|
Abstract: |
Submitted: 7 February 2018; Revised: 28 March 2018; Accepted: 2 April 2018; Published online first: 16 April 2018
The human genome (HG) is comprised of ~23,000 open reading frame (ORF) genes, however, the human proteome is orders of magnitude greater; due to alternate splicing (AS) of ORF genes, errors in transcription or translation, the addition of co- and post-translational modifications (CTM; PTM). A recent guestimate suggested that each ORF within the outbred human population might be translated to generate 100 structurally distinct proteins [1]. Protein and glycoprotein (P/GP) molecules exist in vivo as discreet entities within complex multi-component media, e.g. plasma, cell sap, and exert their function(s) through specific interactions with target/receptor molecules. In health each individual expresses a unique proteome and personal integrity demands immunological tolerance to all self-molecules. Ordered aggregation of monomer molecules may be essential for normal function; however, inappropriate (non-native) aggregation is implicated in the pathogenesis of numerous autoimmune diseases and the generation of autoantibodies [2, 3]. Similarly, denaturation and/or aggregation of P/GP biotherapeutics may render them immunogenic and result in the development of anti-drug/anti-therapeutic antibodies (ADA/ATA).
The thriving biopharmaceutical industry depends on the production of recombinant P/GPs exhibiting an essential structural fidelity with a selected endogenous molecule; therefore, structural variants generated during production, purification, formulation and/or delivery is a major concern as it may equate to potential immunogenicity [2, 3]. Pharmacovigilance must be exercised over the lifetime of an approved drug since incidences of adverse events are reported for drugs that have been long established in the clinic, e.g. insulin [4] and erythropoietin (EPO) [5]. The presence of ADA/ATA is frequently associated with onset of adverse events and/or loss of efficacy [6, 7] and suggests the presence of structurally altered/denatured molecules that are recognized as ‘foreign’ (non-self) by the patient’s immune system, i.e. immunogenic. This mini review enumerates structural parameters that have to be defined and maintained throughout the production, administration and clinical lifetime of recombinant P/GP therapeutics; illustrated for EPO and antibody therapeutics.
Structural heterogeneity: in vivo and ex vivo
Biosynthesis of P/GPs in mammalian cells employs error prone multistep processes and the end product(s) exhibits an inevitable structural heterogeneity. Lack of fidelity with the amino acid sequence encoded by a given ORF may be introduced at multiple stages, e.g. transcription, mRNA translation, miss-incorporation. Additionally, de nova secondary structures may be essential to allow co-translational modifications (CTMs) of the polypeptide as it is extruded from the ribosome tunnel, e.g. the addition of oligosaccharide, N-myristoylation. When released from the ribosome the P/GP transits to the endoplasmic reticulum where it is edited for correct tertiary/quaternary folding and initial oligosaccharide processing; further post-translational modifications (PTMs) are effected during passage through the Golgi apparatus [8–11]; P/GPs may be subject to further modifications throughout their life cycle in vivo, e.g. enzyme cleavage to release secondary bioactive products. It is presumed that all such molecular entities are recognized as ‘self’ by the immune system; therefore the first step in the quest to produce a recombinant P/GP therapeutic is determination of the structure of the natural (endogenous) molecule. However, the techniques employed to isolate and purify P/GPs from body fluids or tissues may result in denaturation and the introduction of non-native chemical modifications (CMs) e.g. deamination; proline isomerisation.
In practice a candidate recombinant P/GP therapeutic is evaluated, structurally and functionally in comparison with the fully characterized endogenous molecule. This approach cannot be realized for a potential recombinant monoclonal antibody (mAb) therapeutic since an endogenous anti-self antibody is not available for comparison. Candidate mAbs are sourced from inbred mice and engineered to generate chimeric or humanized mAbs; from transgenic mice expressing human immunoglobulin genes or random reassociation of human Immuoglobulin (Ig) heavy and light chain expressed within phage display libraries [12, 13]. The choice of production platform is a critical strategic decision since the processes involved in the addition of CTMs, PTMs and CMs are species and cell specific and production of a human P/GP in an alien cell line, e.g. CHO (Chinese hamster ovary) cell line, may result in the introduction of non-self structures and consequent immunogenicity with the generation of ADA/ATA responses [6–8]. Prior to clinical trials a candidate recombinant P/GP therapeutic has to be extensively characterized in comparison with the endogenous molecule, employing multiple orthogonal physicochemical techniques [14, 15]. Patent protection for numerous recombinant P/GP drugs has now expired and many more are approaching expiry, providing opportunities for the production of biosimilar drugs. Candidate biosimilars must be characterized in comparison with the approved innovator drug product [16, 17].
Protein folding: in vivo
Proteins are synthesized, within ribosomes, as a linear sequence (string) of amino acid residues covalently linked through the peptide bond; elements of secondary structure may form, de nova, and can include generation of an acceptor site for the addition of high mannose oligosaccharides N-linked to an asparagine residue present within a glycosylation sequon, i.e. the sequence asparagine-x-serine or threonine (asp-x-ser/thr; N-X-S/T), where x is any amino acid residue other than proline. Following release from the ribosome the protein transits to the endoplasmic reticulum where the high mannose oligosaccharide is truncated and exerts a quality control function for correct folding; miss-folded proteins being marked for proteasomal degradation [8–11]. Multiple PTMs may be effected during passage through the Golgi apparatus including further oligosaccharide processing, phosphorylation, sulphation. In this way a P/GP achieves its native, evolutionary determined, structure that ensures it traffics to the appropriate cellular compartment or is secreted [18–21].
It has been estimated that a protein of 100 amino acid residues undergoing random motion in search of the lowest energy form could pass through 1089 conformations, taking 1066 years, to sample all possible structures; however, within the cell the P/GP passes through intrinsic protein folding pathways to achieve the functional tertiary/quaternary conformation within seconds [22]. Our knowledge and understanding of P/GP structure/function relationships is mostly based on the results of X-ray crystallographic studies and tend to represent proteins as having a fixed (solid) structure [15]. Newer techniques show that proteins are ‘living, breathing’ entities that may exist in conformational equilibria, including intrinsically unstructured regions [23, 24]; ex vivo such regions, may act as focal points for aggregation [2, 3, 9, 23, 24]. Algorisms that attempt to analyse or predict structural parameters of P/GPs as they exist within in vivo environments are in their infancy [10, 24].
Protein folding: ex vivo
Proteins are comprised of amino acid residues that bear non-polar, polar uncharged and charged side chains and may fold to generate molecules having an overall hydrophobic or hydrophilic character. Proteins that are soluble in aqueous media have an overall hydrophilic character whilst hydrophobic amino acid side chains are orientated towards the internal space and form mutual interactions that stabilise structure; however, a scan of the surface exposed side chains may reveal hydrophobic patches that can act as centres for aggregation [2, 9–11]. This potential is underlined by diseases in which P/GP aggregation results in the deposition of insoluble fibrils in tissues, e.g. neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), transmissible spongiform encephalopathies (TSEs), and amyotrophic lateral sclerosis (ALS) [25–31]. Fundamental studies of protein folding and aggregation have focused on the hen egg white lysozyme molecule, the native form of which has high solubility in aqueous media. However, following exposure to denaturing solvents in vitro, followed by restoration to physiologic conditions it can miss-fold to form aggregates and fibrils, similar to pathogenic species seen in disease. Six spontaneous mutations in human lysozyme have been reported and all except one lead to systemic non-neurogenic amyloidosis involving kidney, liver and spleen [27–29]. Prion disease is an extreme example of the propensity for a soluble protein to form fibrils in vivo [30, 31]. In its soluble form the prion protein has a helical structure; however, in the disease state the protein converts to a beta sheet structure that aggregates to form fibrils; the denatured prion protein can act as a ‘catalyst’ to induce normal prion protein to convert to a beta sheet structure.
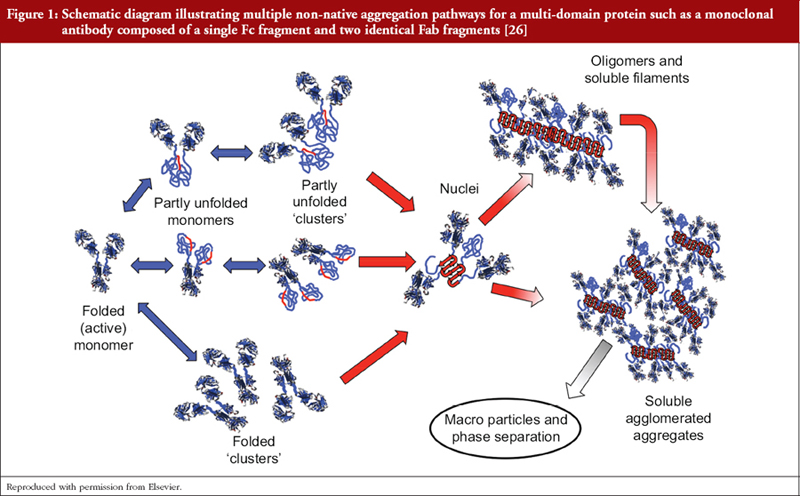
As previously stated we do not have means of determining the fine structure of P/GPs as they exist in vivo and are limited to extrapolation from structural studies of isolated P/GPs purified from human fluids and tissues employing multiple physicochemical techniques that may introduce further structural heterogeneity, e.g. deamidation of asparagine and glutamine residues, oxidation of methionine and tryptophan residues, glycation of lysine [10, 14, 24]. Additionally, proteins may undergo subtle reversible conformational changes that results in momentary exposure of hydrophobic regions that can be mutually attractive with formation of ‘partly unfolded clusters’, i.e. aggregates [3, 25–27], see Figure 1. Such clusters can act as nuclei for the formation of larger aggregates, possibly extending to precipitation. Structural heterogeneity is compounded by differing susceptibilities of individual amino acid residues to modifications depending on its position within the molecule and the immediate microenvironment.
Aggregation prone regions (APRs) may be classified as structural or critical. Structural APRs contribute to the stability of the native protein core structure but may be exposed following denaturation ex vivo and form aggregates under refolding conditions; critical APRs are exposed in the native state and may contribute to physiological protein/protein interactions in vivo and in vitro. Multiple physiochemical techniques and algorithms have been developed to identify APRs and inform protein engineering to reduce a propensity for aggregation [32–34]; a concomitant increases in recombinant proteins productivity has been reported [35]. Since hydrophobic binding contributes to protein/protein interactions APRs may be anticipated as a feature of functional sites and much attention has been focused on the antigen-binding site, i.e. the paratope, of antibody molecules [35, 36]. However, antibodies are multifunctional molecules and the formation of antigen/antibody complexes is an essential prelude to the activation of downstream effector functions activated by interactions of the Fc region with soluble and/or cell bound and ligands, e.g. cellular Fc receptors (FcγR, FcRn), the C1 component of complement [37, 38]. Interaction sites for these ligands have been identified and include the hydrophobic sequence 231-APELLGGPSVFLFPP-245 [15, 20, 37, 38]. Protein engineering has been employed to reduce the propensity for aggregation whilst retaining activation of effector molecules that determines their mechanism of action (MoA).
In health an individual is tolerant to their proteome, however, multiple autoimmune diseases manifest the potential for loss of tolerance to self-molecules or aberrant (mutant) forms of self-molecules arising in vivo. The potential for immunogenicity of biotherapeutics in humans may vary depending on the character of the disease being treated, three broad categories may be identified [39–41]:
The antibody response in humans is comprised of five immunoglobulin (Ig) classes: IgM, IgG, IgA, IgE and IgD; in addition IgG is comprised of four subclasses (IgG1, IgG2, IgG3, and IgG4) and IgA two (IgA1, IgA2) generating nine Ig isotypes [15, 42, 43]; each antibody isotype expresses a unique profile of effector mechanisms. The IgG1 subclass predominates in serum and has been the focus for structure/function studies and the predominant format adopted for approved mAb therapeutics. Following binding to its target, with the formation of antibody/antigen complexes, antibodies of the IgG1 subclass may trigger a cascade of inflammatory effector mechanisms that constitute its MoA. Activation of IgG1 mAbs provides natural protection in the killing and removal of bacteria and other ‘foreign bodies’; recombinant antibody therapeutics specific to cancer cells may similarly be activated, resulting in their killing and removal. Each IgG subclass may be exploited to offer a MoA profile appropriate to differing disease indications. The antibody landscape is developing rapidly, as new engineered constructs are customized to optimize treatment protocols, e.g. antibody fragments that enhance solid tumour penetration, antibody-drug conjugates that are internalized into target cells where drug release is effected [12, 13]. It should be noted that the binding of a divalent antibody to a multivalent antigen, e.g. a cancer cell, results in the formation of an immune complex (IC) that is itself an aggregated form of the antibody. ICs are removed and degraded by leucocytes that are also antigen-presenting cells and may therefore, present peptides derived from the paratope of a mAb [44].
The first GP approved by the European Medicines Agency and US Food and Drug Administration was the murine mAb Muromonab (1986, anti-human CD3 OKT3), produced in mouse hybridoma cells; it was administered to patients undergoing acute rejection of a liver transplant. Whilst successfully suppressing the rejection episode, vigorous anti-mouse IgG antibody responses developed in a majority of patients; excluding the possibility of exposing patients to the therapeutic on a subsequent occasion. Over succeeding years genetic and protein engineering techniques were employed to limit immunogenicity by successively increasing the human IgG character of mAbs and expression of selected IgG-Fc mediated MoA. The commercial mAb therapeutic era may be identified with the development of chimeric mouse/human mAbs comprised of the variable regions of a mouse antibody linked to the constant regions of human IgG1, generating a molecule that is ~30% mouse and ~70% human in structure [6–8, 15]. A significant reduction in immunogenicity resulted and a majority of patients could be repeatedly dosed with these mAbs. Further developments defined the amino acid residues of the mouse antibody that formed the antigen binding site (paratope) and transplanted them into selected human variable regions; generating a ‘humanized’ mAb [6–8, 15]. This technology is being superseded by protocols allowing the generation of ‘fully human’ antibodies. These mAbs are products of rearranged human variable region genes, however, by virtue of the fact that they are selected to be anti-self their unique paratope structure may provoke ADA/ATA responses [12, 13] in an outbred human population.
Meta-analysis of the incidence of ADA for the first approved ‘fully human’ anti-TNF-alpha (TNF-α), antibody (Adalimumab, Humira), generated by phage display, ranged from 1–54 %; when administered across multiple inflammatory diseases [6, 7]. The ADA responses may be transitory and/or of low titre and, with good patient management, do not necessarily result in significant adverse reactions [45]; a threshold for immunogenicity is evidenced by the fact that ADA responses are reduced when patients are concomitantly receiving a mild immunosuppressant, e.g. methotrexate [46]. Antibodies generated from phage display libraries depend on the pairing of VH and VL sequences that express anti-self specificities and would be forbidden in vivo, consequently they may express foreign (non-self) epitopes. The alternative technology for generating fully human antibodies from mice rendered transgenic for human immunoglobulin genes results in a natural pairing of VH and VL sequences and the incidence of ADA for the anti-TNFα Golimumab is reported as 0–19% [6].
A majority of proteins are generated utilizing the standard 20 amino acids linked through the peptide bond between alpha carbon atoms; in contrast oligosaccharides utilize multiple linkages with a potential to generate enormous glycome and glyco-proteome diversity; it is estimated that six sugar residues can be assembled to generate 1012 unique hexasaccharides [47]. The repertoire of sugars utilized varies between species, gender, cell line, etc.; to generate N-linked oligosaccharides, as previously discussed, or oligosaccharides O-linked through serine, threonine or mannose residues. Importantly, CHO and NS0 (murine) cell lines may add immunogenic non-human oligosaccharide structures to intended ‘fully’ human recombinant therapeutics [13, 48, 49]. Protein engineering and gene ‘knock-out’/‘knock-in’ techniques have been employed to modulate the glycoform profile of GPs; as illustrated in this text for EPO and IgG.
Erythropoietin: Recombinant EPO produced in CHO cells was initially shown to exhibit enhanced activity in vitro, in comparison with approved therapeutic isolated from urine. However, trials in vivo revealed a lack of therapeutic efficacy due to its rapid clearance from the circulation. It was later shown the attached oligosaccharides bore terminal galactose sugar residues, rather than the required sialic acid, resulting in clearance in the liver via the asialoglycoprotein receptor. Fractionation of the CHO-derived EPO allowed preparation of an active sialylated glycoform establishing this parameter as a Critical Quality Attribute (CQA); recombinant EPO, Epoetin received regulatory approval in 1989, is comprised of 165 amino acid residues and bears three N-linked and one O-linked oligosaccharide that accounts for ~40% of its mass [50–53].
Successful worldwide use of recombinant EPO followed but in 1999 a cohort of patients in Europe developed pure red cell aplasia (PRCA) (failure of erythrocyte production) due to the generation of ADA that neutralized not only the therapeutic but also endogenous EPO. Investigation showed that ‘minor’ changes had been introduced in the formulation of EPO produced in Europe, in contrast to the US, that were presumed to have resulted in denaturation/aggregation rendering the product immunogenic [51]. This illustrates the structural fragility of P/GPs and the need for pharmacovigilance throughout the lifetime of a drug. Incidences of PRCA continue to be reported around the world and include ‘biosimilar’ EPOs produced by multiple manufacturers and approved by regional or national regulatory authorities [52]. Experiences of Thailand are salutary, as of 1 January 2009, 14 EPO drugs were licensed in Thailand [53]; they originated from various countries and were not biosimilars as defined by the EU/USA/WHO (World Health Organization) requirements. The cost advantage for these versions of EPO resulted in widespread usage but was coincident with an increase in reports of PRCA due to the generation of ADA [53].
Anticipating expiration of patent protection and the advent of biosimilars the innovator company (Amgen) developed an improved (biobetter) product (darbepoeitin alfa), exhibiting increased efficacy and an extended in vivo half-life; it was approved and received patent protection [54, 55]. The improvement was achieved by the introduction of two additional N-linked oligosaccharide attachment sites resulting in the production of glycoforms bearing additional N-linked oligosaccharides expressing terminal sialic acid residues.
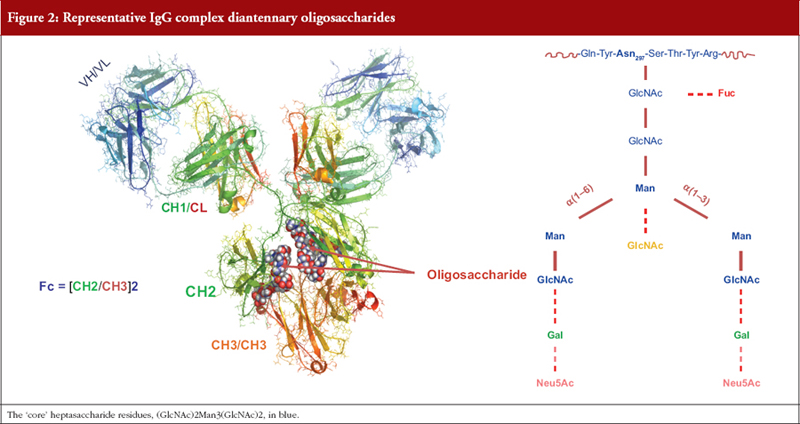
Antibodies: An IgG molecule is comprised of ~1,440 amino acid residues and two N-linked oligosaccharides each comprised of 7 to 13 sugar residues. For decades little account was taken of this ‘minor’ structural feature until it was shown that removal of the oligosaccharide resulted in loss of the ability of ICs to trigger MoAs mediated by activation of FcγR and the C1 complement component, i.e. glycosylation of IgG is a CQA [44, 45]. A minimum requirement for MoA activation is the presence of a seven-residue oligosaccharide on each heavy chain. Differential addition of sugar residues generates a multiplicity of IgG glycoforms that may each modulate the affinity of binding of ICs to effector ligands and hence MoAs, see Figure 2 [44, 45, 56–58].
The glycoform heterogeneity of human serum IgG is not mirrored by the glycoform profile of mAbs produced in CHO, NS0 or Sp2/0 cells; in contrast these cells express a restricted glycoform profile that may include immunogenic non-human glycoforms. The glycoform profile cannot be significantly manipulated by changes in culture conditions, therefore, the contribution of individual glycoforms to MoAs has been investigated by in vitro enzymatic modification of mAb or genetic engineering of the producer cell line. A dramatic outcome from these studies has been the demonstration that IgG antibodies that bear oligosaccharides devoid of fucose residues can exhibit a 10–102 folds increase in their ability to mediate killing of cancer cells by NK (natural killer) cells, similar increases can be achieved for mAb expressing a bisecting N-acetylglucosamine residue. New production CHO cell lines have been established following the ‘knock-out’ of the fucosyltransferase gene or ‘knock-in’ of the bisecting N-acetylglucosamine transferase gene [48, 56–58]. These cell lines have been used to generate approved ‘biobetter’ versions of previously approved mAbs.
An antibody may be protective and deliver therapeutic benefit due to its binding specificity for target, e.g. neutralizing an exogenous bacterial toxin or endogenous TNFα, however, when the target is a bacterium or a cancer cell MoAs that result in killing and removal of debris are essential. [56–58]. The IC formed in turn become targets for leucocytes that bear cell surface receptors (FcγR) specific to the IgG heavy chain Fc region. The cross-linking of multiple FcγR results in leucocyte activation with the release of toxic agents and/or ingestion (phagocytosis), ICs may also activate the C1 component of the complement system to trigger a cascade of enzymatic reactions resulting in the formation of a membrane attack complex that inserts into the cellular membrane with the formation of pores that allow the ingress of water and egress of cellular constituents. Molecules released from the complement cascade also adhere to the IC and engage complement receptors expressed on leucocytes to further enhance cellular activation.
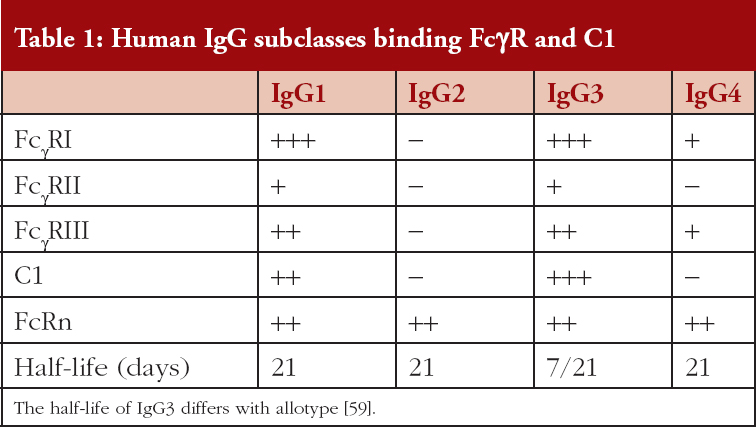
There are three families of FcγR (FcγRI, FcγRII, FcγRIII) that are differentially expressed on leucocytes and bind the IgG subclasses selectively, see Table 1; similarly, the C1 component of complement exhibits selective IgG subclass binding. An important parameter that contributes to mAb efficacy is the long half-lives of ~21 days, for IgG1, IgG2 and IgG4, this allows for extended intervals between administered doses; IgG3 has a shorter half-life of ~7 days [59]. Clearance of IgG is mediated via binding to the neonatal Fc receptor (FcRn) that is expressed on many cell types and is independent of the IgG glycoform, [56–58]. Antibodies of the IgG1 and IgG3 subclass have very similar functional profiles but the IgG2 and IgG4 subclasses exhibit unique profiles. It is important therefore when developing a mAb therapeutic to anticipate the preferred MoA in vivo and generate mAbs of an appropriate IgG subclass. To date, of the 160 mAbs listed in the international immunoglobulins database (IMGT: ImMunoGeneTics) 136 are IgG1, 8 IgG2, 2 IgG3 and 14 IgG4 [58, 60]
It is posited that all recombinant P/GP therapeutics may be immunogenic, at least in a proportion of patients, resulting in loss of efficacy and/or the advent of adverse events. The significance of this outcome should be assessed with respect to the disease being treated, thus cancer and transplant patients will be receiving concomitant cytotoxic drugs that induce various levels of immunosuppression. By contrast patients with chronic diseases undergo long-term exposure to recombinant P/GPs and are at greater risk of developing ADA, that may be circumvented by treatment with mild immunosuppressive agents. Currently, an ever expanding armamentarium of biologicals is being developed that includes engineered IgG molecules that differ in structure to endogenous IgG and/or their fragments. Such manipulations increase the propensity for immunogenicity, however, outcomes may differ between acute conditions for which treatment may be within a relatively short time frame and chronic diseases that require long-term exposure.
Advances in gene sequencing techniques are allowing identification of polymorphisms in ‘susceptibility’ genes that allows for stratification of patients. Stratification can contribute to the development of personalized medicine through identification of cohorts of patients responsive to a given therapeutic whilst similarly identifying patients that are not likely to benefit. Stratification of ‘common’ diseases may identify increasingly small cohorts of patients such that their condition may be classified as an orphan disease, indicative of a need for treatment with expensive customized biologicals, i.e. personalized medicine. This may result in a conflict between the high cost of development of specialist biologicals and the diminished market that stratification identifies. Some ‘respite’ may be offered by the development of biosimilars, however, they are currently providing only ~15–30% reduction in cost. The conflict between our ability to deliver ever expanding therapies for human health care, from conception to death, and to provide equity in delivery will continue and become ever more contentious.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Ponomarenko EA, Poverennaya EV, Ilgisonis EV, Pyatnitskiy MA, Kopylov AT, Zgoda VG, et al. The size of the human proteome: the width and depth. Int J Anal Chem. 2016;2016:7436849.
2. Chiti F, Dobson C. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333-6.
3. Moussa M, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, et al. Immunogenicity of therapeutic protein aggregates. J Pharm Sci. 2016;105(2):417-30.
4. Ghazavi MK, Johnston GA. Insulin allergy. Clin Dermatol. 2011;29(3):300-5.
5. Macdougall IC, Roger SD, de Francisco A, Goldsmith DJ, Schellekens H, Ebbers H, et al. Antibody-mediated pure red cell aplasia in chronic kidney disease patients receiving erythropoiesis-stimulating agents: new insights. Kidney Int. 2012;81(8):727-32.
6. Strand V, Balsa A, Al-Saleh J, Barile-Fabris L, Horiuchi T, Takeuchi T, et al. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. BioDrugs. 2017;31(4):299-316.
7. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13(12):707-18.
8. Jefferis R. Posttranslational modifications and the immunogenicity of biotherapeutics. J Immunol Res. 2016;2016:5358272.
9. Dyson HJ. Making sense of intrinsically disordered proteins. Biophys J. 2016;110(5):1013-6.
10. Hingorani KS, Gierasch LM. Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge. Curr Opin Struct Biol. 2014;0:81-90.
11. Gruebele M, Dave K, Sukenik S. Globular protein folding in vitro and in vivo. Annu Rev Biophys. 2016;45:233-51.
12. Tomszak F, Weber S, Zantow J, Schirrmann T, Hust M, Frenzel A. Selection of recombinant human antibodies. Adv Exp Med Biol. 2016;917:23-54.
13. Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 2012;28:147-75.
14. Parr MK, Montacir O, Montacir H. Physicochemical characterization of biopharmaceuticals. J Pharm Biomed Anal. 2016;130:366–89.
15. Jefferis R. Monoclonal antibodies: mechanisms of action. In: Schiel J, Davis DL, Borisov O, editors. State-of-the-art and emerging technologies for therapeutic monoclonal antibody characterization. Volume 1. Monoclonal antibody therapeutics: structure, function, and regulatory space. Washington, DC: American Chemical Society; 2014.
16. Emmanouilides CE, Karampola MI, Beredima M. Biosimilars: hope and concern. J Oncol Pharm Pract. 2016;22(4):618-24.
17. Wang X, An Z, Luo W, Xia N, Zhao Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell. 2018;9(1):74-85.
18. Stanley P. Golgi glycosylation. Cold Spring Harb Perspect Biol. 2011;3(4). pii: a005199
19. Zhang X, Wang Y. Glycosylation quality control by the Golgi structure. J Mol Biol. 2016;428(16):3183-93.
20. Jefferis R. Glyco-engineering of human IgG-Fc to modulate biologic activities. Curr Pharma Biotechnol. 2016;17(15):1333-47.
21. Beyer B, Schuster M, Jungbauer A, Lingg N. Microheterogeneity of recombinant antibodies: analytics and functional impact. Biotechnol J. 2018;13(1). doi:10.1002/biot.201700476
22. Creighton TE. The protein folding problem. In: Pain RH, editor. Mechanisms of protein folding. Oxford/New York: IRL Press at Oxford University Press; 1994.
23. Chen K, Long DS, Lute SC, Levy MJ, Brorson KA, Keire DA. Simple NMR methods for evaluating higher order structures of monoclonal antibody therapeutics with quinary structure. J Pharm Biomed Anal. 2016;128:398-407.
24. Zhang X, Zhang L, Tong H, Peng B, Rames MJ, Zhang S, Ren G. 3d structural fluctuation of IgG1 antibody revealed by individual particle electron tomography. Sci Rep. 2015;5:9803.
25. Shamsi TN, Athar T. et al. A review on protein misfolding, aggregation and strategies to prevent related ailments. Int J Biol Macromol. 2017;105(Pt 1):993-1000.
26. Roberts CJ. Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol. 2014;32(7):372-80.
27. Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9(3):237-48.
28. Ahn M, Hagan CL, Bernardo-Gancedo A, De Genst E, Newby FN, Christodoulou J, et al. The significance of the location of mutations for the native-state dynamics of human lysozyme. Biophys J. 2016;111(11):2358-67.
29. Sziegat F, Wirmer-Bartoschek J, Schwalbe H. Characteristics of human lysozyme and its disease-related mutants in their unfolded states. Angew Chem Int Ed Engl. 2011;6;50(24):5514-8.
30. Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nat Neurosci. 2012;15(7):936-9.
31. Sigurdson CJ, Bartz JC, Nilsson KP. Tracking protein aggregate interactions. Prion. 2011;5(2):52-5.
32. Meric G, Robinson AS, Roberts CJ. Driving forces for nonnative protein aggregation and approaches to predict aggregation-prone regions. Annu Rev Chem Biomol Eng. 2017;8:139-59.
33. Walsh I, Pollastri G, Tosatto SC. Correct machine learning on protein sequences: a peer-reviewing perspective. Brief Bioinform. 2016;17(5): 831-40.
34. Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: a survey of commercial monoclonal antibodies. MAbs. 2009;1(3):254-67.
35. Obrezanova O, Arnell A, Gómez de la Cuesta R, Berthelot ME, Gallagher TAG, Zurdo J, Stallwood Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. MAbs. 2015;7(2):352-63.
36. van der Kant R, Karow-Zwick RA, Van Durme J, Blech M, Gallardo R, Seeliger D, et al. Prediction and reduction of the aggregation of monoclonal antibodies. J Mol Biol. 2017;429(8):1244-61.
37. Liu B, Guo H, Xu J, Qin T, Xu L, Zhang J, et al. Acid-induced aggregation propensity of nivolumab is dependent on the Fc. MAbs. 2016;8(6):1107-17.
38. Li W, Prabakaran P, Chen W, Zhu Z, Feng Y, Dimitrov DS. Antibody aggregation: insights from sequence and structure. Antibodies. 2016;5(3):19.
39. Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11(2):99-109.
40. Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;2016:1298473.
41. Krishna M, Nadler SG. Immunogenicity to biotherapeutics – the role of anti-drug immune complexes. Front Immunol. 2016;7:21.
42. Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys. 2012;526(2):159-66.
43. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520.
44. Jefferis R. Aggregation, immune complexes and immunogenicity. MAbs. 2011;3(6)503-4.
45. Thomas SS, Borazan N, Barroso N, Duan L, Taroumian S, Kretzmann, et al. Comparative immunogenicity of TNF inhibitors: impact on clinical efficacy and tolerability in the management of autoimmune diseases. A systematic review and meta-analysis. BioDrugs. 2015;29(4):241-58.
46. Fechtenbaum M, Nam JL, Emery P. Biologics in rheumatoid arthritis: where are we going? Br J Hosp Med (Lond). 2014;75(8):448-9, 451-6.
47. Laine RA. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10(12) structures for a reducing hexasaccharide: the Isomer Barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology. 1994;4(6):759-67.
48. Varki A. Biological roles of glycans. Glycobiology. 2017;27(1):3-49.
49. Lalonde ME, Durocher Y. Therapeutic glycoprotein production in mammalian cells. J Biotechnol. 2017;251:128-40.
50. Jelkmann W. Biosimilar recombinant human erythropoietins (“epoetins”) and future erythropoiesis-stimulating treatments. Expert Opin Biol Ther. 2012;12(5):581-92.
51. McKoy JM, Stonecash RE, Cournoyer D, Rossert J, Nissenson AR, Raisch DW, et al. Epoetin-associated pure red cell aplasia: past, present, and future considerations. Transfusion. 2008;48(8):1754-62.
52. Brinks V, Hawe A, Basmeleh AH, Joachin-Rodriguez L, Haselberg R, Somsen GW, et al. Quality of original and biosimilar epoetin products. Pharm Res. 2011;28(2):386-93.
53. Papassiripan M. Clinical experience with EPO products approved via the generic pathway: the experience of Thailand. Generics and Biosimilars Initiative. First MENA Educational Workshop on Similar Biotherapeutic Products/Biosimilars 2015; 1 September 2015; Dubai, UAE. Available from: www.gabi-journal.net/wp-content/uploads/MENA-SBP-2015-Morakot.pdf
54. Sinclair AM. Erythropoiesis stimulating agents: approaches to modulate activity. Biologics. 2013;7:161-74.
55. Egrie JC, Dwyer E, Browne JK, Hitz A, Lykos MA. Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin. Exp Hematol. 2003;31(4):290-9.
56. Jefferis R. Glyco-engineering of human IgG-Fc to modulate biologic activities. Curr Pharm Biotechnol. 2016;17(15):1333-47.
57. Dekkers G, Treffers L, Plomp R, Bentlage AEH, de Boer M, Koeleman CAM, et al. Decoding the human immunoglobulin G-glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front Immunol. 2017;8:877.
58. Hayes JM, Cosgrave EF, Struwe WB, Wormald M, Davey GP, Jefferis R, et al. Glycosylation and Fc receptors. Curr Top Microbiol Immunol. 2014;382:165-99.
59. Braster R, Grewal S, Visser R, Einarsdottir HK, van Egmond M, Vidarsson G, et al. Human IgG3 with extended half-life does not improve Fc-gamma receptor-mediated cancer antibody therapies in mice. PLoS One. 2017;12(5):e0177736.
60. The International Immunogenetics Information System [homepage on the Internet]. [cited 2018 Mar 28]. Available from: http://www.imgt.org/mAb-DB/index#Approval_antibodies
|
Author: Professor Roy Jefferis, PhD, DSc, MRCP, FRCPath, Emeritus Professor of Molecular Immunology, Institute of Immunology and Immunotherapy, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/protein-heterogeneity-and-the-immunogenicity-of-biotherapeutics.html
Copyright ©2024 GaBI Journal unless otherwise noted.