda. He is also an advisor for Allergan, Amgen, Celgene, Pendopharm. He provides research support for AbbVie, Alvine, Atlantic Pharmaceuticals, Amgen, BMS, Boehringer Ingelheim, Celgene, Genentech, GSK, Janssen, Merck, Qu Biologic, RedHill Biopharm, Takeda. He has stock options in Qu Biologic.
Dr Madelaine A Feldman teaches at the Department of Rheumatology at Tulane University Medical School, New Orleans, LA, USA. She is on a speaker bureau for Eli Lilly, and was on a speaker bureau for UCB.
Professor Alejandro Mercedes did not provide a conflict of interest statement.
Provenance and peer review: Not commissioned; internally peer reviewed.
Authors
Adjunct Professor Valderilio Feijó Azevedo, MD, PhD, MBA
Professor of Rheumatology
Federal University of Paraná, 24 Rua Alvaro Alvin, Casa 18 Seminário, Curitiba-Paraná, PR-80740-260, Brazil
Associate Professor Brian Bressler, MD, MS, FRCPC
Founder, The IBD Centre of BC
Director, Advanced IBD Training Program
Clinical Associate Professor of Medicine
Division of Gastroenterology
University of British Columbia
770–1190 Hornby Street
Vancouver, BC V6Z 2K5
Canada
Madelaine A Feldman, MD, FACR
The Rheumatology Group, LLC
Suite 530, 2633 Napoleon Avenue
New Orleans, LA 70115
USA
Professor Alejandro Mercedes, MD
Professor of and Clinical Instructor in Clinical Oncology
Clinical Oncologist
Instituto Nacional del Cáncer Rosa Tavares (INCART)
Calle Correa y Cidró, Zona Universitaria Santo Domingo, Distrito Nacional
Dominican Republic
|
Author for correspondence: Madelaine A Feldman, MD, FACR, The Rheumatology Group, LLC, Suite 530, 2633 Napoleon Avenue, New Orleans, LA 70115, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/clear-naming-traceability-of-biological-medicines-will-protect-patients.html
A white paper: US biosimilars market on pace with Europe
Abstract:
In the US, 28 biosimilars have been approved, with 10 in the last two years. The US is keeping pace with the EU who pioneered biosimilars approvals a decade earlier. Herein, current FDA regulations and hurdles encountered for US biosimilar approval and uptake are discussed.
Submitted: 12 October 2020; Revised: 28 October 2020; Accepted: 29 October 2020; Published online first: 9 November 2020
While many sources claim that Europe is winning the race when it comes to biosimilars, a broader assessment of the landscape reveals a more encouraging story for the US. Although the European Medicines Agency (EMA) pioneered the framework for biosimilar regulation, the US Food and Drug Administration (FDA) is moving at approximately the same pace as EMA based on the number of approvals at the same time after implementation of its regulatory pathway [1].
The biosimilar regulatory framework in Europe was implemented in 2004. Within this framework, the Committee for Medicinal Products for Human Use (CHMP) provides initial assessments for marketing authorization of new medicines that are ultimately approved centrally by EMA [2]. In the US, the Biologics Price Competition and Innovation Act of 2009 (BPCIA Act) created an abbreviated licensure pathway for biosimilars and granted FDA the authority to approve these near-identical biologicals [3]. This legislation was implemented in 2010 as part of the Patient Protection and Affordable Care Act signed by President Barack Obama.
As of September 2020, approximately 10 years after implementation of the biosimilar approval pathway, 28 biosimilars have been approved by FDA, with 18 of those approvals granted in the last two years [4]. In the 10-year time period following the creation of Europe’s biosimilar regulatory pathway, EMA approved 13 biosimilar products (some of which were marketed under several different brands) [5]. From this perspective, the US appears to be on a faster pace than the EU in terms of biosimilar approvals. Currently, there are 45 biosimilars approved in Europe; however, these estimates fall to 35 when products approved in the US as follow-on biologicals via the 505(b)(2) pathway, e.g. somatropin, insulin, teriparatide, or abbreviated new drug application (ANDA) are excluded, see Table 1. Furthermore, Europe’s filgrastim biosimilar Tevagrastim®/Ratiograstim® was approved as Granix® (tbo-filgrastim) in the US via a Biologic License Application (BLA) prior to the implementation of a biosimilar approval pathway and is not included in the US biosimilar count.
Biosimilars have been developed in various therapeutic areas, including oncology and rheumatology, and are based on nine reference products. For some reference products, numerous biosimilars have been developed; for example, five biosimilars of Genentech’s Herceptin® (trastuzumab) and Amgen’s Neulasta® (pegfilgrastim) have been approved in the US and Europe. A summary of the biosimilars landscape in the US and EU is presented in Table 1.
There are currently nine biosimilar applications under CHMP evaluation, including biosimilar candidates for adalimumab (2), bevacizumab (5), pegfilgrastim (1), and trastuzumab (1) [6]. Unlike the transparency on biosimilar filings in Europe, FDA does not provide information on biosimilar candidates under review until they are approved.
Since the creation of a regulatory approval pathway, numerous guidance documents have been developed by both EMA and FDA to guide manufacturers in their development of biosimilar candidates. At least 10 documents have been released by FDA that provide guidance on scientific and quality considerations in the demonstration of biosimilarity and interchangeability [7]. EMA has published three overarching guidelines and nine product-specific guidelines for biosimilars manufacturers that address both non-clinical and clinical issues as well as quality-related issues [8].
Because EMA pioneered biosimilar regulations, the question has commonly been raised: To what extent does the European biosimilar experience translate to the US? Although the US and the EU are on a similar trajectory in terms of the number of biosimilar products approved, these markets differ in several respects. A critical point of divergence between the US and EU biosimilars terrain is the concept of interchangeability. Unique to the US, FDA may designate a product ‘interchangeable’ if it meets additional requirements beyond being biosimilar, which translates to more clinical development that includes switching studies and increased cost from a manufacturer’s perspective. To date, no biosimilar products have interchangeability status. Having a separate designation of interchangeability for an approved biological has been said to give the impression that interchangeable biosimilars are superior in quality to non-interchangeable biologicals ‒ which is not the case. A major focus area for the Association for Accessible Medicines’ Biosimilars Council has been educating physicians and payers on interchangeability designations as they are ‘waiting for interchangeability to really get on board with biosimilars’ [9].
Perceptions of prescribing physicians about the safety of biosimilars is an ongoing issue. There is a need for increased confidence, particularly around switching from a reference product to a biosimilar. Concerns are particularly heightened in the oncology therapeutic area, where treatments can be curative and clinicians fear the loss of efficacy and increased immunogenicity [10]. One of the key concerns around switching to a biosimilar is the negative counterpart of the placebo effect: the nocebo effect, a phenomenon in which negative expectations lead to worsening of symptoms [11]. Some physicians continue to have doubts regarding the rigorous approval process for biosimilars and switching studies that have been performed thus far and transfer these negative concerns to patients through body language or tone of voice when discussing treatment options. Raising awareness about the nocebo effect by educating healthcare professionals on this phenomenon may mitigate its effect in patients receiving biosimilar products [12, 13].
The biosimilars marketplace
While the European biosimilars market has been credited with higher uptake compared to the US market, rates of uptake differ from country to country in Europe and can vary significantly by product class. A report by KPMG commissioned by Medicines for Europe to analyse the procurement of medicines in hospitals in eight European countries highlighted the variability in biosimilar sales against originator in these different Member States [14]. An average of hospital biosimilar volume in March 2019 showed that Denmark achieved 63% overall biosimilar volume, with the UK coming in second at 45%. Germany had 40% biosimilar volume, France had 34%, and Belgium tied with Switzerland for last place among the countries studied at 14%. In a recent assessment of the impact of biosimilar competition in Europe, 16 European countries were reported to have achieved > 90% biosimilar utilization for filgrastim and pegfilgrastim in 2018, while utilization in Ireland was just 27%. Among antitumour necrosis factor biosimilars (adalimumab, etanercept and infliximab), Norway and Denmark had 81% and 96% biosimilar uptake, respectively, while every other country’s utilization was less than 50% [15]. Variations in adoption rates among individual European countries as well as across therapeutic areas are influenced by government involvement, reimbursement structures and tender procurement policies.
In the US, biosimilars have gained significant share in the majority of therapeutic areas in which they have been introduced, ranging on average from 20% to 25% within the first year of launch, with some projected to reach greater than 50% within the first two years [16, 17]. As expected, first-to-market biosimilars tend to capture a greater portion of the segment compared to later entrants. Filgrastim biosimilars have been on the market the longest at five years and have achieved a 72% share, while bevacizumab and trastuzumab biosimilars have approximately 40% share. Rituximab and infliximab have had the most limited adoption, with approximately 20% market share [16].
Biosimilar competition has a significant potential to impact overall drug spending, with the amount of savings per country based on the volume and list price of products prior to biosimilar entry [15, 17]. Manufacturers are reducing healthcare costs by launching biosimilars at a wholesale acquisition cost (WAC) that is generally 15% to 37% lower than the reference product WAC and 3% to 40% below the reference product average sales price (ASP). Manufacturers of originator products have had to adapt their strategies in order to stay competitive, whether it be launching a second-generation product, or a new formulation aimed at reducing side effects. Biosimilar competition and originator manufacturer responses are translating into significant savings. The annualized savings reached US$6.5 billion in the second quarter of 2020, where reference molecules sold US$20.8 billion (annualized) pre-biosimilar [18]. Savings over the next five years as a result of biosimilar alternatives are projected to exceed US$100 billion [17].
Biosimilars include both self-administered drugs obtained at retail pharmacies as well as provider-administrated drugs in inpatient and outpatient settings. In the US, the balance of cost savings to healthcare payers, providers and patients is different for each of these due to differences in payment and cost-sharing arrangements [19]. Most self-administered pharmacy-dispensed biologicals are paid for on a fee-for-service basis and the final amount paid by insurers reflects several transactions, including a confidential rebate payment. On the other hand, the costs associated with provider-administered biologicals are purchased directly from manufacturers and wholesalers or through group purchasing organizations either incorporated into prospective, bundled payments or under fee-for-service arrangements. Access to these biologicals (reference or biosimilar) is determined by formularies that may reflect the highest price concession rather than the lowest list price.
To date, there have been no self-administered biosimilars launched in the US. This is in stark contrast to Europe, where biosimilars administered via self-injection at home are available, including a recently approved subcutaneous form of infliximab [20]. It remains to be seen how self-administered biosimilars will fare in the US system where higher list price drugs may be ‘preferred’ on the formulary based on their profitability for those constructing the formulary.
Overcoming the hurdles
Despite having approved 28 biosimilars in 10 years, the US faces one of its biggest biosimilar hurdles when it comes to launching these products. So far, 18 of 28 biosimilars have been made available to patients, while the others have not been commercialized due to patent-related issues. In an effort to protect their originator products, brand manufacturers block competition by filing numerous follow-on patents. The expense of challenging these patents can serve as a deterrent to potential biosimilar competitors [21]. Brand manufacturers also disincentivize biosimilar utilization by leveraging a ‘rebate trap’ wherein brand manufacturers offer significant rebates for their products that disable competition from biosimilars coming on the market [22].
While there is work to be done to accelerate commercialization of biosimilars, FDA has taken active steps to mitigate some of these barriers to biosimilar utilization. In July 2018, Health and Human Services Secretary Alex Azar announced a Biosimilars Action Plan (BAP) to advance policies that facilitate the development of the biosimilars market and increase competition for biological drugs [23].
Key strategies of FDA’s BAP of 2018:
FDA’s most recent actions addressed biosimilar market competition in a joint statement issued by FDA and the Federal Trade Commission (FTC) that identified four goals to help combat anti-competitive practices [24]. In particular, the agencies noted their concern with false or misleading statements comparing biological reference products and biosimilars, which may be hampering biosimilar uptake by creating negative misperceptions about the safety and efficacy of biosimilar therapies. To further clarify how data and information about biosimilars should be presented in a truthful and non-misleading manner in regulated promotional materials, FDA announced the release of the ‘Draft guidance for industry: promotional labeling and advertising considerations for prescription biological reference and biosimilar products ‒ questions and answers’ and invited comment by stakeholders in the docket. As part of their efforts to promote greater competition, FDA and FTC held a public workshop in March 2020 to discuss their ‘collaborative efforts to support appropriate adoption of biosimilars, discourage false or misleading communications about biosimilars, and deter anticompetitive behaviors in the biological product marketplace’.
Targeting faster reviews, FDA issued new draft guidance in February 2020 detailing how it will speed its review of biosimilar or interchangeable application supplements, which can be used to update the initial biosimilar approval when it is for fewer than all the reference product’s licensed conditions of use [25]. At the time of submission, applicants may decide not to seek licensure of a proposed biosimilar for conditions of use that are protected by patent for the innovator. FDA had previously committed to reviewing and acting on original 351(k) BLA supplements with clinical data within 10 months of receipt; however, the agency now says such supplements will be reviewed and acted upon in a 6-month timeframe.
The US Centers for Medicare and Medicaid Services (CMS) have also revised a number of their payment policies in an effort to promote biosimilar competition. Three policy changes made under Medicare Part B to incentivize biosimilar uptake were: 1) BPCIA required Medicare Part B to pay for biosimilars at the ASP of the biosimilar plus 6% of the innovator product’s ASP [26]; 2) unique payment codes were provided for each biosimilar (reversing policy that grouped drugs under single codes) [27]; and 3) biosimilars were allowed ‘pass-through’ status under the 340B Drug Pricing Program, which meant they could be paid at ASP plus 6% rather than being heavily discounted (–22.5%) [28]. Furthermore, under Medicare Part D, the Bipartisan Budget Act of 2018 changed the treatment of biosimilars in the coverage gap (doughnut hole) discount programme, requiring manufacturers to give discounts for biosimilars [29]. Furthermore, in 2019, there was a lowering of the maximum copay amount on biosimilars to rates commensurate with generic copay amounts for lower income beneficiaries.
In parallel with updated regulatory policies that encourage development of the biosimilars market, a growing body of evidence has amassed to support the clinical safety of biosimilars. In a systematic review of primary data from 90 studies that enrolled 14,225 unique individuals, the nature and intensity of safety signals reported after switching from reference medicines to biosimilars were the same as those already known from continued use of the reference biological [30]. Similar results were observed in a systematic review of 57 switching studies evaluating the efficacy, safety and immunogenicity risk of transitioning between an originator biological and a biosimilar [31]. These results should increase confidence of patients, healthcare professionals and the public in biosimilars, leading to increased acceptance of these safe and effective medicines.
Conclusion
Although it has been suggested that biosimilars are not living up to their promise in the US, the current landscape suggests otherwise. FDA has approved 18 biosimilars in the past two years alone and a total of 28 since the implementation of a regulatory pathway just 10 years ago. Although getting these drugs into the hands of patients has hit some stumbling blocks due to anti-competitive behaviours from brand manufacturers, FDA has been proactive in addressing these ‘shenanigans’ and has created a BAP as well as additional guidances to mitigate such barriers to uptake and ensure the biosimilars market is poised for success. Clinical studies on the impact of switching between reference medicines and biosimilars have underscored the safety of biosimilars and impart confidence in their utilization for both physicians and patients.
In short, the overall outlook on biosimilars is positive. Pharmaceutical Research and Manufacturers of America (PhRMA), an organization representing biopharmaceutical manufacturers, believes that biosimilars are helping to bring down the cost of drugs in the US market and are positioned to achieve further savings. Katie Verb, Director of Policy and Research for PhRMA, says that federal policies are having a positive effect. These sentiments were echoed by Dr Leah Christl, Amgen’s Executive Director for Global Regulatory and Research and Development Policy, who believes the US biosimilars market is showing healthy vital signs.
Funding sources
This paper is funded by the Alliance for Safe Biologic Medicines (ASBM).
The ASBM is an organization composed of diverse healthcare groups and individuals ‒ from patients to physicians, innovative medical biotechnology companies and others ‒ who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Dr Madelaine Feldman is the Chairperson of the Alliance for Safe Biologic Medicines. She has participated in advisory boards for Gilead, Lilly, Pfizer and Samsung. Mr Michael S Reilly, Esq is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002 to 2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Madelaine Feldman, MD, FACR
Michael S Reilly, Esq
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Mehr SR, Brook RA. Biosimilars in the USA: will new efforts to spur approvals and access spur uptake and cost savings? Pharmaceut Med. 2019;33(1):1-8.
2. European Commission. Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending 2001/83/EC on the Community code relating to medicinal products for human use [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2004_27/dir_2004_27_en.pdf
3. Carver KH, Elikan J, Lietzan E. An unofficial legislative history of the Biologics Price Competition and Innovation Act of 2009. Food Drug Law J. 2010;65(4):671-818, ii.
4. U.S. Food and Drug Administration. Biosimilar product information. FDA-approved biosimilar products [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
5. European medicines Agency. Centrally authorized biosimilar medicines [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_bio-similar.
6. European Medicines Agency. EMA/535780/2020. Applications for new human medicines under evaluation by the Committee for Medicinal Products for Human Use. 8 October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/documents/report/applications-new-human-medicines-under-evaluation-chmp-october-2020_en.pdf
7. U.S. Food and Drug Administration. Biosimilar guidances. Current as of 21 June 2019 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances.
8. European Medicines Agency. Scientific guidelines on biosimilar medicinal products. 2020 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/ multidisciplinary/multidisciplinary-biosimilar
9. The Center for Biosimilars. AAM’s Christine Simmon on Biosimilars Council Initiatives and educating around interchangeability. 16 February 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.centerforbiosimilars.com/news/aams-christine-simmon-on-biosimilars-council-initiatives-and-educating-around-interchangeability
10. Konstantinidou S, Papaspiliou A, Kokkotou E. Current and future roles of biosimilars in oncology practice. Oncol Lett. 2020;19(1):45-51.
11. Planès S, Villier C, Mallaret M. The nocebo effect of drugs. Pharmacol Res Perspect. 2016;4(2):e00208.
12. Ebbers HC, Schellekens H. Are we ready to close the discussion on the interchangeability of biosimilars? Drug Discov Today. 2019;24(10):1963-7.
13. Rezk MF, Pieper B. To see or NOsee: the debate on the nocebo effect and optimizing the use of biosimilars. Adv Ther. 2018;35(6):749-53.
14. KPMG. Improving healthcare delivery in hospitals by optimized utilization of medicines: A study into 8 European countries. Commissioned by Medicines for Europe. 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2019/10/20190903_Hospital-Reform-Study_final.pdf
15. IQVIA. White Paper. The impact of biosimilar competition in Europe. Oct 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://ec.europa.eu/docsroom/documents/ 31642/attachments/1/translations/en/renditions/native
16. Amgen. 2020 Biosimilar trends report [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.amgenbiosimilars.com/-/media/Themes/Amgen/amgenbiosimilars-com/Amgenbiosimilars-com/spandf/USA-CBU-80723-2020-Amgen-Biosimilar-Trends-Report.pdf
17. IQVIA. Biosimilars in the United States 2020-2024. Competition, savings and sustainability. October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.iqvia.com/-/media/iqvia/spandfs/institute-reports/iqvia-institute-biosimilars-in-the-united-states.pdf
18. Fein AJ. Drug Channels Institute®; The Booming biosimilar market of 2020. 6 October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.drugchannels.net/2020/10/the-booming-biosimilar-market-of-2020.html
19. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3.
20. GaBI Online – Generics and Biosimilars Initiative. Celltrion launches infliximab biosimilar Remsima SC in Europe. 20 March 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Oct 28]. Available from: www.gabionline.net/Biosimilars/News/Celltrion-launches-infliximab-biosimilar-Remsima-SC-in-Europe
21. Chen BK, Yang YT, Bennett CL. Why biologics and biosimilars remain so expensive: despite two wins for biosimilars, the Supreme Court’s recent rulings do not solve fundamental barriers to competition. Drugs. 2018;78(17):1777-81.
22. U.S. Food and Drug Administration. Speech by Scott Gottlieb: Capturing the benefits of competition for patients.7 March 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/news-events/speeches-fda-officials/capturing-benefits-competition-patients-03072018
23. U.S. Food and Drug Administration. Biosimilars action plan: balancing innovation and competition. July 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/114574/download
24. U.S. Food and Drug Administration. Joint statement of the Food & Drug Administration and the Federal Trade Commission regarding a collaboration to advance competition in the biologic marketplace. 3 February 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/134864/download
25. U.S. Food and Drug Administration. Draft guidance for industry. Biosimilars and interchangeable biosimilars: licensure for fewer than all conditions of use for which the reference product has been licensed. February 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/134932/download
26. Centers for Medicare & Medicaid Services. MLN Matters® Number SE1509. 2014 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/Downloads/SE1509.pdf
27. Centers for Medicare & Medicaid Services. Part B Biosimilar biological product payment and required modifiers. Last modified 2 February 2018 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice/Part-B-Biosimilar-Biological-Product-Payment
28. Centers for Medicare & Medicaid Services. MLN Matters® Number MM11099 Revised. Update of the Hospital Outpatient Prospective Payment System (OPPS). Jan 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM11099.pdf
29. Congress.Gov. 115th Congress. H.R.1892 ‒ Bipartisan Budget Act of 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.congress.gov/bill/115th-congress/house-bill/1892
30. Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463-78.
31. McKinnon RA, Cook M, Liauw W, Marabani M, Marschner IC, Packer NH, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32(1):27-52.
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-white-paper-us-biosimilars-market-on-pace-with-europe.html