Medicines regulation in the MENA region and the importance of the World Health Organization’s INN proposal of Biological Qualifier
Author byline as per print journal: Peter J Pitts, BA; Michael S Reilly, Esq
|
Abstract:
The World Health Organization should finalize its Biological Qualifier guidance. Distinguishable naming will allow quick and accurate tracing of the manufacturer, should adverse events occur and improve patient safety by reducing confusion and mishaps. This will ensure that developing nations, including those in the MENA region, have access to high quality, affordable medicines.
|
Submitted: 1 June 2018; Revised: 29 October 2018; Accepted: 30 October 2018; Published online first: 2 November 2018
Introduction
When it comes to monitoring the quality, safety and efficacy of biological medicines, distinguishable naming is imperative because biosimilar therapies are similar to, but not exactly the same as, existing biological medicines. Since no biosimilar is perfectly identical to its innovator parent, every biological – whether reference product or biosimilar – must be fully distinguishable from other biologicals to permit quick and accurate tracing of its manufacturer, should an adverse event be observed. Precise naming of all biologicals will improve patient safety by reducing confusion and mishaps in prescribing and holding manufacturers accountable. Also, differential nomenclature helps enable national health authorities to collect and compare real-world data that measure the clinical effects of biologicals including biosimilars. Insights from such data, over time, will enable us to better measure a drug’s effectiveness in delivering successful health outcomes for patients.
The World Health Organization (WHO) must finalize their Biologic Qualifier guidance. It is this organization that has the responsibility to ensure that developing nations of the world have access to affordable, quality medicines. Safety is mission critical and the Biological Qualifier is a potent tool on behalf of global public health.
The primacy of medicines quality
Over the last few years we have travelled to many countries around the world, visiting with medicines regulators from Australia, Europe, Latin America and the Middle East. Although a wide range of topics were discussed with these regulators, one of the most pressing issues for all was the urgency of access to quality medicine.
This paper will focus on medicines regulation in the Middle East and North Africa, specifically Algeria, Egypt, Iraq, Jordan, Kuwait, Lebanon, Morocco, Oman, Qatar, Saudi Arabia, Turkey and the United Arab Emirates (UAE). We will also discuss the importance of the World Health Organization’s leadership around establishing a global nomenclature policy that will help maintain the quality, safety and efficacy of biological and biosimilar medicines for that region.
Without quality, safety and effectiveness are non-starters and access is without meaning. Without quality, healthcare spending is not just wasteful – but harmful. Without quality it is all about ‘lowest price tenders’ without any consideration for value. Without quality, regulation is a sham.
Medicines quality in the Middle East
Medicine quality is on the minds of leaders in the Middle East. At the Second Annual Arab Conference on Food and Drugs (Sharm El Sheikh, Egypt, 11-13 April 2015), a conference of regional regulatory agencies, Dr Rasha Ziada (Egyptian Ministry of Health) made the important point that if a pricing authority does not take outcomes into consideration; it will lead to overall price distortions. Amen! Dr Ola Ghaleb (Ministry of Health UAE), spoke about the UAE’s strategy of performance-based risk-sharing arrangements. Outcomes are now capitalized and bolded in the international lexicon of healthcare policy. The necessary precursor to positive outcomes is quality.
A significant number of the conference presenters discussed the value of sharing pharmaceutical economic data across borders, but there was not an equal counterbalancing discussion of the value of sharing clinical data for approvals and outcomes-based decision-making processes. The opportunity to enhance regulatory capacity and product quality through collaboration and information sharing is significant. Unfortunately, cost is too often the primary focus while other priorities such as quality are left to languish.
There was certainly an effort (both on many of the panels as well as during the breaks and after hours) to stress the urgency of the quality agenda. The good news is that speaker after speaker (sometimes in passing and other times passionately) made the point that it must not just be about ‘getting the lowest price’, but also appropriately pricing the most clinically effective treatments. Cost savings without quality are no bargain.
The crisis in drug quality is very real. In Saudi Arabia, according to Alhawassi et al. [1]:
The list of essential medicines from WHO is also considered essential in primary health care in Saudi Arabia. Yet, unfortunately, many medications from this list are among the most widely substandard and counterfeited [2]. For example, one study conducted in Saudi Arabia showed that amoxicillin has already been identified as substandard [3]. Consequently, one of the central aims of advancing pharmaceuticals and patient care in Saudi Arabia is the ‘safe use’ of quality medications [4].
Access to medicine alone is not enough; medicine quality is an essential element of patient care. Quality – even for relatively common and easily evaluated medicines like amoxicillin – is a challenge and must be a policy priority. As put forth by Alhawassi et al. [1]:
One initiative of the Saudi 2030 vision plan should be to advance patient care through a more robust, safety/quality-centred culture together with a more collegial relationship between local and international drug manufacturers and Saudi regulatory authorities. Such an enhanced working relationship would result in a higher quality care to the public (Saudi, 2030 [5]). This concept of aggressive attention to better patient care through greater attention to quality and safety is only now emerging in developed and less developed countries.
The observation of scientist W Edwards Deming applies today as it did decades ago. ‘Change is not required. Survival is not mandatory’.
As in the West, generic drugs provide greater access to medicine for millions of patients in the Middle East. As in other parts of the developing world, assuring quality through robust regulatory oversight is often at counter-point with available human and financial resources. As in every part of the world, Middle Eastern health officials (from national ministries of health to local inspectorates) recognize the imperative that ‘the most expensive drug is the one that does not work’. No nation can afford to buy low quality products.
Countries around the world are struggling to adequately monitor the quality of medicines available to their citizenry. From more regular manufacturing inspections, to risk-based investigations into the sourcing of ingredients, to a rethinking of post-marketing surveillance (pharmacovigilance), there is not one single solution.
Attention to quality cannot end at product approval. This dimension is clearly elucidated from the Jordan Food and Drug Administration (JFDA). In a recent journal article Dr Hayel Mohammad Obeidat, JFDA’s Director General writes, ‘We believe that 21st century pharmacovigilance must also include tighter and more regularly monitored post-approval bioequivalence measures. It is a new and difficult task and calls for better validated methodologies for both data collection and signal prioritization. It is the responsibility of JFDA to take the leadership role and help educate our various constituencies to the importance of 21st century Phase IV monitoring and interventions [6]’.
What we need are standards and systems that recognize the situation as it exists and provide both a path for convergence with global best practices and immediate tactical programmes that can address the true situation on the ground. In brief, regions such as the Middle East require tactical, pragmatic regulation that recognizes the asymmetries inherent in an evolving regulatory ecosystem. Global institutions can play an important role in facilitating this.
Quality, pharmacovigilance and biosimilars
A key issue driving the development of 21st century regulatory pharmacovigilance strategies is the need for updated post-marketing surveillance of biosimilars. Biological medicines have revolutionized the treatment of many serious and life-threatening diseases; as patents for these products expire around the world, biosimilars are becoming available. Biologicals are very complex medicines made using living cells and cannot be copied exactly, thus copies are called biosimilar, not generic drugs. Appropriate approval standards, specific to biological medicines, are a threshold requirement for all medicines that are deemed ‘biosimilar’. The problem of alleged copies approved outside of a scientifically sound biosimilar framework is a serious safety problem and a topic for another paper. However, even in the context of sound scientific standards, vigilance cannot stop at approval for any biological.
Issues related to the particularities of biologicals (sources, process, quality requirement and new safety profile) require sophisticated new thinking.
Fundamentally, all of the players in the pharmacovigilance ecosystem will have problems characterizing biosimilar issues since we do not have an existing, validated predictive models of potential ‘hot spot’ products, base ingredients and/or suppliers. Consequently, pharmacovigilance for biologicals will have to evolve at the same time as new medicines are launched into this space. Small numbers and the novelty of biological products and their safety profiles – alone and in combination with other medicines – for manufacturers, medical providers and patients will likely render monitoring challenging. This is where global institutions can step in.
We are in a situation of post-marketing ‘indetermination’ and the first step should be to develop new epidemiological approaches that based on a better understanding of the differences between the concepts of ‘generic’ and ‘biosimilar’. We understand there can be different safety profiles for generics (based on differing bioequivalence ranges, excipient and active pharmaceutical ingredient sourcing). When it comes to biosimilar pharmacovigilance, however, variability-induced iatrogenesis concerns, differences between batches by multiple manufacturers, and the elastic definition of ‘similarity’ is not a question of ‘safety profile’, but rather of ‘concept’ [7].
Biosimilar nomenclature and the unique role of WHO
It is into this maelstrom that the steady hand of WHO is needed. And nowhere is this more true or urgent than in the current debate over biosimilar medicines generally and product nomenclature specifically.
WHO has published a draft proposal for a global system to assign ‘biological qualifiers’ (BQs) to biologicals and biosimilars [8]. A BQ is a random four-letter code assigned to a biological manufactured at a specified site. WHO said the scheme would be voluntary for each regulatory authority and applicable retroactively. The qualifier would not be part of a biological’s International Nonproprietary Name (INN), although WHO’s INN expert group would oversee the scheme. WHO said the proposed scheme is intended to avoid separate national qualifier systems. It will also permit less developed national regulatory systems to institute a globally consistent protocol that will help to guarantee the quality, safety and efficacy of biological and biosimilar medicines.
Consider Lebanon, ‘the hospital of the Middle East’. Minister Ghassan Hasbani, Lebanon’s Deputy Prime Minister and Minister of Health, is revamping the medicines tendering programme for Lebanon and one of the key tenets being weighed in the new national decision-making process is value. As His Excellency said from the podium, ‘It is not only a cost, it is an investment’. And, as with any investment, it is impossible to understand the cost without proper consideration of the return.
Minister Hasbani recognizes that biosimilars represent an important tool in expanding access to patients in Lebanon. But, as in the West, it must be access combined with quality and safety.
When it comes to health care, clarity is better than confusion, especially when it comes to drug safety — the sine qua non of medicines regulation. And that means clarity in biosimilar nomenclature.
What is so important about a biosimilar’s name? Patient safety. According to the US Food and Drug Authority, distinct and precise nomenclature for all biologicals, innovator and biosimilar, will promote accurate prescribing and facilitate accurate attribution of adverse events [9].
Distinguishable naming is imperative because biosimilar therapies are similar to, but not exactly the same as, existing biological medicines. Since no biosimilar is perfectly identical to its innovator parent, every biological – whether reference product or biosimilar – must be fully distinguishable from other biologicals to permit quick and accurate tracing of its manufacturer, should an adverse event be observed. This facilitates manufacturer accountability.
On a global level, an INN [10] is used to identify the active ingredient in a drug, which in the case of a chemical/generic drug is equivalent. Biosimilars are not identical to their innovator parents; they are ‘highly similar’.
The differences, however, are crucially important since there is the potential for all biologicals to elicit dangerous immune responses. For this reason, if biosimilars use identical INNs and prescribing, dispensing or adverse event records identify products only by INNs, global regulators cannot recognize precisely which product is causing a problem. Testimony to WHO showed that there is a high level of ambiguity in attributing adverse events to a specific manufacturer when products share the same non-proprietary name [11]. Efforts to include other identifiers, such as batch number, that would distinguish between products and manufacturers routinely fail [12]. The addition of a unique suffix to the non-proprietary name provides a distinguishing feature that can be used to enhance traceability in a marketplace with multiple similar options.
Precise naming will improve patient safety by reducing confusion and mishaps in prescribing and dispensing; biosimilars are not identical to the reference product or one another, thus switching from one product to another may not be appropriate. Also, differential nomenclature helps enable national health authorities to collect and compare real-world data that measure the clinical effects of biologicals including biosimilars. Insights from such data, over time, will enable us to better measure a drug’s effectiveness in delivering successful health outcomes for patients.
WHO must finalize their BQ guidance. It is this organization, after all, that has the responsibility to ensure that developing nations of the world have access to affordable, quality medicines. Safety is mission critical and the BQ is a potent tool on behalf of global public health.
Concluding thoughts on the global regulatory fraternity
Our experience with healthcare regulators worldwide has reinforced our belief that health care and health policy professionals devoted to ensuring timely access to innovative medicines, quality generic drugs and biosimilars. It is not easy, and it is not only a job – it is a personal public health mission.
There are many issues surrounding the introduction of biosimilars into the global healthcare ecosystem: safety, effectiveness, interchangeability, potential adverse medical events, appropriate regulatory labelling and physician prescribing guidelines. But, even so, biosimilars are here. They are safe and effective. They are less costly. And they deserve a seat at the therapeutic table.
When it comes to biosimilar nomenclature, it is important for WHO to look … backwards. According to the 10th century Arab physician, Ibn Sina, ‘The time of action must be observed, so that essence and accident are not confused’.
Funding
This paper is funded by Alliance for Safe Biologic Medicines.
Competing interests: Mr Peter J Pitts declares no conflict of interest. Mr Michael S Reilly, Esq, is the Executive Director of and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Peter J Pitts, BA
Former Associate Commissioner, United States Food and Drug Administration
Visiting Professor, Université Paris Descartes Medical School
President, Center for Medicine in the Public Interest
757 Third Avenue, 20/F
New York, NY 10017, USA
Michael S Reilly, Esq
Executive Director
Alliance for Safe Biologic Medicines
PO Box 3691, Arlington
VA 22203, USA
References
1. Alhawassi TM, Abuelizz HA, Almetwasi M, Mahmoud MA, Alghamdi A, Alruthia YS, et al. Advancing pharmaceuticals and patient safety in Saudi Arabia: a 2030 vision initiative. Saudi Pharm J. 2018;26(1):71-4.
2. Caudron JM, Ford N, Henkens M, Macé C, Kiddle-Monroe R, Pinel J. Substandard medicines in resource-poor settings: a problem that can no longer be ignored. Trop Med Int Health. 2000;13(8):1062-72.
3. Salomon JA, Vos T, Hogan DR, Gagnon M, Naghavi M, Mokdad A. Common values in assessing health outcomes from disease and injury: disability weights measurement study for the Global Burden of Disease Study 2010. Lancet. 2012:15;380(9859):2129-43.
4. Australian Commission on Safety and Quality in Health Care. Medication Safety; Safety and Quality improvement Guide, Australia 2012 [internet]. 2012 [cited 2018 Oct 29]. Available from: https://www.safetyandquality.gov.au/wp-content/uploads/2012/10/Standard4_Oct_2012_WEB.pdf
5. Wikipedia. Saudi Vision 2030 [homepage on the Internet]. [cited 2018 Oct 29]. Available from: https://en.wikipedia.org/wiki/Saudi_Vision_2030
6. Al Kayyali L, Al Haqaish W, Bawaraesh N, Pitts PJ. The Jordan Food and Drug Administration: a culture of quality and continuous improvement. J Comm Biotechnol. 2014;20(4).
7. Pitts PJ, Louet HL, Moride Y, Conti RM. 21st century pharmacovigilance: efforts, roles, and responsibilities. Lancet Oncol. 2016;17(11):e486-e492.
8. World Health Organization. Essential medicines and health products. International Nonproprietary Name [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/en/
9. U.S. Food and Drug Administration. Labeling for biosimilar products. [homepage on the Internet]. [cited 2018 Oct 29]. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm493439.pdf
10. World Health Organization. Essential medicines and health products. Biological qualifier [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/inn_bio_bq/en/
11. World Health Organization. 63rd Consultation on International Nonproprietary Names for Pharmaceutical Substances. Geneva, 18-21 October 2016 [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/63rd_Executive_Summary.pdf
12. World Health Organization. 64th Consultation on International Nonproprietary Names for Pharmaceutical Substances. Geneva, 4-7 April 2017 [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/64th_Executive_Summary.pdf
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/medicines-regulation-in-the-mena-region-and-the-importance-of-the-world-health-organizations-inn-proposal-of-biological-qualifier.html
The need for distinct nomenclature for originator and biosimilar products
Author byline as per print journal: Michael Sarshad, BSc, MBA; Rosanne Campbell, BComm, PGdip, MSc; Peter J Pitts, BA; Jacqueline anderpuye- Orgle, MSc, PhD
|
Abstract:
As the number of biosimilar product approvals continues to grow, it will become even more important to collect pharmacovigilance data that are accurate and attributable to the specific product. Different countries and regions have taken various approaches to ensuring accurate product traceability. Meanwhile, how biosimilars are named has become complex and inconsistent globally. The US Food and Drug Administration’s (FDA) recently published final guidance states that a distinct, four-letter suffix should be added to the non-proprietary name. This approach has its critics who contain that it causes confusion for prescribers and will create an artificial barrier inhibiting prescribing. Through a literature review, the authors explore the impact that introducing such a suffix has on safety reporting, product-specific tracking, prescriber comfort with biosimilars, and biosimilar uptake. The findings from outside of the US suggest that differential naming facilitates pharmacovigilance and better product traceability and helps to minimize inadvertent substitution. Also, based on the evidence presented, it would seem that a system using International Nonproprietary Names (INNs) is essential for increasing physician comfort with the use of biosimilars. Furthermore, early data belie the notion that the US differential naming approach hinders the possibility of a favourably biosimilar marketplace and the ability for a biosimilar sponsor to compete successfully.
|
Submitted: 7 November 2018; Revised: 21 December 2018; Accepted: 27 December 2018; Published online first: 31 December 2018
Introduction/Study Objectives
The availability of biological drugs is an important advancement in treating complex diseases. However, the inherent complexities in developing and manufacturing biologicals have inevitably meant that their use is costly, which in turn places a high burden on the healthcare system. In the US, the per cent of pharmaceutical spending attributable to biologicals has increased from 13% in 2006 to 27% in 2016 [1]. In 2007, the average daily cost of a biological was 22 times that of a small molecule drug [2]. Biosimilars present the possibility of lowering these costs while offering additional treatment options for patients.
The pace of biosimilar approvals in the US has been accelerating, with the US Food and Drug Administration (FDA) approving five products in 2017, two of which were for the blockbuster originator biologicals Humira® and Remicade® [3]. Currently, there are 12 biosimilar products approved in the US and 22 distinct biosimilar products approved in the EU, see note. In fact, in the first eight and a half years after the regulatory pathway was established, FDA approved more biosimilars to more reference products (12 unique biosimilars to eight reference products) than the EU (five unique biosimilars to two reference products). As the number of biosimilar approvals continues to grow, it will become even more important to collect pharmacovigilance data that are accurate and attributable to the specific product. The World Health Organization (WHO) proposed an approach of assigning all biological products a 4-6 character alfa-numeric biological qualifier (BQ) code. The BQ would consist of four random consonants and an optional 2-digit checksum (calculated from the letters and their order in the BQ code). In October 2017, WHO announced that it would not proceed with BQ code system.
Different countries and regions have taken various approaches to ensuring accurate product traceability; the EU passed legislation requiring the recording of brand name, batch number and lot number, while the US and Japan pursued the use of distinguishable non-proprietary names. The latter has been criticized by some who believe it may negatively impact the uptake of biosimilars.
How biosimilars are named has become complex and inconsistent globally. FDA’s final guidance on the naming of biologicals (reference products and their biosimilars), with the addition of a distinct four-letter suffix to the non-proprietary name, was a major step forward in supporting more accurate pharmacovigilance. Those in favour of the suffix contend that the distinct nomenclature allows for better product-specific tracking of safety and real-world effectiveness. This in turn promotes prescriber and patient comfort with biosimilars, thus serving an important role in the overall adoption of biosimilars and enhancing a robust marketplace.
Those who argue against differential naming of biosimilars, including the US Federal Trade Commission (FTC), state that it causes confusion for prescribers, as distinguishable non-proprietary names will make physicians believe there is a difference between the originator and the biosimilar. This, they suggest, would create an artificial barrier by discouraging prescribing, with the consequence of slowing uptake and potentially of inhibiting cost savings.
The recent FDA guidance on biosimilar naming and an FTC statement in a recent Department of Health and Human Services (HHS) document, American Patients First: The Trump Administration blueprint to lower drug prices and reduce out-of-pocket cost, prompted a review of published evidence to examine the impact of a distinct four-letter suffix as part of a biological product and a global naming standard for biosimilars [4, 5].
Methods
An ad-hoc literature review was conducted between January and October 2018 to gather evidence related to the various stakeholders impacted by introducing a distinct suffix as part of biological product non-proprietary names. These stakeholders included regulatory agencies, physicians, payers, patients, government/affiliated agencies and advocacy groups. PubMed was searched for evidence related to differential nomenclature published in the last five years (2013–2018). Additional sources of evidence included ClinicalTrials.gov, pharmacovigilance databases, naming guidelines and surveys. Literature was reviewed to identify the link between differential nomenclature and better safety reporting, real-world effectiveness and prescriber comfort with biosimilars.
Results
Better safety reporting and real-world effectiveness
In Europe, guidelines on good pharmacovigilance practices (GVP) have been developed for biological medicinal products and require that prescriptions for originator biologicals and biosimilars specify brand names [6]. This helps to reduce ambiguous adverse event (AE) reporting, i.e. an inability to link an AE to a specific product [originator or biosimilar]. A review of the European Eudravigilance database found that during the period March 2017 to February 2018, around 25% of the AEs reported for infliximab were ambiguous even after implementing GVP, with no link to originator or biosimilar, see Figure 1. While these guidelines are beneficial, adopting the current FDA system of a distinct four-letter suffix for biologicals can further reduce ambiguity.
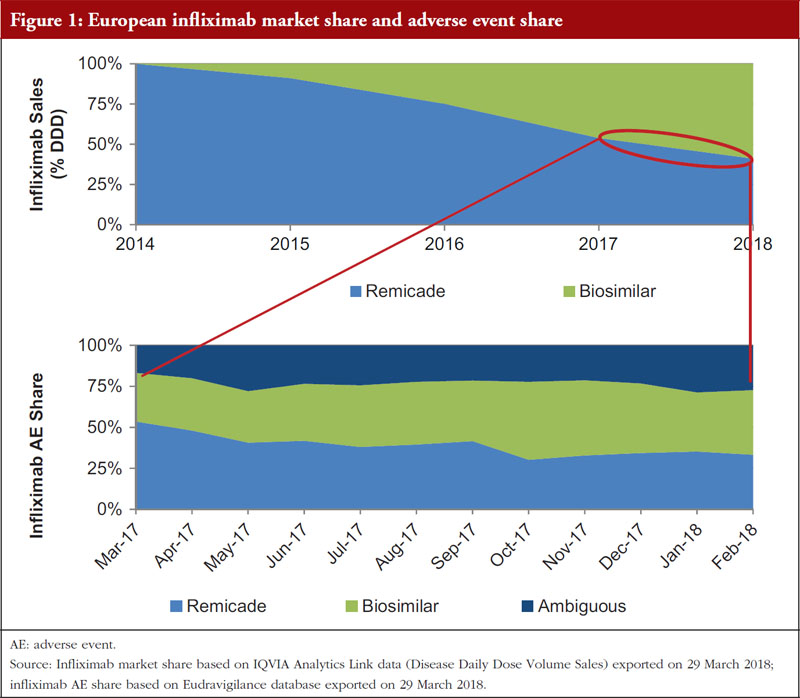
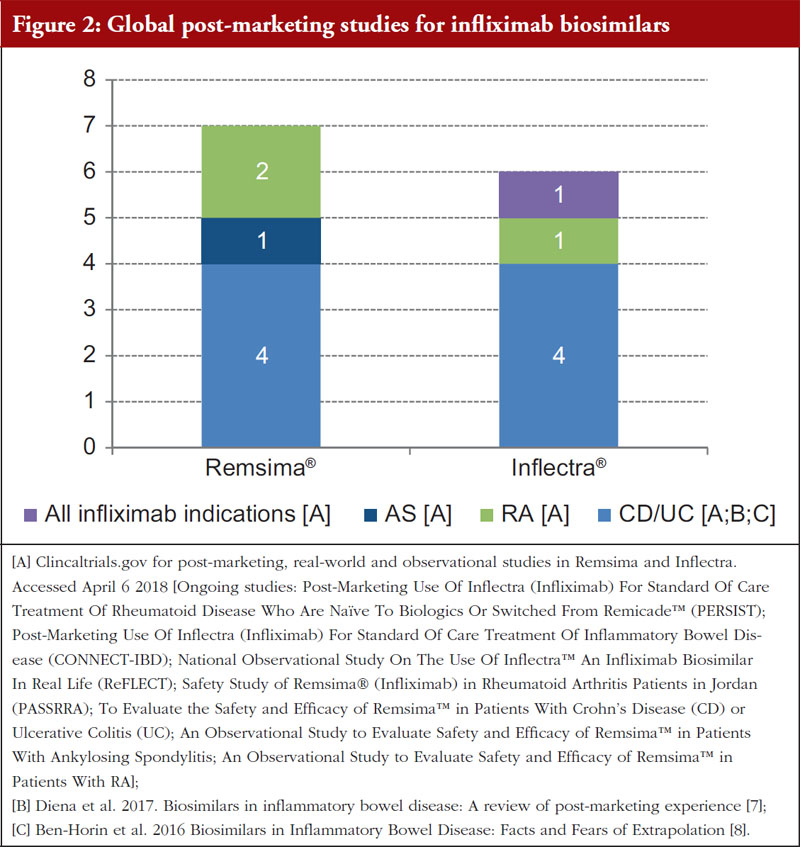
However, the distinction between infliximab biosimilar products using differential scientific nomenclature has been sufficient to facilitate post-marketing studies, particularly in inflammatory bowel disease (IBD) indications, as limited data were available at the time of regulatory approval. The Clinicaltrials.gov site was reviewed to establish the number of ongoing, global real-world and observational studies conducted post-marketing for infliximab biosimilars, see Figure 2. The Clinicaltrials.gov database was selected as it includes research studies conducted in 205 countries. In addition, evidence from two published sources, Diena et al. (2017) and Ben-Horin et al. (2016), which reviewed Inflectra® and Remsima® post-marketing studies, see Figure 2 [7, 8], was incorporated.
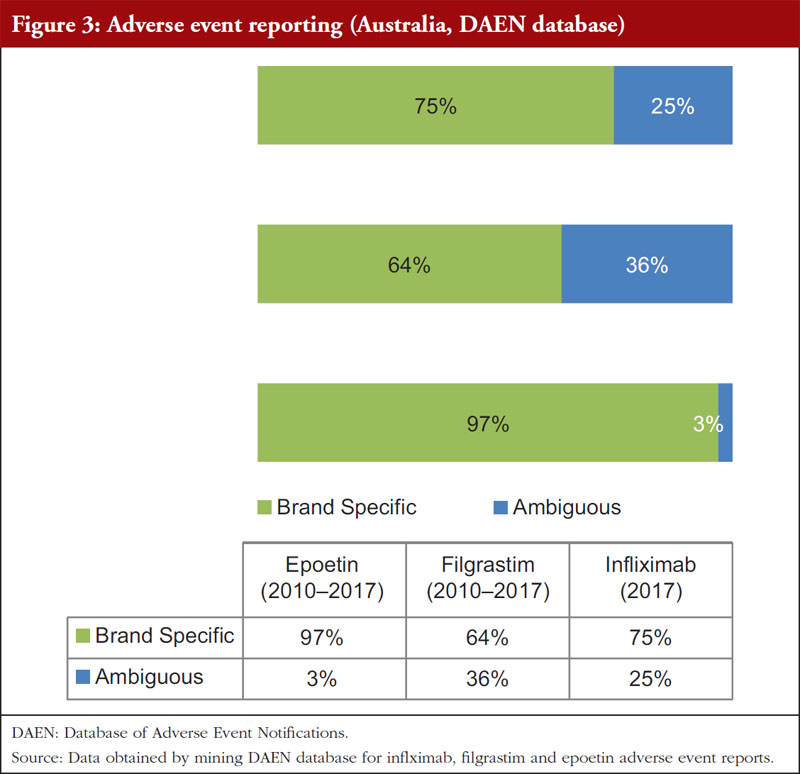
Furthermore, a review of the Australian Therapeutic Goods Administration (TGA) Database of Adverse Event Notifications (DAEN) was conducted to determine if there was a difference in AE reporting between the biosimilars infliximab, filgrastim and epoetin. In Australia, infliximab and filgrastim biosimilars do not have distinct Australian Biological Names (ABN). However, the TGA has used a distinct ABN for epoetin lambda, a biosimilar of epoetin alpha, due to apparently different glycosylation patterns [9]. The data highlight that infliximab (25%) and filgrastim (36%) were associated with higher rates of ambiguous reporting compared to epoetin (3%), see Figure 3.
Prescriber comfort with biosimilars
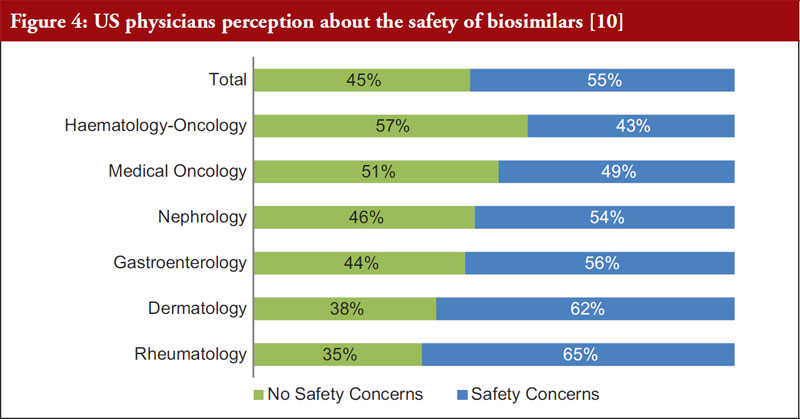
It is relevant to understand physician perceptions of the safety of biosimilars. FDA can only approve a biosimilar if it is highly similar to, and has no clinically meaningful differences from, the originator biological. However, in a survey of US physicians across a variety of specialties, over 50% of respondents expressed safety concerns with FDA-approved biosimilars, see Figure 4 [10].
The Biosimilars Forum conducted a survey from November 2015 to January 2016 of US physicians who frequently prescribe biologicals (Reported in Cohen et al. [10]). The aim of the study was to investigate physician awareness, knowledge and perception of biosimilars. Figure 5 indicates that the vast majority of respondents (75%) considers it necessary to seek additional information on biosimilars prior to prescribing them.

In 2015, the Alliance for Safe Biologic Medicines (ASBM) conducted a survey of US Healthcare Professionals (HCPs) to establish their perspective on biosimilar naming. In this survey, 66% of respondents supported distinct non-proprietary naming for originator biologicals and biosimilars, see Figure 6 [11]. Similarly, a 2017 ASBM survey of Canadian healthcare providers found that 68% of respondents wanted distinct non-proprietary naming for all biologicals [12].

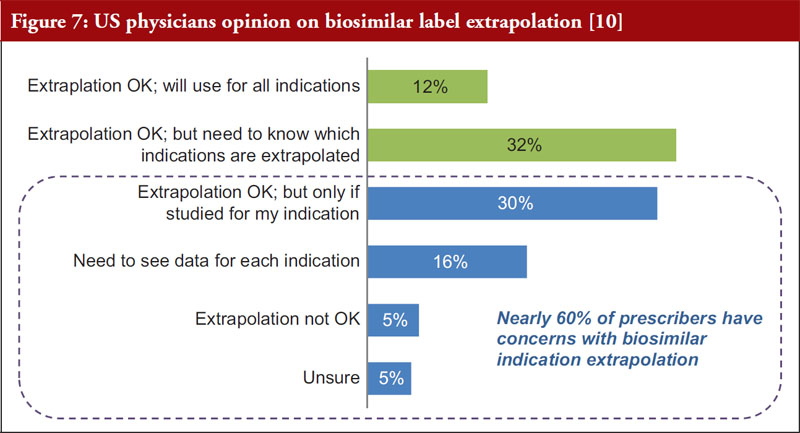
There are cases in which the biosimilar is approved in an indication based on the extrapolation of other clinical data. A 2016 survey of US physicians (Cohen et al. [10]) established that roughly 60%, see Figure 7, of respondents expressed concerns with using biosimilars in indications where approval was extrapolated from other clinical data [10]. By promoting effective pharmacovigilance, differential nomenclature may help to alleviate physician concerns with the concept of extrapolation.
Discussion
Due to the inherent complexities associated with developing and manufacturing biosimilar products, differentiating them is essential to both product uptake and product-specific tracking. As highlighted in the introduction, those who argue against distinct biological naming maintain that it does not support biosimilar competition and creates an environment unconducive to a biosimilar manufacturer’s commercial success. However, based on the evidence presented, distinct nomenclature would seem to be essential for accurate pharmacovigilance and to increasing physician comfort with the use of biosimilars, thereby promoting biosimilar uptake and creating a viable competitive market.
Biosimilar uptake
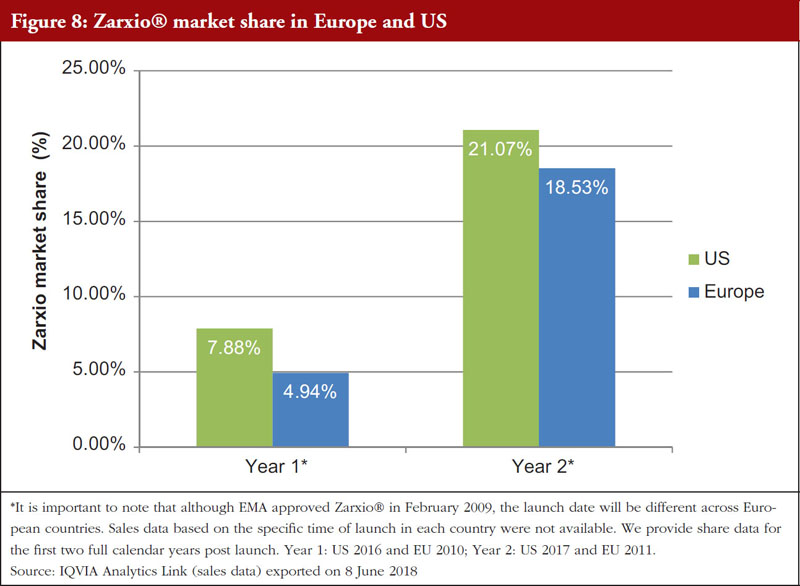
Having identified the need for a global naming standard, different countries have taken varying approaches to implementation. Some may argue that the system used by FDA may hinder biosimilar uptake, however, market data have proven otherwise. While it is not currently possible to make any causal inferences in this paper, anecdotal evidence suggests that distinct non-proprietary names have not deterred uptake. One example of note is the launch of the biosimilar Zarxio®. Zarxio®, a biosimilar of filgrastim, was approved in Europe in 2009 and in the US in 2015. The FDA-designated suffix in the US is filgrastim-sndz, whereas in the EU, all biosimilars are identified by a new brand name with no suffix to the scientific name. A comparison of market share in the first two full calendar years after approval reveals that Zarxio® had slightly better uptake in the US versus in Europe, see Figure 8.
This suggests that the US differential naming approach of including a suffix does not hinder the possibility of a favourable biosimilar marketplace and the ability for a biosimilar sponsor to compete successfully.
As demonstrated in Figure 8, Zarxio® uptake more than doubled in the second full calendar year post-launch.
The recent US HHS blueprint (To Lower Drug Prices and Reduce Out-of-Pocked Costs) stated that physician and patient confidence in biosimilars is critical to increased acceptance of biosimilars [5]. The evidence suggests that additional information and data about biosimilars could help physicians become more comfortable with using them. In an ASBM (Alliance for Safe Biologic Medicines) survey of Canadian physicians, only 54% of respondents stated that should two biological medicines have the same INN, the implication is that they are identical [12]. In a survey of US physicians, 76% of respondents indicated that they had the greatest interest in learning more about the totality of evidence related to safety, efficacy and potency of biosimilars [10]. FDA’s current naming policy (a distinct four-letter suffix as part of a biological product) facilitates better pharmacovigilance and product traceability, which can promote physician comfort with prescribing biosimilars. Assigning distinct non-proprietary names for biosimilars also supports the development of real-world pharmacovigilance data for biosimilars and allows for that information to be disseminated to physicians who often request data to make an informed decision on product therapy choice. Increased comfort in prescribing biosimilars can contribute to a more robust and competitive marketplace if it impacts adoption positively.
Product-specific tracking and physician comfort
A lack of unique names across biological products complicates the reporting and tracking of AEs; allowing for better pharmacovigilance and safety tracking would help to alleviate some of these concerns. Unique non-proprietary names of biosimilars can facilitate access to information on individual products, which will lead to better physician understanding of biosimilars and ultimately comfort in prescribing them.
Biosimilar label extrapolation is an area of concern for some physicians. The data suggest that differential nomenclature may promote product-specific tracking in post-marketing studies for extrapolated indications, as described in Figure 2. Prescribers may be more comfortable with label extrapolation if they have confidence in the traceability of different biosimilar products and their specific studies.
The evidence suggests that differential nomenclature will facilitate traceability, helping to demonstrate the real-world safety and efficacy of biosimilar products. This supports the development of high quality products and continuous investment in manufacturing, as manufacturers can be held accountable for the products distributed. Establishing a reputation for high quality manufacturing eases entry into the biosimilar market, encourages adoption, and promotes marketplace competition. Developing high quality, traceable biosimilar products will contribute to formulary inclusion, enabling payers to better leverage competitive market forces to control the cost of care.
In summary, it is believed that findings outside of the US support the notion that a differential naming policy facilitates pharmacovigilance and better product traceability and will also help to minimize inadvertent substitution for drugs distributed via retail pharmacies. This supports manufacturer accountability and the development of high quality products in addition to more attributable measurement of efficacy and safety outcomes for biosimilars.
Conclusion
The number of available biosimilars will continue to grow, therefore, it is essential to have a system that allows these products to be differentiated and traced. The evidence collected in this review demonstrates that a distinct four-letter suffix as part of a biological product may reduce the ambiguity in AE reporting, facilitate post-marketing studies in indications with limited data at the time of regulatory approval and alleviate physician concerns related to biosimilar safety and extrapolation. Distinguishable suffixes for biologicals will allow better product tracking and awareness around product effectiveness/outcomes, which ultimately will strengthen physician and patient comfort with biosimilars. Thus, it will enable a competitive and sustainable biosimilar marketplace while promoting biosimilar uptake.
Competing interests: Michael Sarshad, Rosanne Campbell and Peter J Pitts have received funding for this research from Amgen.
Jacqueline Vanderpuye-Orgle was employed by Amgen and held Amgen stock during this research. She is currently employed by Parexel.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Michael Sarshad, BSc, MBA
Senior Engagement Manager
Commercial Strategy & Planning, Consulting Syneos Health
Rosanne Campbell, BComm, PGdip, MSc
Syneos Health
10 Bloomsbury Way, London WC1A 2SL, UK
Peter J Pitts, BA
Former Associate Commissioner, United States Food and Drug Administration
Visiting Professor, Université Paris Descartes Medical School President, Center for Medicine in the Public Interest 20/F, 757 Third Avenue, New York, NY 10017, USA
Jacqueline Vanderpuye-Orgle, MSc, PhD
Parexel International
Glendale Adventist Medical Center, Suite 140, 1560 E Chevy Chase Drive, Glendale, CA 91206, USA
Note: Distinct biosimilar products approved in EU include: (1) products in the EU that are considered ‘transition’ products in the US, i.e. biological products approved pursuant to new drug applications (NDAs), including section 505(b)(2) NDAs, e.g. growth hormones, insulins; and (2) products licensed in the EU under multiple brand authorizations.
References
1. Morton FS, Boller LT. Enabling competition in pharmaceutical markets. Hutchins Center Working Paper #30. May 2017.
2. Statement of Ed Weisbart, MD, Chief Medical Officer, Express Scripts in Hearing Before the Subcommittee on Health of the Committee on Energy and Commerce. May 2, 2017.
3. U.S. Food and Drug Administration. FDA-Approved Biosimilar Products [homepage on the Internet]. [cited 2018 Dec 21. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580432.htm
4. US Federal Trade Commission. Statement of the Federal Trade Commission to the Department of Health and Human Services Regarding the HHS blueprint to lower drug prices and reduce out-of-pocket costs [homepage on the Internet]. [cited 2018 Dec 21]. Available from: https://www.ftc.gov/policy/advocacy/advocacy-filings/2018/07/statement-federal-trade-commission-department-health-human
5. Federal Register. Notice by the US Health and Human Services Department. HHS blueprint to lower drug prices and reduce out-of-pocket costs. May 2018 [homepage on the Internet]. [cited 2018 Dec 21]. Available from: https://www.federalregister.gov/documents/2018/05/16/2018–10435/hhs-blueprint-to-lower-drug-prices-and-reduce-out-of-pocket-costs
6. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP). Product- or population-specific considerations II: biological medicinal products. 4 August 2016 [homepage on the Internet]. [cited 2018 Dec 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/08/WC500211728.pdf
7. Deiana S, Gabbani T, Annese V. Biosimilars in inflammatory bowel disease: A review of post-marketing experience. World J Gastroenterol. 2017;23(2):197-203.
8. Ben-Horin S, Vande Casteele N, Schreiber S, Lakatos PL. Biosimilars in inflammatory bowel disease: facts and fears of extrapolation. Clin Gastroenterol Hepatol. 2016;14(12):1685-96.
9. Power DA. Licensing and prescribing biosimilars in Australia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(3):152-4. doi:10.5639/gabij.2013.0203.030
10. Cohen H, Beydoun D, Chien D, Lessor T, McCabe D, Muenzberg M, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther, 2017;33(12):2160-72.
11. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
12. Industry Standard Research. ASBM Biosimilars. Canada prescribers and biosimilars. Biosimilars, familarity, knowledge, attitudes and beliefs. 2017.
|
Author for correspondence: Michael Sarshad, BSc, MBA, Syneos Health Consulting, Suite 270, 1315 Lincoln Boulevard, Santa Monica, CA 90401, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/the-need-for-distinct-nomenclature-for-originator-and-biosimilar-products.html
Medicines regulation in the MENA region and the importance of the World Health Organization’s INN proposal of Biological Qualifier
Author byline as per print journal: Peter J Pitts, BA; Michael S Reilly, Esq
Abstract:
The World Health Organization should finalize its Biological Qualifier guidance. Distinguishable naming will allow quick and accurate tracing of the manufacturer, should adverse events occur and improve patient safety by reducing confusion and mishaps. This will ensure that developing nations, including those in the MENA region, have access to high quality, affordable medicines.
Submitted: 1 June 2018; Revised: 29 October 2018; Accepted: 30 October 2018; Published online first: 2 November 2018
Introduction
When it comes to monitoring the quality, safety and efficacy of biological medicines, distinguishable naming is imperative because biosimilar therapies are similar to, but not exactly the same as, existing biological medicines. Since no biosimilar is perfectly identical to its innovator parent, every biological – whether reference product or biosimilar – must be fully distinguishable from other biologicals to permit quick and accurate tracing of its manufacturer, should an adverse event be observed. Precise naming of all biologicals will improve patient safety by reducing confusion and mishaps in prescribing and holding manufacturers accountable. Also, differential nomenclature helps enable national health authorities to collect and compare real-world data that measure the clinical effects of biologicals including biosimilars. Insights from such data, over time, will enable us to better measure a drug’s effectiveness in delivering successful health outcomes for patients.
The World Health Organization (WHO) must finalize their Biologic Qualifier guidance. It is this organization that has the responsibility to ensure that developing nations of the world have access to affordable, quality medicines. Safety is mission critical and the Biological Qualifier is a potent tool on behalf of global public health.
The primacy of medicines quality
Over the last few years we have travelled to many countries around the world, visiting with medicines regulators from Australia, Europe, Latin America and the Middle East. Although a wide range of topics were discussed with these regulators, one of the most pressing issues for all was the urgency of access to quality medicine.
This paper will focus on medicines regulation in the Middle East and North Africa, specifically Algeria, Egypt, Iraq, Jordan, Kuwait, Lebanon, Morocco, Oman, Qatar, Saudi Arabia, Turkey and the United Arab Emirates (UAE). We will also discuss the importance of the World Health Organization’s leadership around establishing a global nomenclature policy that will help maintain the quality, safety and efficacy of biological and biosimilar medicines for that region.
Without quality, safety and effectiveness are non-starters and access is without meaning. Without quality, healthcare spending is not just wasteful – but harmful. Without quality it is all about ‘lowest price tenders’ without any consideration for value. Without quality, regulation is a sham.
Medicines quality in the Middle East
Medicine quality is on the minds of leaders in the Middle East. At the Second Annual Arab Conference on Food and Drugs (Sharm El Sheikh, Egypt, 11-13 April 2015), a conference of regional regulatory agencies, Dr Rasha Ziada (Egyptian Ministry of Health) made the important point that if a pricing authority does not take outcomes into consideration; it will lead to overall price distortions. Amen! Dr Ola Ghaleb (Ministry of Health UAE), spoke about the UAE’s strategy of performance-based risk-sharing arrangements. Outcomes are now capitalized and bolded in the international lexicon of healthcare policy. The necessary precursor to positive outcomes is quality.
A significant number of the conference presenters discussed the value of sharing pharmaceutical economic data across borders, but there was not an equal counterbalancing discussion of the value of sharing clinical data for approvals and outcomes-based decision-making processes. The opportunity to enhance regulatory capacity and product quality through collaboration and information sharing is significant. Unfortunately, cost is too often the primary focus while other priorities such as quality are left to languish.
There was certainly an effort (both on many of the panels as well as during the breaks and after hours) to stress the urgency of the quality agenda. The good news is that speaker after speaker (sometimes in passing and other times passionately) made the point that it must not just be about ‘getting the lowest price’, but also appropriately pricing the most clinically effective treatments. Cost savings without quality are no bargain.
The crisis in drug quality is very real. In Saudi Arabia, according to Alhawassi et al. [1]:
Access to medicine alone is not enough; medicine quality is an essential element of patient care. Quality – even for relatively common and easily evaluated medicines like amoxicillin – is a challenge and must be a policy priority. As put forth by Alhawassi et al. [1]:
The observation of scientist W Edwards Deming applies today as it did decades ago. ‘Change is not required. Survival is not mandatory’.
As in the West, generic drugs provide greater access to medicine for millions of patients in the Middle East. As in other parts of the developing world, assuring quality through robust regulatory oversight is often at counter-point with available human and financial resources. As in every part of the world, Middle Eastern health officials (from national ministries of health to local inspectorates) recognize the imperative that ‘the most expensive drug is the one that does not work’. No nation can afford to buy low quality products.
Countries around the world are struggling to adequately monitor the quality of medicines available to their citizenry. From more regular manufacturing inspections, to risk-based investigations into the sourcing of ingredients, to a rethinking of post-marketing surveillance (pharmacovigilance), there is not one single solution.
Attention to quality cannot end at product approval. This dimension is clearly elucidated from the Jordan Food and Drug Administration (JFDA). In a recent journal article Dr Hayel Mohammad Obeidat, JFDA’s Director General writes, ‘We believe that 21st century pharmacovigilance must also include tighter and more regularly monitored post-approval bioequivalence measures. It is a new and difficult task and calls for better validated methodologies for both data collection and signal prioritization. It is the responsibility of JFDA to take the leadership role and help educate our various constituencies to the importance of 21st century Phase IV monitoring and interventions [6]’.
What we need are standards and systems that recognize the situation as it exists and provide both a path for convergence with global best practices and immediate tactical programmes that can address the true situation on the ground. In brief, regions such as the Middle East require tactical, pragmatic regulation that recognizes the asymmetries inherent in an evolving regulatory ecosystem. Global institutions can play an important role in facilitating this.
Quality, pharmacovigilance and biosimilars
A key issue driving the development of 21st century regulatory pharmacovigilance strategies is the need for updated post-marketing surveillance of biosimilars. Biological medicines have revolutionized the treatment of many serious and life-threatening diseases; as patents for these products expire around the world, biosimilars are becoming available. Biologicals are very complex medicines made using living cells and cannot be copied exactly, thus copies are called biosimilar, not generic drugs. Appropriate approval standards, specific to biological medicines, are a threshold requirement for all medicines that are deemed ‘biosimilar’. The problem of alleged copies approved outside of a scientifically sound biosimilar framework is a serious safety problem and a topic for another paper. However, even in the context of sound scientific standards, vigilance cannot stop at approval for any biological.
Issues related to the particularities of biologicals (sources, process, quality requirement and new safety profile) require sophisticated new thinking.
Fundamentally, all of the players in the pharmacovigilance ecosystem will have problems characterizing biosimilar issues since we do not have an existing, validated predictive models of potential ‘hot spot’ products, base ingredients and/or suppliers. Consequently, pharmacovigilance for biologicals will have to evolve at the same time as new medicines are launched into this space. Small numbers and the novelty of biological products and their safety profiles – alone and in combination with other medicines – for manufacturers, medical providers and patients will likely render monitoring challenging. This is where global institutions can step in.
We are in a situation of post-marketing ‘indetermination’ and the first step should be to develop new epidemiological approaches that based on a better understanding of the differences between the concepts of ‘generic’ and ‘biosimilar’. We understand there can be different safety profiles for generics (based on differing bioequivalence ranges, excipient and active pharmaceutical ingredient sourcing). When it comes to biosimilar pharmacovigilance, however, variability-induced iatrogenesis concerns, differences between batches by multiple manufacturers, and the elastic definition of ‘similarity’ is not a question of ‘safety profile’, but rather of ‘concept’ [7].
Biosimilar nomenclature and the unique role of WHO
It is into this maelstrom that the steady hand of WHO is needed. And nowhere is this more true or urgent than in the current debate over biosimilar medicines generally and product nomenclature specifically.
WHO has published a draft proposal for a global system to assign ‘biological qualifiers’ (BQs) to biologicals and biosimilars [8]. A BQ is a random four-letter code assigned to a biological manufactured at a specified site. WHO said the scheme would be voluntary for each regulatory authority and applicable retroactively. The qualifier would not be part of a biological’s International Nonproprietary Name (INN), although WHO’s INN expert group would oversee the scheme. WHO said the proposed scheme is intended to avoid separate national qualifier systems. It will also permit less developed national regulatory systems to institute a globally consistent protocol that will help to guarantee the quality, safety and efficacy of biological and biosimilar medicines.
Consider Lebanon, ‘the hospital of the Middle East’. Minister Ghassan Hasbani, Lebanon’s Deputy Prime Minister and Minister of Health, is revamping the medicines tendering programme for Lebanon and one of the key tenets being weighed in the new national decision-making process is value. As His Excellency said from the podium, ‘It is not only a cost, it is an investment’. And, as with any investment, it is impossible to understand the cost without proper consideration of the return.
Minister Hasbani recognizes that biosimilars represent an important tool in expanding access to patients in Lebanon. But, as in the West, it must be access combined with quality and safety.
When it comes to health care, clarity is better than confusion, especially when it comes to drug safety — the sine qua non of medicines regulation. And that means clarity in biosimilar nomenclature.
What is so important about a biosimilar’s name? Patient safety. According to the US Food and Drug Authority, distinct and precise nomenclature for all biologicals, innovator and biosimilar, will promote accurate prescribing and facilitate accurate attribution of adverse events [9].
Distinguishable naming is imperative because biosimilar therapies are similar to, but not exactly the same as, existing biological medicines. Since no biosimilar is perfectly identical to its innovator parent, every biological – whether reference product or biosimilar – must be fully distinguishable from other biologicals to permit quick and accurate tracing of its manufacturer, should an adverse event be observed. This facilitates manufacturer accountability.
On a global level, an INN [10] is used to identify the active ingredient in a drug, which in the case of a chemical/generic drug is equivalent. Biosimilars are not identical to their innovator parents; they are ‘highly similar’.
The differences, however, are crucially important since there is the potential for all biologicals to elicit dangerous immune responses. For this reason, if biosimilars use identical INNs and prescribing, dispensing or adverse event records identify products only by INNs, global regulators cannot recognize precisely which product is causing a problem. Testimony to WHO showed that there is a high level of ambiguity in attributing adverse events to a specific manufacturer when products share the same non-proprietary name [11]. Efforts to include other identifiers, such as batch number, that would distinguish between products and manufacturers routinely fail [12]. The addition of a unique suffix to the non-proprietary name provides a distinguishing feature that can be used to enhance traceability in a marketplace with multiple similar options.
Precise naming will improve patient safety by reducing confusion and mishaps in prescribing and dispensing; biosimilars are not identical to the reference product or one another, thus switching from one product to another may not be appropriate. Also, differential nomenclature helps enable national health authorities to collect and compare real-world data that measure the clinical effects of biologicals including biosimilars. Insights from such data, over time, will enable us to better measure a drug’s effectiveness in delivering successful health outcomes for patients.
WHO must finalize their BQ guidance. It is this organization, after all, that has the responsibility to ensure that developing nations of the world have access to affordable, quality medicines. Safety is mission critical and the BQ is a potent tool on behalf of global public health.
Concluding thoughts on the global regulatory fraternity
Our experience with healthcare regulators worldwide has reinforced our belief that health care and health policy professionals devoted to ensuring timely access to innovative medicines, quality generic drugs and biosimilars. It is not easy, and it is not only a job – it is a personal public health mission.
There are many issues surrounding the introduction of biosimilars into the global healthcare ecosystem: safety, effectiveness, interchangeability, potential adverse medical events, appropriate regulatory labelling and physician prescribing guidelines. But, even so, biosimilars are here. They are safe and effective. They are less costly. And they deserve a seat at the therapeutic table.
When it comes to biosimilar nomenclature, it is important for WHO to look … backwards. According to the 10th century Arab physician, Ibn Sina, ‘The time of action must be observed, so that essence and accident are not confused’.
Funding
This paper is funded by Alliance for Safe Biologic Medicines.
Competing interests: Mr Peter J Pitts declares no conflict of interest. Mr Michael S Reilly, Esq, is the Executive Director of and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Peter J Pitts, BA
Former Associate Commissioner, United States Food and Drug Administration
Visiting Professor, Université Paris Descartes Medical School
President, Center for Medicine in the Public Interest
757 Third Avenue, 20/F
New York, NY 10017, USA
Michael S Reilly, Esq
Executive Director
Alliance for Safe Biologic Medicines
PO Box 3691, Arlington
VA 22203, USA
References
1. Alhawassi TM, Abuelizz HA, Almetwasi M, Mahmoud MA, Alghamdi A, Alruthia YS, et al. Advancing pharmaceuticals and patient safety in Saudi Arabia: a 2030 vision initiative. Saudi Pharm J. 2018;26(1):71-4.
2. Caudron JM, Ford N, Henkens M, Macé C, Kiddle-Monroe R, Pinel J. Substandard medicines in resource-poor settings: a problem that can no longer be ignored. Trop Med Int Health. 2000;13(8):1062-72.
3. Salomon JA, Vos T, Hogan DR, Gagnon M, Naghavi M, Mokdad A. Common values in assessing health outcomes from disease and injury: disability weights measurement study for the Global Burden of Disease Study 2010. Lancet. 2012:15;380(9859):2129-43.
4. Australian Commission on Safety and Quality in Health Care. Medication Safety; Safety and Quality improvement Guide, Australia 2012 [internet]. 2012 [cited 2018 Oct 29]. Available from: https://www.safetyandquality.gov.au/wp-content/uploads/2012/10/Standard4_Oct_2012_WEB.pdf
5. Wikipedia. Saudi Vision 2030 [homepage on the Internet]. [cited 2018 Oct 29]. Available from: https://en.wikipedia.org/wiki/Saudi_Vision_2030
6. Al Kayyali L, Al Haqaish W, Bawaraesh N, Pitts PJ. The Jordan Food and Drug Administration: a culture of quality and continuous improvement. J Comm Biotechnol. 2014;20(4).
7. Pitts PJ, Louet HL, Moride Y, Conti RM. 21st century pharmacovigilance: efforts, roles, and responsibilities. Lancet Oncol. 2016;17(11):e486-e492.
8. World Health Organization. Essential medicines and health products. International Nonproprietary Name [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/en/
9. U.S. Food and Drug Administration. Labeling for biosimilar products. [homepage on the Internet]. [cited 2018 Oct 29]. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm493439.pdf
10. World Health Organization. Essential medicines and health products. Biological qualifier [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/inn_bio_bq/en/
11. World Health Organization. 63rd Consultation on International Nonproprietary Names for Pharmaceutical Substances. Geneva, 18-21 October 2016 [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/63rd_Executive_Summary.pdf
12. World Health Organization. 64th Consultation on International Nonproprietary Names for Pharmaceutical Substances. Geneva, 4-7 April 2017 [homepage on the Internet]. [cited 2018 Oct 29]. Available from: http://www.who.int/medicines/services/inn/64th_Executive_Summary.pdf
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/medicines-regulation-in-the-mena-region-and-the-importance-of-the-world-health-organizations-inn-proposal-of-biological-qualifier.html