Author byline as per print journal: Professor Paul J Declerck, PhD; Paul W Tebbey, PhD
|
Abstract: |
Submitted: 5 May 2016; Revised: 23 June 2016; Accepted: 24 June 2016; Published online first: 7 July 2016
Biological therapeutics are highly sensitive to formation of structural variants during manufacturing processes. These structural variants can affect clinical performance. Here, we review important recently published technical data on adalimumab consistency and manufacturing experience from a clinical perspective.
Adalimumab (Humira®, AbbVie, Inc) is a recombinant human immunoglobulin (Ig) G1 anti-tumour necrosis factor-α (TNF-α) monoclonal antibody originally approved for the treatment of rheumatoid arthritis in the US in 2002, and subsequently approved for the management of Crohn’s disease, ulcerative colitis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, and hidradenitis suppurativa [1]. Since its approval in the US, many other countries have approved its use for multiple indications [2, 3].
As is the case for many biologicals, adalimumab is a large glycoprotein that is a heterogenous mixture of structural isoforms [4–6]. Glycosylation, the addition of sugar residues by the host organism during manufacturing process, is a major post-translational protein modification resulting in different structural isoforms [7, 8]. In addition to glycosylation, enzymatic cleavage of C-terminal lysines contributes to the heterogeneity of monoclonal antibodies [9]. The formation and nature of these structural variants are highly sensitive to manufacturing processes, and changes in any of the manufacturing steps may lead to differences in the specific isoforms that are present.
Differences in glycosylation profile and cleavage of C-terminal lysines can affect the tertiary (i.e. the overall 3-dimensional shape of the protein) and quaternary structure (i.e. interaction of protein subunits) of a biological agent [10]. Thus, it is important to characterize the key quality attributes, e.g. physicochemical and structural features, of a biological drug substance and constantly monitor these during production, using robust analytical methods to help ensure product consistency, including the pattern and percentage of each structural isoform.
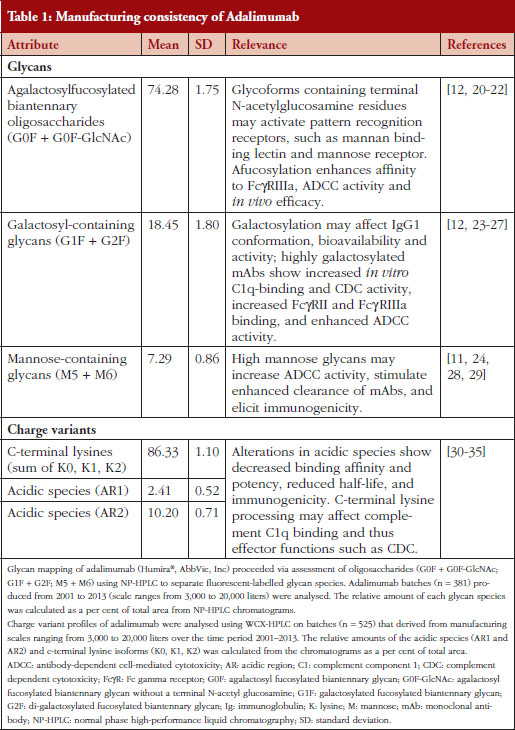
A recent product quality analysis evaluated 544 total batches of adalimumab manufactured between 2000 and 2013 [6]. Molecular charge, as measured by the presence of acidic species and C-terminal lysine variants, and glycosylation pattern were the key properties used to demonstrate the comparability and consistency of the production process, see Table 1 [6]. The quantitation of the sum and overall lysine variant profile demonstrated consistency between the batches over the course of 13 years [6]. Additionally, a high degree of consistency for the sum of lysine variants was observed between five different bioreactor sizes used for production [6]. Comparison of the identity and quantity of adalimumab oligosaccharides profile from 381 different batches indicated a high level of consistency between the batches over time and throughout the production scale changes, see Table 1 [6]. Furthermore, the consistency of the charged species and glycosylation patterns of adalimumab was supported by biological data, demonstrating that its interaction with and intrinsic binding affinity for soluble TNF-α ligand remained stable during this period [6].
It has been clearly demonstrated that the manufacturing process for adalimumab produced a consistent product over an extended (> 10 years) period of time [6], even as necessary changes to the manufacturing processes and production scale were introduced. It is paramount for manufacturers of biological products to maintain tight control of the drug substance and its production to ensure effective and safe use in patients. In the case of adalimumab, the need for a high level of consistency and product quality may be magnified, in part, because of the diverse patient populations being treated, and the underlying etiologies of the different chronic immune-mediated inflammatory diseases for which adalimumab is indicated.
Variations in glycosylation pattern or other post-translational modifications can result in subtle changes in the conformation of a biological that can potentially alter its solubility, stability, efficacy, or immunogenicity [8, 10]. Studies have demonstrated that variations in glycosylation can lead to significant changes in the circulating levels of the biological subsequent to altered pharmacokinetics, and alter its distribution to specific tissues and organs [7]. For example, therapeutic IgGs containing high-mannose glycans in the Fc region are cleared more rapidly in humans than other glycan forms [11]. Furthermore, some residues, such as galactose-containing glycans, can induce conformational changes that expose portions of the molecule previously hidden from the immune system and possibly lead to an antigenic response [8, 12].
Immunogenicity is a primary area of safety for biologicals and is often carefully monitored [13]. Immunogenic response results in the development of non-neutralizing (sometimes termed binding) or neutralizing anti-drug antibodies against the biological product. Non-neutralizing antibodies bind biological molecules on sites unrelated to target binding; however, non-neutralizing antibodies can reduce drug bioavailability through increased clearance, indirectly decreasing efficacy [14]. Neutralizing antibodies, on the other hand, bind biologicals on sites that interfere with target binding, reducing or sometimes halting efficacy [14]. Immunogenic response can have a number of outcomes ranging from adverse events, such as hypersensitivity reactions, i.e. reactions to drug administration, to reduced serum levels and neutralized activity of the biological; in other cases, it has no apparent effect on efficacy or safety of the product [9].
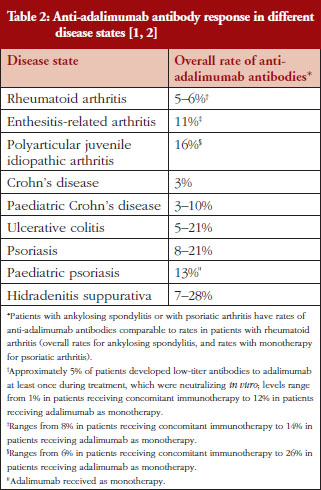
The presence of anti-adalimumab antibodies after treatment varied between 3% and 28% among different disease states, see Table 2 [1, 2, 15–17]. Because the measured incidence of anti-drug and neutralizing antibodies is highly dependent on the assay type and because multiple factors (such as assay sensitivity and specificity, methodology, disease state, sample handling, and timing of sample collection) affect the results, comparison of the incidence of antibodies between studies or between different biologicals is misleading [14]. Nevertheless, presence of anti-adalimumab antibodies, even at moderate concentration, can impact patients’ response to treatment. In studies of rheumatoid arthritis and Crohn’s disease, anti-adalimumab antibodies were associated with lower adalimumab serum levels [16, 17]. Furthermore, a higher percentage of treatment non-responders had anti-adalimumab antibodies compared with treatment responders in one study [16]. Because adalimumab is used for the treatment of patients with chronic immune-mediated inflammatory diseases, it may be important to establish and maintain a long-term immunologic equilibrium in patients treated with adalimumab. By providing a consistent product with key attributes maintained within a narrow window of variability from batch to batch, the risk of potential changes in the antigenicity of adalimumab over time is minimized.
Changes to a tightly controlled manufacturing process that may lead to variations in epitopes could disrupt the immunologically tolerated state of a patient to their initial therapeutic. Likewise, switching a stable patient to a related but different molecule, as would be the case when using a biosimilar of the initial therapeutic made by a different manufacturer, may increase antigenicity. Well-established mechanisms of tolerance include clonal deletion, receptor editing, clonal anergy, blockade of memory response, and competitive tolerance [18]. Overall, immune responses are unpredictable and immunological response to treatment with biological therapies, such as adalimumab, can vary greatly between individual patients and disease states. Furthermore, patients can develop anti-drug antibodies with clinical impact ranging from no effects to secondary loss of response. The immune response to adalimumab is a dynamic process in which anti-drug antibodies can develop and then disappear over time. Whether a stable patient who undergoes a non-medical switch between an initial therapeutic and its biosimilar (which would expose the immune system to distinct antigens) can maintain or subsequently regain tolerance is unknown. Appropriate investigation is recommended because of the paucity of available clinical data that pertain to the phenomenon of switching [19]. Importantly, data regarding the use of multiple biosimilar molecules in a single patient are also lacking.
The comparison of over 500 batches of adalimumab from 2000 to 2013 demonstrated that the key physicochemical and functional quality attributes of adalimumab have remained within a narrow range during this long time frame. Thus, patients who have been or are currently treated with adalimumab have received a consistent product during the course of their treatments, have developed immune tolerance to the antigens consistently delivered, and ultimately have achieved stability in terms of clinical response.
AbbVie funded these studies, contributed to their design, and was involved in the collection, analysis and interpretation of the data, and in the writing, review and approval of the publication. Medical writing support was provided by Patrick Little, PhD, and Maria Hovenden, PhD, of Complete Publication Solutions, LLC (North Wales, PA, USA).
Funding source: AbbVie, Inc funded the analyses.
Competing interests: Professor Paul Declerck participated at advisory board meetings for AbbVie, Amgen and Hospira and is on the Speakers’ Bureau of AbbVie, Celltrion, Hospira, Merck Serono and Roche. Dr Paul W Tebbey is an employee of AbbVie, Inc and therefore receives a salary and may own AbbVie stock or stock options.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor Paul Declerck, PhD
Department of Pharmaceutical and Pharmaceutical Sciences, KU Leuven, University of Leuven, O & N II, 49 Herestraat, BE-3000 Leuven, Belgium.
Paul W Tebbey, PhD
AbbVie, Inc, US Medical Affairs, Biotherapeutics, AP32-2, 1 N Waukegan Road, North Chicago, IL 60064, USA.
References
1. Humira® adalimumab. North Chicago, IL: AbbVie Inc. 2015 [homepage on the Internet]. [cited 2016 Jun 23]. Available from: https://www.humira.com/?cid=ppc_ppd_ggl_franchise_brand_2015_humira_Phrase_64X1790908
2. European Medicines Agency. Humira summary of product characteristics. 2015 [homepage on the Internet]. [cited 2016 Jun 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000481/WC500050870.pdf
3. AbbVie Corporation. Humira product monograph. 2015 [homepage on the Internet]. [cited 2016 Jun 23]. Available from: http://docplayer.net/334609-Product-monograph-humira-adalimumab-40-mg-in-0-8-ml-sterile-solution-50-mg-ml-subcutaneous-injection.html
4. Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28(4):363-72.
5. Sekhon BS, Saluja V. Biosimilars: an overview. Biosimilars. 2011;1:1-11.
6. Tebbey PW, Varga A, Naill M, Clewell J, Venema J. Consistency of quality attributes for the glycosylated monoclonal antibody Humira® (adalimumab). MAbs. 2015;7(5):805-11.
7. Sola RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs. 2010;24(1):9-21.
8. Revers L, Furczon E. An introduction to biologics and biosimilars. Part II: Subsequent entry biologics: biosame or biodifferent? Can Pharm J. 2010;143(4):184-91.
9. Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22(11):1383-91.
10. Dorner T, Strand V, Castaneda-Hernandez G, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322-8.
11. Goetze AM, Liu YD, Zhang Z, et al. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21(7):949-59.
12. Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics. 2010;9(8):1716-28.
13. Gils A, Vande Casteele N, Poppe R, et al. Development of a universal anti-adalimumab antibody standard for interlaboratory harmonization. Ther Drug Monit. 2014;36(5):669-73.
14. Shankar G, Pendley C, Stein KE. A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nat Biotechnol. 2007;25(5):555-61.
15. van de Putte LB, Atkins C, Malaise M, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508-16.
16. Bartelds GM, Wijbrandts CA, Nurmohamed MT, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66(7):921-6.
17. Baert F, Kondragunta V, Lockton S, et al. Antibodies to adalimumab are associated with future inflammation in Crohn’s patients receiving maintenance adalimumab therapy: a post hoc analysis of the Karmiris trial. Gut. 2016 Jul;65(7):1126-31.
18. Stewart JJ, Agosto H, Litwin S, et al. A solution to the rheumatoid factor paradox: pathologic rheumatoid factors can be tolerized by competition with natural rheumatoid factors. J Immunol. 1997;159(4):1728-38.
19. Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12(11):1473-85.
20. Ward E, Mittereder N, Kuta E, et al. A glycoengineered anti-CD19 antibody with potent antibody-dependent cellular cytotoxicity activity in vitro and lymphoma growth inhibition in vivo. Br J Haematol. 2011;155(4):426-37.
21. Herbst R, Wang Y, Gallagher S, et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther. 2010;335(1):213-22.
22. Kanda Y, Yamada T, Mori K, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology. 2007;17(1):104-18.
23. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. 2003;325(5):979-89.
24. Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci. 2015;104(6):1866-84.
25. Hodoniczky J, Zheng YZ, James DC. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol Prog. 2005;21(6):1644-52.
26. Thomann M, Schlothauer T, Dashivets T, et al. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PloS one. 2015;10(8):e0134949.
27. Kurogochi M, Mori M, Osumi K, et al. Glycoengineered monoclonal antibodies with homogeneous glycan (M3, G0, G2, and A2) using a chemoenzymatic approach have different affinities for FcgammaRIIIa and variable antibody-dependent cellular cytotoxicity activities. PloS one. 2015;10(7):e0132848.
28. Brady LJ, Velayudhan J, Visone DB, et al. The criticality of high-resolution N-linked carbohydrate assays and detailed characterization of antibody effector function in the context of biosimilar development. MAbs. 2015;7(3):562-70.
29. Alessandri L, Ouellette D, Acquah A, et al. Increased serum clearance of oligomannose species present on a human IgG1 molecule. MAbs. 2012;4(4):509-20.
30. Harris RJ, Kabakoff B, Macchi FD, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl. 2001;752(2):233-45.
31. Liu H, Ponniah G, Zhang HM, et al. In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs. 2014;6(5):1145-54.
32. Scallon BJ, Tam SH, McCarthy SG, Cai AN, Raju TS. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol. 2007;44(7):1524-34.
33. Raju TS, Lang SE. Diversity in structure and functions of antibody sialylation in the Fc. Curr Opin Biotechnol. 2014;30:147-52.
34. Vlasak J, Bussat MC, Wang S, et al. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem. 2009;392(2):145-54.
35. van den Bremer ET, Beurskens FJ, Voorhorst M, et al. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. MAbs. 2015;7(4):672-80.
|
Author for correspondence: Professor Paul Declerck, PhD, Department of Pharmaceutical and Pharmaceutical Sciences, KU Leuven, University of Leuven, O & N II, 49 Herestraat, BE-3000 Leuven, Belgium |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/importance-of-manufacturing-consistency-of-the-glycosylated-monoclonal-antibody-adalimumab-humira-and-potential-impact-on-the-clinical-use-of-biosimilars.html
| Abstract: Biopharmaceuticals are medicines whose active drug substance is made by living cells. Copies of these drugs, called biosimilars, are not identical to their reference drug and therefore specific regulatory requirements for registration apply. Whereas pharmaceutical quality evaluation requires a full dossier and a detailed comparative analysis to the reference drug, non-clinical and clinical requirements are much less extensive compared to the requirements for an innovator. Limited clinical experience and their complex nature exclude biosimilars from being considered interchangeable with the reference drug.. |
Submitted: 20 April 2011; Revised manuscript received: 22 September 2011; Accepted: 14 October 2011
Biopharmaceuticals, also called ‘biological medicinal products’ or ‘biological medicines’, are medicines whose active drug substance is made by a living organism or derived from a living organism by means of recombinant DNA and/or controlled gene expression methods. These products are polypeptides, (glyco-)proteins, and/or nucleic acids and their molecular characteristics are much more complex than traditional chemical drugs.The final biopharmaceutical product is influenced by many variables, such as the type of expression system, e.g. bacteria, yeast, and mammalian cells; the growth conditions, the purification process, the actual formulation and the conditions during storage and transport. Post-translational modifications occur during cellular synthesis, such as glycosylation, phosphorylation, sulphation, methylation, acetylation and hydroxylation which may affect biological activity and which results in an intrinsic molecular heterogeneity. It can be calculated, theoretically, that these modifications may result in more than one million product-related variants. Since this structural variability is substantial and can be very subtle, the currently available analytical techniques are insufficient to fully characterise the end product. In contrast, ‘traditional low molecular weight chemical drugs’ are produced by well-controlled and highly reproducible chemical reactions and are molecules with a small, well-defined and stable chemical structure, which can be completely characterised by analytical methods.The heterogeneity of biopharmaceuticals is further increased by the fact that these products are quite sensitive to ‘external’ conditions. The latter can affect product integrity and stability, leading, for example, to varying degrees of peptide denaturation, aggregation, oxidation, and degradation. Such modifications are less likely to occur in traditional non-biopharmaceutical drugs because they are much smaller and can be better controlled and are more predictable by nature [1-3].Importantly, and in contrast to traditional chemical drugs, biopharmaceuticals are potentially immunogenic. In this respect it is important to note that subtle structural differences, for example, consequent to small differences in the number and type of product variants, may significantly affect the immunogenic potential of the drug product [4-6]. Additionally, product- or process-related impurities can provoke an immune response [2, 7].The main differences between low molecular weight (chemical) drugs and biological drugs are summarised in Table 1.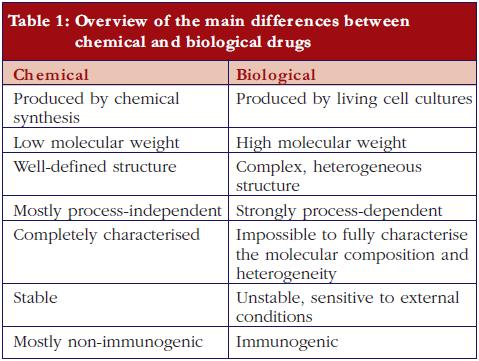
After the expiration of patent(s) for the first approved biopharmaceuticals, ‘copying’ and marketing of these biological substances can be offered by other biotech companies and might possibly, as with generics, reduce cost to patients and social security systems. However, biopharmaceuticals are made by living cells. Because of their intrinsic complexity and because no two cell lines, developed independently, can be considered identical, biopharmaceuticals cannot be fully copied. This is recognised by the European regulatory authorities and has resulted in the establishment of the term ‘biosimilar’ in recognition of the fact that, whilst biosimilar products are similar to the original product, they are not exactly the same [8, 9].European legislation has included specific guidelines for the approval of biosimilars since 2005. The Australian Therapeutic Goods Administration has adopted the European guidelines. Canadian health authorities have recently published a guidance document for approval of biosimilars, mainly based on the European guidelines [10]. Adaptation of US legislation concerning biosimilar approval processes is still under development. In March 2010, the Public Health Service Act was amended to create an abbreviated approval pathway for biosimilars [11]. Also WHO has issued guidelines for the evaluation of biosimilars [12].Thus a biosimilar, sometimes called ‘similar biological medicinal product’ or ‘follow-on biologic’ (Japan and USA) or ‘subsequent entry biologic’ (Canada), is a medicine that is similar to a biopharmaceutical that has already been authorised (the ‘reference product’). Since the active substance of a biosimilar is similar but not identical to the active substance of the reference product the regulatory requirements for approval of generics are inadequate to demonstrate the quality, efficacy, and safety of biosimilars.For a generic low molecular weight chemical drug, it is sufficient to demonstrate comparable quality, for example, content and purity, and a comparable clinical pharmacokinetic (PK) profile, i.e. relative bioavailability/bioequivalence, with a reference (innovator) product to obtain regulatory approval [13]. For biosimilars, EMA not only requires comparative quality and clinical PK studies, but also non-clinical studies, clinical pharmacodynamic (PD) studies, and limited toxicology studies, as well as comparative clinical efficacy and tolerability studies [8, 14, 15]. However, non-clinical PKs, safety profile, reproduction toxicology, mutagenicity and carcinogenicity studies are not mandatory for approval of a biosimilar, in contrast to what is required for reference biopharmaceuticals. A comparison of the European regulatory requirements for a marketing authorisation application of a biosimilar versus a reference is shown in Table 2.The guidelines for quality requirements for a biosimilar product claiming to be similar to an approved and marketed biopharmaceutical product published by the Committee for Medicinal Products for Human Use (CHMP) [14] states that the active substance in the biosimilar should be similar to the one in the reference product. Demonstration of similarity requires comparability exercises versus the chosen reference product. This implies the use of appropriately selected state-of-the-art analytical methods that are able to detect ‘slight differences relevant for quality evaluation.’ These comparability exercises also include the comparative evaluation of physicochemical parameters, biological activity using relevant bioassays and a qualitative and quantitative comparative assessment of purity and impurity profiles. The guideline also indicates that ‘… it is not expected that the quality attributes in the similar biological and reference medicinal products will be identical …’. Indeed, the quality attributes of the final biological product inherently vary with the type of host cell, the growth conditions, the purification process, the formulation, and the storage conditions. However, the CHMP requires that any difference in the quality attributes between the biosimilar and its reference product, such as variability in post-translational modifications or differences in impurity profiles should be justified in relation to its potential impact on efficacy and tolerability. The existence of differences in quality attributes between a biosimilar and the reference product is reported in the public assessment reports (EPAR) (visit www.ema.europa.eu) made available upon approval of the biosimilar. For example, for a biosimilar of epoetin alfa, differences are reported with respect to glycosylation (higher levels of phosphorylated high-mannose-type structures, lower levels of N-glycolylneuraminic acid and diacetylated neuramic acids) and oxidation (lower levels of the oxidised variant). For a biosimilar of somatropin, differences in impurities are reported as well as a higher level of deamidated variants.The general non-clinical and clinical requirements for a biological medicinal product claiming to be similar to an approved biopharmaceutical were also published by the CHMP in 2006 [15]. These are much less comprehensive compared to the requirements for an innovator, see Table 2, and evaluation is mainly based upon data obtained by comparative studies—biosimilar versus reference. In addition to the general non-clinical and clinical guidelines, product class-specific annexes to these guidelines have also been adopted for biosimilars containing recombinant interferon alfa, recombinant granulocyte-colony stimulating factor, recombinant somatropin, recombinant insulin, low molecular weight heparins, and recombinant erythropoeitins as the active substance. Draft guidelines for biosimilars containing monoclonal antibodies [16], as well as concept papers for biosimilars containing recombinant interferon beta [17] and recombinant follitropin [18] have been released for consultation.In general, non-clinical comparative tests should comprise in vitro studies, e.g. receptor-binding studies or cell-based assays, as well asin vivo PD studies. In addition, given the immunogenic potential of biopharmaceuticals, at least one repeat dose toxicity study should be performed including toxicokinetic measurements such as determination of antibody titres, cross-reactivity and neutralising capacity[6]. Such tests are particularly useful to detect the presence of hostcell proteins and/or impurities in the product. However, in contrast to the reference products, the approval process of biosimilars does not require safety pharmacology, reproduction toxicology, mutagenicity and carcinogenicity studies, see Table 2. Clinical studies required for regulatory approval of a biosimilar should include comparative PK and PD studies in healthy volunteers, followed by comparative efficacy and tolerability trials. The guideline states specifically, ‘… the clinical comparability exercise is a stepwise procedure that should begin with PK and PD studies followed by clinical efficacy and safety trial(s) or, in certain cases, PK/PD studies for demonstrating clinical comparability.’ The latter studies are usually performed only in the most sensitive and most relevant target patient population(s). In some cases, PK/PD studies alone might be considered sufficient [15] and in other cases, for example, where it is assumed that the mechanism of action of the drug is only dependent on its interaction with one single binding partner as the target, therapeutic similarity demonstrated in one indication may be extrapolated to other indications of the reference product. Extrapolation, however, remains a matter of debate especially when different indications imply the use of significant different doses [19], or different patient populations, for example, children versus adults, or when extrapolation to use in healthy individuals is concerned, for example, use of filgrastim for stem cell mobilisation and collection in healthy donors.On the other hand, the guidelines put special emphasis on assessment of the clinical tolerability of a biosimilar because of its potential immunogenicity. Indeed, differences in quality attributes between the biosimilar and its reference product cannot always be detected during the quality control process, and their clinical consequences cannot always be predicted from non-clinical animal studies [6]. Hence, clinical trials that extensively evaluate the tolerability and immunogenicity of the biosimilar are indispensable. These assessments require optimal antibody testing, characterisation of the observed immune response, and evaluation of the correlation between antibodies and their effects on PKs, PDs, efficacy and tolerability. It is also important to realise that for one product the risk of immunogenicity may differ depending on the therapeutic indication. In most cases, pre-approval data over at least a six-month period are requested and a post-approval commitment to provide data up to 12 months. Since immunogenicity is a long-term event, gathering of immunogenicity data after marketing authorisation remains an important prerequisite. Consequently, within the authorisation procedure, the company applying for regulatory approval of a biosimilar, as for any newly approved biopharmaceutical, should also provide plans for post-marketing surveillance including a risk management programme.
Clinical studies required for regulatory approval of a biosimilar should include comparative PK and PD studies in healthy volunteers, followed by comparative efficacy and tolerability trials. The guideline states specifically, ‘… the clinical comparability exercise is a stepwise procedure that should begin with PK and PD studies followed by clinical efficacy and safety trial(s) or, in certain cases, PK/PD studies for demonstrating clinical comparability.’ The latter studies are usually performed only in the most sensitive and most relevant target patient population(s). In some cases, PK/PD studies alone might be considered sufficient [15] and in other cases, for example, where it is assumed that the mechanism of action of the drug is only dependent on its interaction with one single binding partner as the target, therapeutic similarity demonstrated in one indication may be extrapolated to other indications of the reference product. Extrapolation, however, remains a matter of debate especially when different indications imply the use of significant different doses [19], or different patient populations, for example, children versus adults, or when extrapolation to use in healthy individuals is concerned, for example, use of filgrastim for stem cell mobilisation and collection in healthy donors.On the other hand, the guidelines put special emphasis on assessment of the clinical tolerability of a biosimilar because of its potential immunogenicity. Indeed, differences in quality attributes between the biosimilar and its reference product cannot always be detected during the quality control process, and their clinical consequences cannot always be predicted from non-clinical animal studies [6]. Hence, clinical trials that extensively evaluate the tolerability and immunogenicity of the biosimilar are indispensable. These assessments require optimal antibody testing, characterisation of the observed immune response, and evaluation of the correlation between antibodies and their effects on PKs, PDs, efficacy and tolerability. It is also important to realise that for one product the risk of immunogenicity may differ depending on the therapeutic indication. In most cases, pre-approval data over at least a six-month period are requested and a post-approval commitment to provide data up to 12 months. Since immunogenicity is a long-term event, gathering of immunogenicity data after marketing authorisation remains an important prerequisite. Consequently, within the authorisation procedure, the company applying for regulatory approval of a biosimilar, as for any newly approved biopharmaceutical, should also provide plans for post-marketing surveillance including a risk management programme.
In general, when copies of chemical drugs (generics) have been approved, approval has been based on demonstrated bioequivalence compared to the reference product. Having an identical structure and a proven bio-equivalence implies that the generics and reference product, as well as any two generics, are interchangeable. For biopharmaceuticals, however, the situation is completely different since two independently developed biopharmaceuticals demonstrated to be bio-equivalent will not have identical pharmaceutical quality attributes and therefore cannot be considered interchangeable in the absence of evidence gathered from adequately designed clinical studies. Indeed, potential differences in immunogenicity can only be observed in large study populations and switching between biological preparations from different origins may increase the risk of antibody development. On the other hand, it should also be realised that, in contrast to various generics of the same reference product which can be considered identical, two biosimilars, independently developed and compared to the same reference product cannot be considered biosimilar to each other. It is obvious that demonstration of similarity between biosimilar A and reference product on the one hand and between biosimilar B and reference product on the other hand does not allow any conclusions with respect to a possible similarity between the two biosimilars, i.e. the degree of similarity between A and B. Thus, from a scientific point of view as well as for the sake of patient safety, biopharmaceuticals, irrespective of their regulatory status as biosimilar or reference, should not be considered interchangeable in the absence of solid clinical data. This is also enforced in the new US Health Care Reform Bill, which clearly states that more data are required for a product to be labelled interchangeable rather than the mere fact of being biosimilar [20]. It must be stressed that if interchangeability has been proven between two biopharmaceuticals, e.g. between two biosimilars or between a biosimilar and its reference, this remains strictly valid only for the two specific products that were evaluated.It may also be of interest to note that, in this context, it is often claimed that approved manufacturing changes form the proof for a generalisation of interchangeability for biosimilars. For a variety of reasons this argument contains some major flaws. Firstly, all independently developed biosimilars have so far been proven to be different from their reference product with respect to particular quality aspects. In contrast, in most cases, manufacturing changes are accompanied by the demonstration that the majority, if not all, quality attributes remain within preset specification limits. In cases where manufacturing changes would have resulted in a significant qualitatively different composition, demonstration of clinical safety will be required [21]. Secondly, any company producing a biopharmaceutical must handle hundreds of various (process-)specific quantitative and qualitative criteria that need to be taken into account in the comparison of the products obtained before and after the manufacturing change. Therefore, a comparison (‘before’ versus ‘after’) can be made at various stages in the process. This is in contrast to the comparability exercises for biosimilars which only involve the end product. Thirdly, introduction of a manufacturing change will, at most result in only one switch for the patient in only one direction, i.e. a drug produced by the ‘old process’ to a drug produced by the ‘new process’, not vice versa. Fourthly, approval of a manufacturing change should not be interpreted as a proof that both versions can be safely switched back and forward. Taken together, even though it needs to be realised that some manufacturing changes could result in safety concerns, the putative safety risk associated with a manufacturing change can, in general, be considered a few orders of magnitude smaller compared to that associated with the differences between two independently developed biopharmaceuticals [22]. It should be stressed that a risk assessment for any biopharmaceutical always needs to be considered on a case-by-case basis.
The production of biopharmaceuticals involves complex processes and includes the development of an engineered cell line, the production of the active substance through large scale culturing of cells, the purification of the protein including a wide variety of downstream processing steps, and its formulation. Consequently, any two independently developed biopharmaceuticals starting from the same DNA sequence will be characterised by particular differences in composition.Approval of biosimilars is contingent upon a full and detailed demonstration of pharmaceutical quality, a comparative analysis with a reference product, limited non-clinical and clinical evaluations, and a post-approval follow-up. In the absence of specific data concerning interchangeability, any measures taken, e.g. by health insurance companies and/or reimbursement authorities, to control budgets by stimulating the use of less expensive biopharmaceuticals should contain a mechanism that prevents switching between products in a patient.For patientsBiopharmaceuticals are complex medicines produced by living cells. Copies of approved biopharmaceuticals have been introduced recently. Because of their intrinsic complexity such copies are similar but not identical to the reference medicine and are therefore called ‘biosimilars’. Approval of biosimilars requires a full quality analysis including a detailed comparison to the reference whereas non-clinical and clinical evaluations are less extensive. It is important to keep in mind that inherently related to the complex nature of biopharmaceuticals, similarity is not equal to interchangeability. Therefore, switching between similar biopharmaceuticals in a patient should be prevented.
References
1. Crommelin DJ, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm. 2003;266:3-16.
2. Crommelin D, Bermejo T, Bissig M, et al. Pharmaceutical evaluation of biosimilars: important differences from generic low-molecular-weight pharmaceuticals. Eur J Hosp Pharm Sci. 2005;11(1):11-7.
3. Sekhon BS, Saluja V. Biosimilars: an overview. Biosimilars. 2011;1:1-11.
4. Kessler M, Goldsmith D, Schellekens H. Immunogenicity of biopharmaceuticals. Nephrol Dial Transplant. 2006;21(5):9-12.
5. Sauerborn M, Brinks V, Jiskoot W, Schellekens H. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol Sci. 2010;31:53-9.
6. European Medicines Agency [homepage on the Internet]. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. 2007 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf
7. Schellekens H, Casadevall N. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol. 2004;251 Suppl 2:II4-9.
8. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products. 2006 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
9. Declerck PJ. Biotherapeutics in the era of biosimilars: what really matters is patient safety. Drug Saf. 2007;30:1087-92.
10. Health Canada [homepage on the Internet]. Guidance for sponsors: Information and submission requirements for subsequent entry biologics. 2010 [cited 2011 Dec 11]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/seb-pbu/notice-avis_seb-pbu_2010-eng.php
11. US legislation H.R. 1548: Pathway for Biosimilars Act. [cited 2011 Dec 11]. Available from: http://www.govtrack.us/congress/bill.xpd?bill=h111-1548
12. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 2010 [cited 2011 Dec 11]. Available from: http://www.who.int/ biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
13. Meredith PA. Potential concerns about generic substitution: bioequivalence versus therapeutic equivalence of different amlodipine salt forms. Curr Med Res Opin. 2009;25:2179-89.
14. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues. 2006 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003953.pdf
15. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2006 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
16. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products containing monoclonal antibodies Draft. 2010 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/11/WC500099361.pdf
17. European Medicines Agency [homepage on the Internet]. Concept paper on similar biological product containing recombinant interferon beta. 2010 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089208.pdf
18. European Medicines Agency [homepage on the Internet]. Concept paper on similar biological medicinal products containing recombinant follicle stimulation hormone. 2010 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089208.pdf
19. Declerck P, Darendeliler F, Goth M, Kolouskova S, Micle I, Noordam C, et al. Biosimilars: controversies as illustrated by rhGH. Curr Med Res Opin. 2010;26:1219-29.
20. Sensabaugh SM. Requirements for biosimilars and interchangeable biological drugs in the United States. Drug Inf J. 2011;45(2):155-62.
21. The international conference on harmonisation of technical requirements for registration of pharmaceuticals for human use 2004, ICH Harmonised tripartite guideline comparability of biotechnological/biological products subject to changes in their manufacturing process Q5E. [cited 2011 Dec 11]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf
22. Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2006;22:1383-91.
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
| Author: Professor Paul J Declerck, PhD, Laboratory for Pharmaceutical Biology, Faculty of Pharmaceutical Sciences, Katholieke Universiteit Leuven, Campus Gasthuisberg, O&N2, PO Box 824, 49 Herestraat, BE-3000 Leuven, Belgium |
Source URL: https://gabi-journal.net/biologicals-and-biosimilars-a-review-of-the-science-and-its-implications.html
Copyright ©2024 GaBI Journal unless otherwise noted.