|
Abstract: |
Submitted: 27 January 2014; Revised: 21 April 2014; Accepted: 22 April 2014; Published online first: 5 May 2014
Biosimilars (known as subsequent entry biologics [SEBs] in Canada) present new challenges in the regulation of biotherapeutic products. New and unexpected situations are encountered with some frequency, necessitating novel modes of problem solving. Here, we explain some of the principles used in decision making around SEBs. In this context, it is critical to discuss both interchangeability and extrapolation of indications as these are the two most controversial elements in decision making in this area. We also explain the approach taken by Canada, with some unique features related to extrapolating indications and clinical uses.
The advent of biosimilars followed the expiration of the patents for biological drugs. Interest had started to grow in the manufacturing of ‘copies’ of the active medicinal ingredients of some biologicals, as well copies of their final dosage forms. This new category of biological drug was named subsequent entry biologicals (SEBs) in Canada, and known in other countries as biosimilars. It is expected that these products will be cheaper [1] than the original products on which they are based. Bioequivalence studies have long been considered sufficient to prove the safety and effectiveness of generic drug products, but biosimilars (SEBs) are not generics, they are biological products that require a specified set of studies due to their unique properties, both physico-chemical and pathophysiological. It is important to note that, contrary to generic pharmaceuticals, the only a priori characteristic of a biosimilar is that, from the chemistry and manufacturing perspective, it must be highly similar [2–5] to the reference product chosen as a comparator (RBP: reference biological product).
The term SEB is used throughout this paper, except when there is a need to clarify that SEBs and biosimilars are terms that may be used interchangeably.
Biological products are complex and can vary with the batch produced, the site of production and multiple other factors, some intangible. As a consequence, the safety and efficacy parameters of a biological product may vary from batch to batch. At the same time, it is assumed that the pharmacodynamic (PD) effect of the biological stays consistent [3, 5–7].
SEBs are similar (or highly similar) to biological products already authorized for market or, in most instances, already marketed. The product to which an SEB is compared is called a reference biological product or drug (RBP or RBD). The quality attributes are comparable or highly similar and the PD action(s) of the product remain consistent with those of the product to which it is compared [5–8]
In general, a generic drug is compared with the first product of a class. SEBs may be compared to one of several original products in the class; SEBs cannot be used as reference products for new SEBs.
SEBs have some additional distinctive characteristics compared with generic drug products:
The premise underlying the development of SEBs is that they are biological products that enter the market subsequent to an existing biological and are highly similar (biosimilar) to a comparator. As such, information in the public domain may be used to shorten the development period for that drug/drug product, thereby reducing development costs. In Canada, SEB products fall under the same provisions as new drugs: Division 8 of the Drug Regulations [5]. It must be demonstrated that the active medicinal ingredient of the SEB is similar, from the chemistry and manufacturing perspective, to the reference product, and the final dosage form is equivalent to that of the reference product. As a result, no SEB can be authorized for market with bioequivalence as the sole study type. However the SEB can, and usually is, developed via a shortened clinical development pathway. This has still some advantages as far as costs are concerned.
Before developing an SEB, sponsors would do well to understand clearly, and be able to implement appropriately, the concepts of similarity.
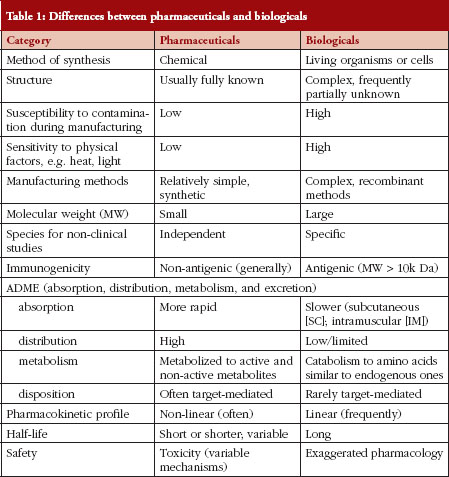
There are many differences between pharmaceuticals and biologicals, varying from their method of manufacture/synthesis to their pharmacokinetic properties, their routes of administration and the manner in which their safety and efficacy are measured. A summary of these differences is provided in Table 1.
The differences outlined in Table 1 explain why decision making regarding SEBs is so complex. In fact, many of these characteristics – of pharmaceuticals versus biologicals – appear diametrically opposed. Experience in Canada has shown that sponsors/manufacturers of biosimilars use the unique features of each biological drug product in an unexpected way when choosing their RBP. There are examples where a concentration that is not marketed and has never been authorized in Canada is being used with the argument that the concentration does not matter. This is unwise because biologicals are commonly administered by the subcutaneous route, and practical experience has shown that the kinetic profile of a biological administered by the subcutaneous route is concentration-dependent. In other instances, subtle changes to a molecule have been introduced so as not to breach intellectual property laws; however, those changes can influence the therapeutic and immunological properties of a biological agent in manners that are unforeseen.
In general, there is an acceptance by regulatory agencies that SEBs cannot be identical copies of innovator products. The differences between a pharmaceutical and a biological require specific regulatory approaches, tailored to the concept of similarity. Most countries have created regulations specific to biosimilars. These regulations have taken various approaches with concepts ranging from approximations to the generics pathway, to hybrids between the pathways for pharmaceuticals and biologicals and, as in the case of Canada, a practical approach that takes into consideration the unique properties of biologicals. These properties are also applied in a flexible way, based on the science and its evolution. Regulatory and legal considerations are also brought in the equation when deciding on a particular product.
In Canada, SEBs fall under the same provisions as those of new drugs. The difference between an SEB and a new drug is that an SEB is developed by comparison with a reference product of similar structure and mechanism of action previously authorized and marketed in Canada.
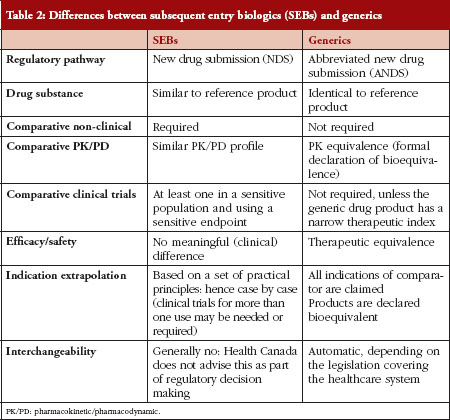
In order to understand the pathway chosen in Canada, there needs to be a comparison between SEBs and generics, see Table 2.
Table 2 outlines the differences between generic drug products and SEBs, and explains some of the concerns regarding automatic substitution or interchangeability between two biological products. Concerns surround the correlates of quality, safety, immunogenicity, clinical and post-market traceability of biosimilars. It must be emphasized that two biologicals cannot be exactly the same. That is, the demonstration that two drug products are identical, which is the a priori requirement for a drug product to be a generic copy of another, cannot be proven for biological drugs. The US Food and Drug Administration (FDA), however, could declare some biosimilars as substitutable or interchangeable based on their fingerprint characteristics: in those instances no clinical studies would be required.
The approaches taken by different jurisdictions are documented in their respective guidelines dealing with biosimilars. In addition, the WHO has published a guidance document on biosimilars where the approach taken is very similar to that taken by Canada [14].
The meaning of substitutability and interchangeability may be defined in various ways. Although an SEB may be used instead of the reference biological, exchanging a reference product during a therapeutic course and switching between that reference and its SEB raises many concerns and could put the patient at risk. Substitution and interchangeability must be thought about carefully, for the following reasons:
Differences in interchangeability and substitutability between the European Medicines Agency, US FDA and Health Canada are outlined in Table 3. Under US legislation, interchangeable or interchangeability means that a biological product is biosimilar to the reference product and there is an expectation that the two products will produce the same clinical result in any given patient. When a given biological product is administered more than once to an individual, the risk of alternating or switching between the biosimilar and its reference product (in terms of safety or efficacy) is no greater than the risk of using the reference product without such alternation or switch. This, it can be noted, is a very high bar.
In the US, some states are moving, or proposing to move, to a vote on amendments to their generic-equivalent laws to include guidance on biosimilars. These amendments propose to establish that pharmacies can only substitute if the FDA licenses a biosimilar as interchangeable, and only then if the physician does not specify: ‘brand medically necessary’.
The extrapolation of indications, based on a limited number of studies, is an important factor to discuss because most savings in the development of SEBs can be made by extrapolating several of the indications of the comparator.
While the indication of SEBs in Canada must be the same as those of the reference product, some jurisdictions, such as EMA, have considered the extrapolation of uses to all those approved for a class of products.
Since pharmacokinetic/pharmacodynamic (PK/PD) factors may differ between indications and uses for any given biological therapeutic, extrapolation must be considered very carefully and should be based on a set of principles so as to ensure that the extrapolation is carried out in a consistent manner for all SEBs using the same principles for all.
As noted above, an SEB is not identical to the reference product chosen. While science and scientific and therapeutic principles play a role in both interchangeability/substitutability and extrapolation of indications, each country has a unique approach, based on national and local laws and practice issues and perceptions. In addition to regulations that govern interchangeability/substitutability of SEBs and the extrapolation of their indications, there exist administrative processes that differ in every country regarding the substitution of a prescribed product with another ‘equivalent product’.
It is also recognized that local medical practice and standards of practice contribute to the use of a drug which, in the case of a new drug such as an SEB, carries several unknowns. That is why in many, if not most, countries automatic substitution of SEBs for the reference product is not recommended and why Canada has taken a cautious approach to extrapolation of indications and uses.
It is hoped that with increased experience, some of these uncertainties and misgivings will be overcome.
Competing interests: None of the authors declare any competing interests.
Provenance and peer review: Commissioned; externally peer reviewed.
Jian Wang, MD, PhD
Chief, Clinical Evaluation Division, Centre for Evaluation of Radiopharmaceuticals and Biotherapeutic, Biologics and Genetic Therapies Directorate, Health Canada
Patrick Bedford, BSc, MA
Senior Policy Analyst, Office of Policy and International Collaboration, Biologics and Genetic Therapies Directorate, Health Canada
References
1. U.S. Senate. Biologics Price Competition and Innovation Act of 2009, Public Law 111–148 –Mar. 23, 2010 124 Stat. 703;351.
2. U.S. Senate. The Patient Protection and Affordable Care Act, Public Law 111–148–Mar. 23, 2010 124 Stat. 119.
3. U.S. Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. February 2012 [homepage on the Internet]. 2012 Feb 8 [cited 2014 Apr 21]. Available from: http://www.basinc.com/services/gen/BiosimilarsScientific.pdf
4. Health Canada. Fact sheet: subsequent entry biologics in Canada [homepage on the Internet]. 2006 Jul 26 [cited 2014 Apr 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/activit/fs-fi/fs-fi_seb-pbu_07–2006-eng.php
5. Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs). 5 March 2010 [homepage on the Internet]. 2010 Mar 8 [cited 2014 Apr 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf
6. U.S. Food and Drug Administration. Guidance for industry: quality considerations in demonstrating biosimilarity to a reference protein product. February 2012 [homepage on the Internet]. 2012 Feb 8 [cited 2014 Apr 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
7. European Medicines Agency. Guideline on similar medicinal products. CHMP/437/04. 30 October 2005 [homepage on the Internet]. 2006 Jun 15 [cited 2014 Apr 21]. Available from: https://www.tga.gov.au/pdf/euguide/chmp043704final.pdf
8. Health Canada. Questions & answers to accompany the final guidance for sponsors: information and submssion requirements for subsequent entry biologics (SEBs). 2014 [cited 2014 Apr 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/
applic-demande/guides/seb-pbu/01–2010-seb-pbu-qa-qr-eng.php
9. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005. 22 February 2006 [homepage on the Internet]. 2006 Sep 28 [cited 2014 Apr 21]. Available from: http://www.triskel.com/2%20Guideline%20biotech%20derived%20proteins.pdf
10. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues. EMEA/CHMP/BWP/49348/2005. 22 February 2006 [homepage on the Internet]. 2006 Dec 5 [cited 2014 Apr 21]. Available from: https://www.tga.gov.au/pdf/euguide/bwp4934805en.pdf
11. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. EMEA/CHMP/BMWP/14327/2006. 13 December 2007 [homepage on the Internet]. 2008 Jan 10 [cited 2014 Apr 21]. Available from: https://www.tga.gov.au/pdf/euguide/bmwp1432706en.pdf
12. European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products). EMA/837805/2011. 27 September 2012 [homepage on the Internet]. 2012 Sep 27 [cited 2014 Apr 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
13. Health Canada. Health Canada letter to Provincial/Territorial Drug Plans Directors: Interchangeability/Substitutabiity of Subseqnent Entry Biologics (SEBs), 29 July 2010.
14. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 19 to 23 October 2009 [homepage on the Internet]. 2010 Jun 4 [cited 2014 Apr 21]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
|
Author for correspondence: Agnes V Klein, MD, DPH, Director, Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Canada, AL 0700C, Room 0372, Tunney’s Pasture, Ottawa, Ontario, K1A 0K9, Canada |
Copyright © 2014 Pro Pharma Communications InternationalDisclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/subsequent-entry-biologics-biosimilars-in-canada-approaches-to-interchangeability-and-the-extrapolation-of-indications-and-uses.html
Copyright ©2024 GaBI Journal unless otherwise noted.