Author byline as per print journal: Professor John-Joseph Borg, PhD; Yolanda Elias Gramajo, MD; Professor Andrea Laslop, MD; Robin Thorpe, PhD, FRCPath; Jian Wang, MD, PhD
|
Introduction: Biosimilars have the potential to improve access to medicines for many across the globe. However, work is required to ensure adequate regulation, pharmacovigilance and education about biosimilars. Colombia implemented biosimilars regulation in 2017 and a 3rd Colombian Educational Workshop was organized by GaBI and the Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) in 2019 to follow up on progress and provide a forum for further discussion. |
Submitted: 9 April 2020; Revised: 8 June 2020; Accepted: 15 June 2020;; Published online first: 29 June 2020
Biosimilars or similar biotherapeutic products (SBPs), are a relatively new class of biotherapeutic agent. With appropriate regulatory and pharmacovigilance procedures in place, these products have the potential to improve access to medicines worldwide. These drug products are being developed and are entering markets across the globe. With sufficient trust in and uptake of these products, healthcare costs could be reduced dramatically.
Europe has been at the forefront of developing biosimilars regulation in recent years and now an internationally agreed system of regulatory approval is needed. Similarly, pharmacovigilance of biosimilars needs to be done on a global level. There are also questions regarding the interchangeability of biosimilars and a global understanding of this should be laid out. To achieve consensus on these points, open discussion across national borders and between different stakeholder groups is key.
To facilitate discussion concerning quality assessment of biologicals/biosimilars in Colombia, in 2016 and 2017, an educational workshop [1] and a follow-up meeting [2], were organised by the Generics and Biosimilars Initiative (GaBI) in collaboration with the National Food and Drug Surveillance Institute of Colombia (Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA). Together, these meetings provided a forum to exchange knowledge on best practice and enabled the status of biosimilars quality assessment in Colombia to be viewed with clarity [3].
Following the implementation of biosimilar regulation in Colombia in 2017, a third meeting on the regulatory assessment of biosimilars in Colombia was held in 2019. This interactive workshop provided a forum for regulators, clinicians, pharmacists, academics and healthcare professionals from Colombia who are involved in biological/biosimilar medicines evaluation, to share knowledge and exchange information with expert speakers from Canada, Europe and the US. They engaged in active discussions that concerned the current status of biosimilars regulation in Colombia; standards and the stepwise approach of quality and immunogenicity assessment of biologicals/biosimilars; structure–function relationship of biological medicines; switching; pharmacological (pharmacokinetic (PK)/pharmacodynamic (PD)) and clinical studies of biosimilars; extrapolation of indications; post-marketing pharmacovigilance practices of biologicals/biosimilars to ensure patient safety; and future educational needs and how to improve the uptake of biosimilars.
Overall, the meeting aimed to bring experts together to clarify biosimilars regulatory concepts and concerns, in order to improve biosimilar regulation and increase their uptake in Colombia.
On 30 April 2019, GaBI held the 3rd Colombian Educational Workshop on Regulatory Assessment of Biosimilars. As with previous workshops [1–3], the aim was to review and explain the current status of biosimilars regulation in Colombia. The format was similar to that followed in prior scientific meetings and educational workshops as reported in GaBI Journal [4–6]. There were a number of expert speaker presentations followed by Q&A and an in-depth panel discussion. The presentations are downloadable from the GaBI website [7]. All participants were encouraged to engage in active discussion about the presentations given by expert speakers and to discuss selected case studies.
Presentations were in English or Spanish with simultaneous translations into Spanish or English, respectively.
The workshop began with a welcoming speech from Dr Lucia Ayala, Director of Medical Devices and Technologies at INVIMA. This included a brief introduction to the Colombian regulatory approach to biosimilars. The Austrian Medicines and Medical Devices Agency’s (AGES MEA) Head of Scientific Office, Professor Andrea Laslop, then provided a second welcome address. She celebrated the longstanding collaboration between INVIMA and GaBI and briefly outlined the workshop objectives (as summarized in the Introduction).
Biosimilars in Colombia: a year after regulation implementation
Mr Aurelio Enrique Mejía Mejía, Director of Medicines and Health Technologies at Colombia’s Ministerio de Salud y Protección Social, and Johanna Andrea García Cortes, Professional Specialist at INVIMA, gave presentations which discussed the implementation of Colombia’s Biotechnology Decree 1782/2014. Their presentations entitled ‘Colombia’s biological/biosimilar regulation: a year after its implementation’ and &lsqup;Biosimilar regulation in Colombia: one year later’, respectively, can be found on GaBI website [7].
In a brief summary, the two presentations outlined details of the 2014 Biotechnology Decree 1782. This laid out the requirements and procedures of INVIMA to evaluate the quality, safety and efficiency of biological medications and aimed to enable the appropriate approval and commercialization of such products in Colombia in the future. The two presentations discussed the decree and its impact following implementation in August 2017. In the first year of implementation, it has led to more therapeutic options being available to patients and an increase in competition in Colombia’s biological drug market. At present, 23 biosimilars are under review in Colombia, of which trastuzumab has undergone all regulatory processes and achieved marketing authorization. In addition, there are regulations in place to ensure that biosimilars enter the market meeting global standards of safety and efficacy. The next step is to improve the education of healthcare professionals to help increase the uptake of biosimilar products across the country.
Quality assessment of biologicals/biosimilars – most relevant quality attributes: case study on monoclonal antibody
Professor John-Joseph Borg, Malta Medicines Agency’s Director of Post-Licensing, discussed the quality assessment of biologicals/biosimilars in the European Union (EU) [8].
Professor Borg outlined the basic concept of a biosimilar:
He noted that, due to the complexity of biosimilars, it is not appropriate to follow the same approach as with simple generic drugs when addressing bringing biosimilars to market. As such, comparability exercises are required for biosimilars to be authorized in the EU.
In the EU, a complete and appropriate quality dossier is required for a biological product to be approved (Module 3). For a biosimilar, this must also include a comprehensive comparability exercise (quality, non-clinical, clinical), see figure 1. He added that establishing comparability is a stepwise approach and non-clinical and clinical data are required before product developers should move on to address the next step and address any residual uncertainty. Licensing decisions are then made based on the entire data package which includes quality, non-clinical and clinical parameters that demonstrate similarity to a reference product. All licensing is done on a case-by-case basis.
So, to build an application dossier for a biosimilar, a product developer will need a full Module 3, plus the comparability exercises (there should be multiple data points with all data being recorded over a period of time), followed by non-clinical studies and then clinical studies. If a product meets the requirements and is approved, conditions can then be attached to approval in the post-authorization phase.
Professor Borg further discussed the comparability evaluation and noted that all aspects mentioned above must be taken into account. This evaluation includes details of the nature and level of knowledge of the product, i.e. its complexity, structure-activity relationship, relationship between therapeutic and endogenous proteins and the mode of action. He also discussed aspects of immunogenicity and highlighted that this is an issue for all biological products. If a reference causes immunogenicity, so will a biosimilar. The risk factors for immunogenicity are both product and patient related.
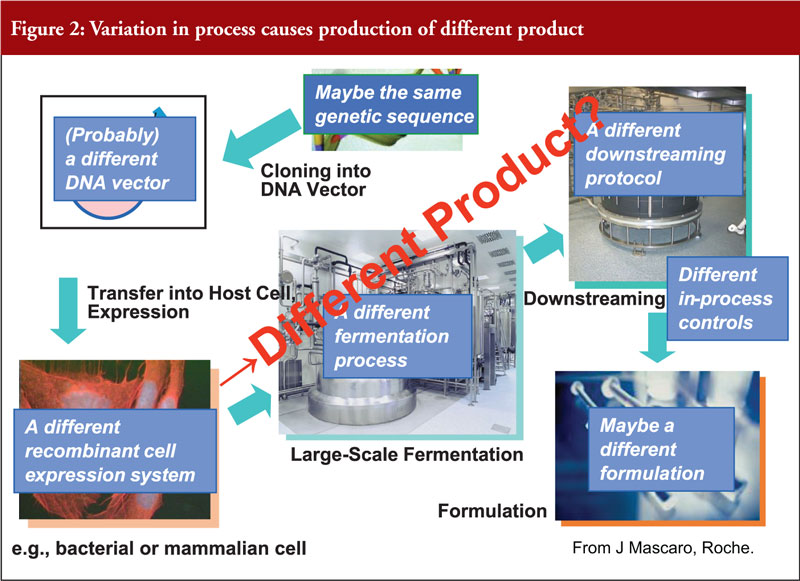
Another key issue when considering biosimilars is that any variation in process can cause a different product to be produced, see figure 2. These variations can occur at any point during processing. Therefore, a regulator needs to know about every change made during production/processing and any effects that changes may, or may not, have. The sourcing and testing of the reference product must also be done over a period of time to account for any differences in the reference.
Overall for biologicals, the quality is highly dependent on the manufacturing process which is very complex. For a product to be accepted, it must meet quality criteria and be validated with safety and efficacy data. Professor Borg noted that there are additional critical quality aspects for a biosimilar and he went through a non-exhaustive list of these, see Table 1.
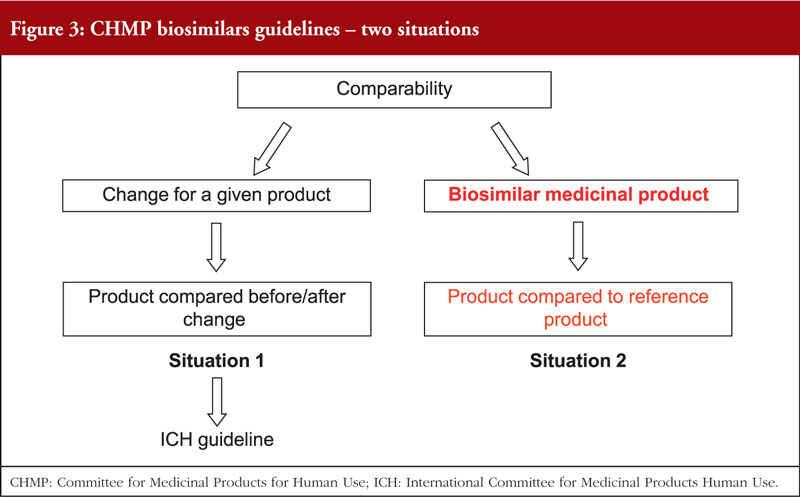
The EU Committee for Medicinal Products for Human Use (CHMP) guideline for comparability was discussed, see figure 3. This included notes on the origin of the reference product and how, when comparing a reference to a biosimilar, the same reference product (from the same country of origin) should be used over time.
Professor Borg gave an overview of the chemistry, manufacturing and controls (CMC) documents for monoclonal antibodies (mAbs) derived from a monoclonal cell line. This included a description of the development and production of mAbs and their characterization (physicochemical characterization; immunochemical properties; biological activity; information on purity, impurity and contaminants; and on quantity). The talk was finalized with an outline of Remsima biosimilar comparability where he included a comprehensive list of the studies comparing the physicochemical and biological activity of the Remsima biosimilar and the Remicade originator.
Pharmacological studies (PK/PD) to assess biosimilar medicinal products
Dr Jian Wang, Division Chief of the Clinical Evaluation Division, Biologics and Genetic Therapies Directorate, Health Canada, discussed biosimilar pharmacological (PK/PD) studies.
Biosimilars are similar to their brand-name reference products and not the same due to being made from different cell lines and by different manufacturing processes. To support regulatory approval, guidance/policy requests that comparisons between the biosimilar and its reference are made. These include, see Figure 4:
Clinical studies are needed to ensure that residual uncertainty from quality assessment does not cause clinically meaningful differences in efficacy, safety and/or immunogenicity in the sensitive population. Dr Wang stated that the main goal of clinical PK/PD studies is to rule out unacceptable PK/PD differences that could indicate the presence of significant structural and functional differences between a biosimilar and a reference product.
Regarding comparative PK studies, Dr Wang noted that these comparative clinical PK studies should be conducted in a setting that is reflective of the clinical situation and/or is sensitive to detect differences between the biosimilar and the reference. They should also be planned based on the characteristics of the reference, including its mode of action, safety profile and PK properties. In general, PK studies can be done on healthy volunteers. However, these individuals may not always reflect the PK parameters of patients. As such, comparative PK studies can also be conducted in the patient population.
The same principles of study design, statistical methods and criteria of acceptance for small molecules are used as a general guidance for biologicals. When the route of administration is intravenous (IV) which does not include an absorption phase, some additional parameters from elimination phase will be included for equivalence assessment.
In some aspects of study design, the single dose cross-over design is the most sensitive PK study design, for short half-life biologicals. However, this design can be limited by the properties of the biological such as having a long half-life or following the formation of antidrug antibody (ADA). In specific cases, parallel and/or multi-dose designs can be considered.
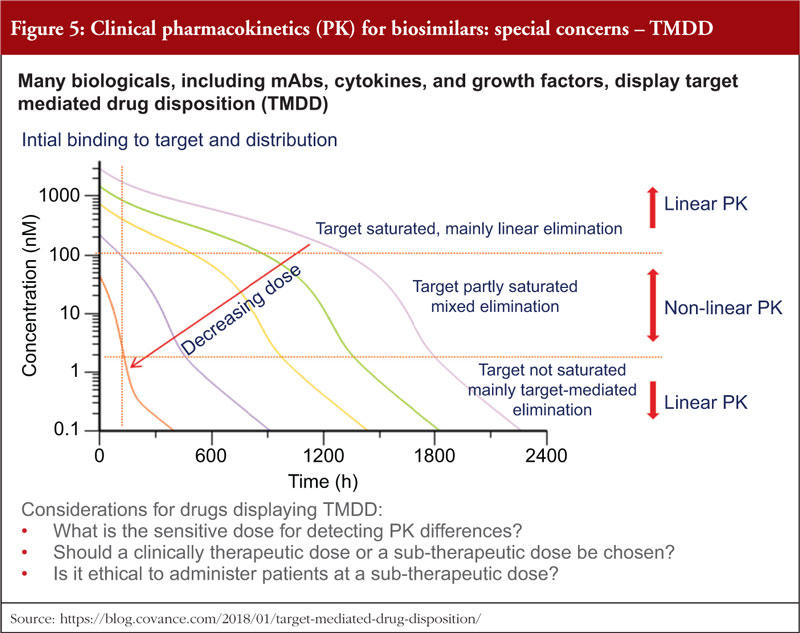
Many biologicals, including mAbs, cytokines, and growth factors, display target mediated drug disposition (TMDD), see figure 5. For such biologicals, three considerations must be made when conducting comparative PK/PD studies:
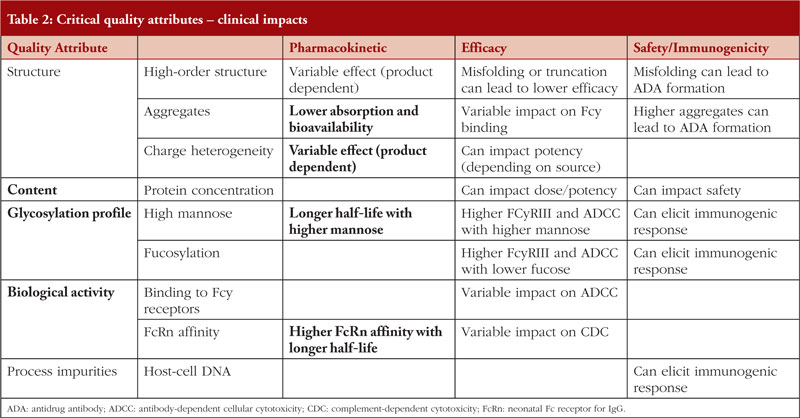
Critical quality attributes can influence the PK of a mAb and therefore, may have a direct impact on biosimilarity, see Table 2. In terms of immunogenicity, most biologicals induce some level of ADAs and these may have undesirable clinical effects on PK, efficacy and/or safety, including immunogenicity. It was also noted that PopPK studies are being used in demonstrating comparability for biosimilar mAbs as supportive studies.
Finally, on comparative PD studies, Dr Wang stated that PD studies are desirable, if feasible, and can help reduce residual uncertainty. PD parameters should be investigated as part of the phase III trial. He then discussed PD surrogates and aspects of PD sensitivity (including clinical, assay and dosing). Also, it is necessary to measure the baseline endogenous levels of a biological in blood plasma if the biological product is produced endogenously. In conclusion, for products with a reliable PD marker, a high quality and sensitive PD study (usually combined with PK) may be better than an efficacy study in terms of detecting differences in efficacy between a biosimilar and its reference product.
Head-to-head clinical studies and biosimilarity studies to assess biosimilar medicinal products
Dr Yolanda Elias Gramajo, Senior Clinical Evaluator, Clinical Trials Division, Health Canada, discussed head-to-head clinical studies and biosimilarity studies to assess biosimilar medicinal products.
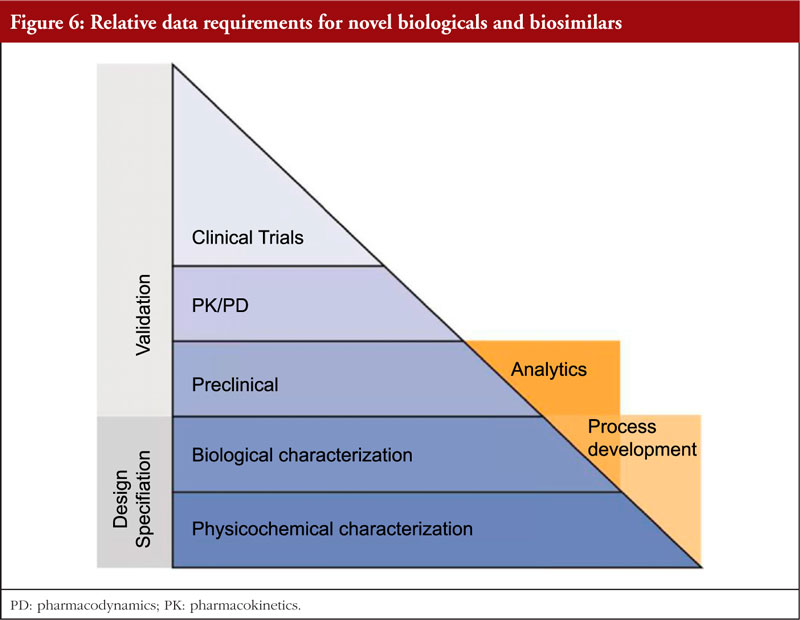
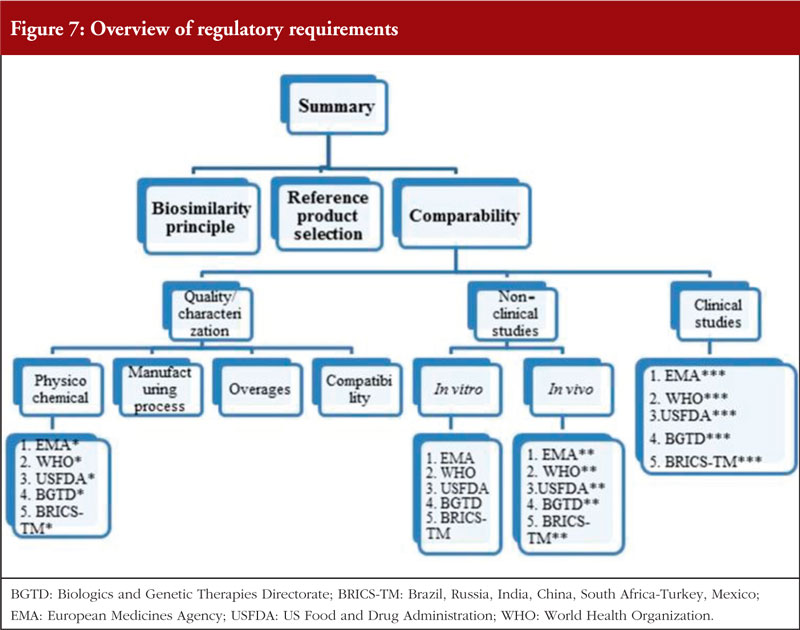
Dr Elias Gramajo began by outlining that the pathway for biosimilar development and approval is designed to demonstrate similarity to its reference product with respect to quality, safety and efficacy. This uses a stepwise approach that is often depicted as a pyramid, see figure 6. This includes analytical, non-clinical and clinical studies. She gave a brief overview of the general regulatory requirements and the requirements for biosimilar development, see figure 7.
As part of the clinical programme for biosimilar development, comparability exercises are required. The comparative exercise objective is to demonstrate similarity in PK/PD, efficacy, safety and immunogenicity between the biosimilar and the reference product. In general, clinical trials are required (phase III for at least one indication) but in some cases PK/PD studies can suffice.
When it comes to the clinical development programme for biosimilar products, it is preferred that clinical trials follow an equivalence design. Here, a trial has the primary objective of showing that the response to two or more treatments differs by an amount which is clinically unimportant. This is usually demonstrated by showing that the true treatment difference is likely to lie between a lower and upper equivalence margin of clinically acceptable differences. She outlined some key considerations for comparative clinical trials including that generally 90%–95% confidence interval (CI) equivalence margin in the PP population (per-protocol) is acceptable, randomized design, double-blind/adequately powered, power and sample size sufficient to detect difference, dose and route consistent with reference.
Sensitive clinical study populations were then discussed, and that the comparative clinical study should be conducted in a sufficiently sensitive population that is representative of the authorized indications to detect differences between the biosimilar and its reference. The endpoint should be considered to improve the detection of potential differences between the biosimilar and the reference within the sensitive population.
When it comes to safety, immunogenicity is the most important aspect. Dr Elias Gramajo presented a number of points relevant to this including ADA formation. She also outlined the immunogenicity assessment strategy which includes screening assays, confirmatory assays, neutralizing assays and PK/clinical impact assessment. In all cases a biosimilar should never be more immunogenic than its reference in terms of ADA incidence or concentration.
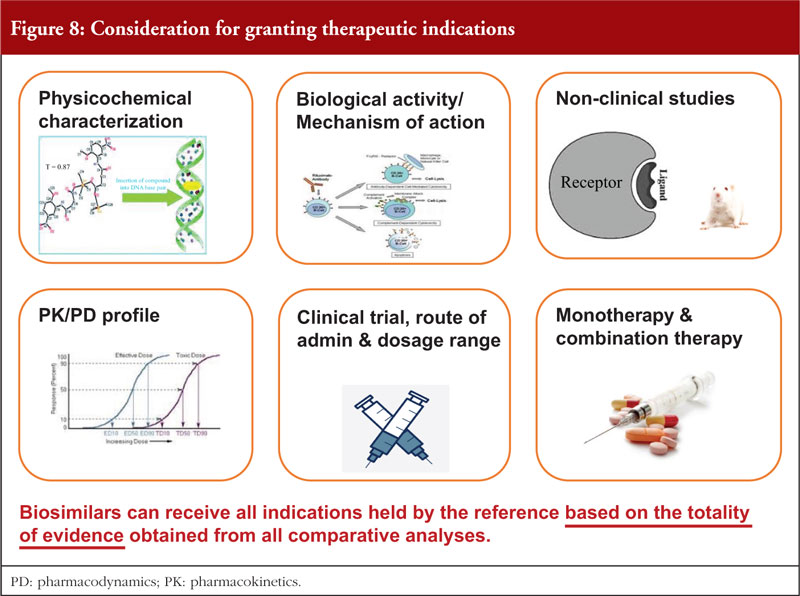
In conclusion, when a biosimilar is authorized it can be granted all therapeutic indications held by the reference based on the totality of evidence obtained from all comparative analyses, see figure 8.
Extrapolation of biosimilars
Professor Andrea Laslop, Chair of the workshop, delivered a presentation on the principles of extrapolation of indications in the EU. Biosimilar extrapolation is when a biosimilar can be used for any indication that the reference product is approved for, even if the biosimilar itself has not been directly studied in a comparative clinical trial for that indication.
Professor Laslop outlined some general considerations for extrapolation, which is an important feature of the biosimilar approval process but that it continues to cause contention. Many EU biosimilar guidelines have been set up to address the issue and to date, extrapolation has been implemented in all biosimilar product approvals. She highlighted the fact that the concept is not new and has been used for biosimilars and generics and also in paediatric indications and other special populations. In addition, extrapolation can occur when there have been changes to the manufacturing processes of biologicals.
Concerning changes in the manufacturing process, comparability exercises are carried out to ensure the product efficacy and safety is not altered. When such changes are made, the product will be different and thus by definition, it will be a biosimilar (typically clinical data are not required to approve manufacturing changes).
Mechanism of action (active site) is key to extrapolation, see Table 3. This is mediated by functional molecular moieties in a disease-specific manner, which can be characterized by sensitive assays. If the same mechanism of action (MoA) or the same receptors are involved for the function of the biological/biosimilar product in different diseases, then extrapolation is straightforward. However, in some cases additional non-clinical or clinical data may be required, e.g. different active site, different receptor, different safety profile.
She noted that the overarching guideline on biosimilars states: if biosimilarity has been demonstrated in one indication, extrapolation to other indications of the reference product could be acceptable with appropriate scientific justification. Likewise, the general guideline on biosimilars containing biotechnology-derived proteins as the active substance, non-clinical and clinical issues, also says that extrapolation could be acceptable, but that in case of unclear relevance of the safety and efficacy data from one indication for another one, additional data will be required. In all cases extrapolation is only considered in light of the totality of evidence.
Professor Laslop presented a number of cases where extrapolation had occurred and justified the reasons for extrapolation in each example [9, 10].
In conclusion, extrapolation is essential for the success of biosimilars and must be done on the basis of the totality of data available. It is not done automatically and requires scientific justification. This is a common process in drug development and is not exclusive to biosimilars. Extrapolation is expected if comparability of the product, with respect to a reference, has been demonstrated on all levels. In addition, post-approval information for extrapolated indications is helpful and, to date, has shown the success of extrapolation.
Immunogenicity studies for assessing biosimilar products
Dr Robin Thorpe, Co-chair of the workshop, delivered a presentation on immunogenicity studies. This was similar to that given at the GaBI MENA 2018 meeting [6].
In conclusion, it was noted that immunogenicity issues occur all along the life cycle of a product and particularly when:
And that assessment requires:
Two further presentations were given by representatives from Amgen. ‘Biologicals and biosimilars – the complexity of structure and function’ was delivered by Dr Jennifer Liu, Director of Analytical Sciences, and ‘Considerations for product specific pharmacovigilance of multisource biologicals’ was delivered by Dr Thomas Felix, R & D Policy Director.
After the presentations, there was the opportunity for discussion about the topics covered. The key discussion points are summarized below.
The regulatory pathway in Colombia
The current status of biosimilars regulation in Colombia/The regulatory pathway in Colombia
Following the two presentations given on ‘Biosimilar’s regulation in Colombia: one year on’, there were some queries that required further clarification.
Dr Arley Gómez López expressed concern about the fact that Colombia does not have certain infrastructure in place for adequate drug analysis. Mr Mejía Mejía responded that the current regulation is in place to generate incentives and allow for the development of the infrastructure that is required. He emphasized that it is important to understand that the process is gradual. Having clarity regarding the standards, which has now been achieved, will allow companies to start to develop the capabilities in order to accomplish exactly the types of studies that are needed. Overall, Colombia’s Ministry of Health intends to give clarity regarding the norms so that they can be developed from the conditions. He also agreed that it is necessary to develop public and/or private initiatives to facilitate the development of the infrastructure.
Comparisons between the Colombian and other regulatory agencies
When asked about improved access to medicines in Colombia through commercial mechanisms, Mr Mejía Mejía stated that, at present, only hepatitis C medicines are bought through centralized purchasing. By focusing on the regulation of biotechnological medicines, it is hoped that access to medicines will be improved through the availability of greater therapeutic options which promote competition and reduce costs. Now that we have regulation in place, products are going through the approval process. When products are approved by INVIMA, they can start being prescribed by doctors and be covered by the health system. Eventually, in the case where the government and the ministry allow it, they can also be subject to centralized purchase or negotiations.
According to data in the presentations given, there seemed to be a lot of biosimilar applications in Colombia compared to the EU. Dr García Cortes advised that, when it comes to expensive medicinal products, it is good that there are a lot of biosimilar applications as this will promote competition and decrease prices. INVIMA’s Ms Alejandra Mayra Gomez Leal added that many biosimilars have been approved by the European Medicines Agency (EMA) and not the US Food and Drug Administration (FDA) and that Colombia is open to applications and whether or not they have been approved in other places may only be used as a reference.
There was a concluding comment by Andrea Carolina Reyes Roja, pharmaceutical chemist working at Misión Salud, a Colombian NGO. She stated that Misión Salud has followed the regulation process for biotechnological medicines in Colombia for more than 10 years. She congratulated the government for the emphasis it has put on the utilization of the abbreviated pathway as a public policy. Other health agencies, such as EMA, FDA and the World Health Organization (WHO), have a ‘questions and answers’ document published in October of 2018 which reaffirms the possibility of reducing clinical studies when supported by the vast knowledge of the molecules in analytical studies of high complexity and limited to PK/PD studies. The clarity that the Colombian government has provided on this issue will increase access and reduce the costs of medicines.
Following the presentation given by Professor Borg, Dr García Cortes wanted to know if Europe is thinking about removing the bridging studies. She noted that in Colombia they have found cases where there are significant differences between the product in the US and the European counterpart. In such cases, it is difficult to evaluate the comparability, so should bridging studies be removed? And in these circumstances should it be the US or EU product? And if only one reference product is taken, whether it be the European or the US. In response, Professor Borg noted that in the EU the reference product needs to be sourced in the EU to comply with comparability requirements. In addition, Andrea Carolina Reyes Roja stated that for the quality comparison, Europe requests the comparison to the European reference product and does not accept head-to-head comparison to a US-derived reference. She pointed out that this is different from the clinical development programme, where under certain conditions bridging exercises can be accepted. For example, a clinical study of the US reference could be used if it is also demonstrated that both the US and the EU reference product are sufficiently similar on the quality level.
Stepwise approach – PK/PD trials, comparability studies
When to use PK and PD studies and data
Ms Alejandra Mayra Gomez Leal was interested to know more about mAb studies and PK data collection. For the majority of mAb studies as presented by Dr Wang, the primary evaluation was for the efficacy aspects, and as a secondary objective, they evaluate the PK. Dr Wang explained that normally the first PK measurements will be done after the first dose of medication, and this essentially gives a single-dose PK. However, depending on how many samples can be collected, the study must be very well designed to collect multiple samples at different time points so that a PK profile can be created. In addition, and in general, PK data are collected at multiple dosing time points, so it is possible to have single-dose and multi-dose PK data.
Associate Professor Claudia Patricia Vaca González noted in her conclusion, that Dr Wang said that when there is a good indicator of PDs it can be more desirable to have a PK/PD study than a clinical efficacy study. She wondered how generalized this statement was, considering there is a growing interest from the agencies and countries to have competition and competitive products on the market. This approach could improve the speed of introduction of these products into the market. Dr Wang responded by noting that this statement is not possible to generalize to all biologicals. He reiterated that for products with well-known, well-established PD surrogates, you can generally use comparative PK/PD studies. However, agencies may also ask for a longer-term safety study which may not be powered to demonstrate equivalence and the results of which cannot be inferred from the comparative PK/PD study.
The science and rational of clinical trials
Rejection after clinical trials
There was a query regarding the number of biosimilars that were rejected in Canada, following clinical trials. Dr Wang stated that, when it comes to biosimilars and clinical trials, it is not purely a science issue, but also a regulatory issue. A biosimilar should be as good, or as bad, as the reference. If it is better, it becomes a judgement call. How much better is it? And is that going to cause safety concerns or not? In biologicals, it is often the case that you will have better efficacy but a worse safety profile. So, a proper benefit-risk assessment must be made and within the considerations of the country’s regulations. Overall, the situation is quite complicated, it is not the case that products go through all steps prior to clinical trials, without hitch, and then get rejected on the basis of trial results.
The importance/discussion of pharmacovigilance, naming issue
Improving and maintaining pharmaco-vigilance
Associate Professor Claudia Patricia Vaca González made some comments on the naming of biologicals. She said that WHO allowed countries the freedom to establish differentiation from the common international names of products. This came after lengthy discussions about both the inconvenience of having differentiation in nomenclature which causes confusion between patients and doctors; and following concerns expressed by different countries that this name differentiation could be used to reduce competition when generic or biosimilar medicines enter the market. In addition, she noted that nomenclature is very important when it comes to pharmacovigilance and tracing of the active principle. It is important to have information related to the product batch and the ability to trace a product through its logistic chain. She stated that health systems in all countries must improve upon traceability to improve pharmacovigilance.
Dr Felix responded that the concept of active ingredient traceability is very important. This is how small molecules are traced around the world. However, active ingredient traceability and batch level traceability are not two things that necessarily go together. In most countries, it is not possible to find the manufacturer of a product with only the non-proprietary name product batch number.
Professor Laslop agreed that it is important to have separate pharmacovigilance reporting for each individual biosimilar product. In Europe, pharmacovigilance data on the non-proprietary name would not be collected alone. The European implementation pathway instead requires prescription according to brand name. That is, in Europe, biosimilars are never prescribed according to non-proprietary name but rather by specific brand name together with the batch number, and this is how a product can be traced.
Dr Felix added that Europe has not implemented a specific naming approach and some countries outside Europe see this as a greenlight to do the same. However, they do not take into account certain aspects of European legislation in place to improve traceability which include brand level prescribing, good pharmacovigilance guidance and education of healthcare professionals.
With respect to this discussion, Professor Borg noted that new regulation is coming into force in the EU with respect to the falsified medicines directive. All batch numbers will be recorded where dispensed. Ideally in the future, all will be linked centrally at EMA with the help of IT software. Dr Felix explained that the directive is a European legislation which is in place to prevent the ability of counterfeit medications entering into the European system. As such, there is very high accuracy in the traceability of biological products and all products that enter Europe. In the future, this has potential to be a powerful tool and to improve patients’ safety, particularly if data input into the system occurs at the dispensing level.
A final comment was made by a member of the audience regarding pharmacovigilance and residual uncertainties. He expressed concern that these medicines were being released to the market despite the existence of uncertainties. In response, Dr Felix stated that, for all medicines, regulatory approval pathways are built on the best available evidence at a given point in time. Even for an innovative product, there is still some residual uncertainty that might exist, and the study of products must continue after they are approved. This is the case, particularly when they are used in patient populations that were excluded in clinical trials or inpatient populations that were not studied originally. In all circumstances, the collection of aggregate information is required to show that the benefit continues to outweigh risks. This is going to be especially important in the next wave of innovative medications that are being developed for patients with rare diseases which are based on clinical trial data sets that are much smaller. These will be promising in terms of their clinical benefit but due to their nature, there will be all the more reason for post-approval traceability of these products and continued measurement of the benefit-risks related to them.
Immunogenicity
After Dr Thorpe’s presentation on immunogenicity, Professor José Orozco asked if it is possible for a biosimilar to be less immunogenic. Dr Thorpe explained that there are instances where immunogenicity, at least appears, to be lower for a biosimilar. Often this is due to assay artefact problems and the real problem is that when assays are carried out the antigen must be selected with extreme care. The most logical way of doing this is to measure the antibodies against the product that the patients have received. The patients who have received a biosimilar, the new biosimilar, get screened against the biosimilar and the ones who have received the innovator get screened against that. However, this is labour intensive. As such, it is common to use just one antigen – and all the patients get screened against one antigen. This is allowed in the EU if the antigen is the biosimilar itself, as here you increase the possibility of detecting antibodies against a biosimilar which gives you a possible, slightly higher apparent immunogenicity of the biosimilar. In other circumstances, immunogenicity can be reduced in a biosimilar. This can be due to assay effects or may be because of high purity of the biosimilar (compared to older reference product made with less advanced technology). When a biosimilar has less immunogenicity, this seems good, however, this should ideally be justified in the assessment in terms of purity or assay issues.
There was a panel discussion chaired by Professor Laslop and co-chaired by Dr Gramajo.
The Regulatory panel discussion was introduced by Professor Laslop. The panel included Arley Gómez López, Research Director from the University Research Foundation ‘de la Salud’; Juan Fernando Juez Castillo, INVIMA; and Judy Hasleidy Martínez Martínez, INVIMA. Professor Laslop opened discussions and noted that the workshop presentations had thus far brought to light the two main pillars for assessment of biosimilars: the quality comparison (which includes non-clinical data) and the comprehensive clinical comparison.
Reducing the biosimilar assessment process
Mr Castillo noted that it is evident that we still have a lot to learn when it comes to biosimilars and their assessment. There is a great deal of complexity when it comes to biosimilars and their production. Therefore, it is very important to have a very strict process for analyses in Colombia. For now, he believes that shortening any analysis could result in a fall into reductionism, which is to be avoided. However, it may be necessary to have public and/or private concessions as neither the universities nor the government alone currently have the capacity to realize these types of complex analyses adequately.
When it comes to reducing the comparability exercise and the clinical studies for biosimilars, Mr Castillo also noted that this should only be done on a case-by-case basis. It is not possible to generalize and say that a certain category of products should follow a certain abbreviated ‘third pathway’ with less information required.
To emphasize this he noted that some mAb patents are expiring and, despite extensive characterization, there are still many uncertainties about the molecules. However, this is not the case for molecules like insulins which are well understood.
Dr Martínez Martínez noted that in Colombia the concept of a third pathway for certain drug products with reduced trials has been discussed. She explained that, Colombia’s Decree 1782 aims to ensure molecules are precisely characterized, have robust pharmacovigilance and good safety information. These molecules undergo clinical trials until enough information is known to prove their safety (either in Colombia or in other countries). In some cases, smaller biological molecules, such as insulins, heparins and filgrastim, can be well characterized and do not require as many clinical trials. Such molecules could be considered in the third pathway. However, to date, not many products have followed this third pathway. Overall, INVIMA is working with comparability to try and reduce the clinical and preclinical trials needed for some products but this should never impact on product safety. At present, INVIMA has not worked on the specific criteria for the different pathways. Laboratories can present the information through the pathway they chose based on the information and justifications that they selected throughout the trial. In general, if a characterization does not seem sufficient to the regulator, then more information is requested.
Professor Laslop added that this third pathway approach is similar to those adopted in Europe, Canada and the US. Here, there is potential to waive full clinical trials for certain less complex biosimilar molecules. However, if Colombia wants to adopt a similar approach to those nations, the guidelines and definitions need to be very clear. This is particularly important in terms of molecule complexity and what is, and is not, to be included in each of the different approval pathways.
In addition, Ms García Cortes noted that she did not support the idea of having a list of molecules in a third pathway. Biologicals should be assessed on a case-by-case basis, running through the three steps. These steps investigate the complexity of the molecule, the complexity of its MoA and the complexity of its production. Changes in production method such as a change in vial, can lead to massive adverse effects in the product that are not due to the molecule itself. As such, creating a list of molecules to be included in a third pathway does not seem plausible as their effectiveness is also dependent of the complexity of their action mechanism and production method.
A member of the audience noted that a third pathway should exist to give clarity in cases where it is not necessary to get to the end of the clinical trials and safety and efficacy studies. In such cases, PK/PD studies are carried out and the medicines are widely known and have robust pharmacovigilance. However, it is imperative not to compromise safety and INVIMA performs exhaustive evaluations to ensure this.
Another audience member also noted that research ethics should be part of the regulation because it ensures that patients’ rights are observed.
The applicability of the assessment pyramid
When a biosimilar undergoes evaluation for assessment of comparability, it should generally follow the stepwise approach outlined in the assessment pyramid. However, in many cases, not all stages of the pyramid are reached and undertaken prior to product approval. As such, it was proposed that this pyramid should be changed. Professor Laslop stated that she would not be in favour of removing or altering the pyramid as it is still the default pathway for biosimilar development, biosimilar assessment and, ultimately, biosimilar approval. In Europe, the possibility for abridged clinical assessments is exactly defined in product specific guidelines.
Dr Gramajo noted that the situation is the same in Canada. All assessments are done on a case-by-case basis. Those developing biosimilars should consult with agencies at an early stage to determine what information is needed and which trials are necessary. As such, the overall pyramid is still relevant.
Clinical trials
Dr Thorpe stated that classically, to prove clinical efficacy, a classical efficacy trial should be carried out. This is the standard and remains valid. However, there can be occasions when there is a better way of establishing clinical efficacy than performing a classical clinical trial. For example, with G-CSF (filgrastim) it is possible to determine PD and PK measurements more accurately than you can measure clinical responses. Clinical efficacy trials can still be carried out, but to the best of his knowledge, when these were carried out in Europe with filgrastim, the outcome was the same as shown by PK/PD studies. However, this does not mean that clinical trials do not need to be carried out with other products. The omission of clinical trials always needs to be justified.
According to Mr Castillo, if one arrives at the PK/PD step and there is little uncertainty, then this is when there seems little sense in continuing to clinical trials. They are unlikely to provide additional information. However, if there are many uncertainties following PK/PD studies it is unethical to continue to clinical trials. He believes that too many clinical trials are requested in the EU and North America. Many of these trials are huge, requiring many patients over a prolonged period of time and sample sizes required could exceed those needed for innovator products. Mr Castillo again affirmed that such decisions should be made on a case-by-case basis. He also highlighted that in Colombia, clinics are struggling to find enough patients with which to carry out large clinical trials. This is due to how the healthcare system is constructed. However, he hopes that this is changing so that more continuous studies can be done to assess long-term efficacy and trace adverse effects.
WHO reliance assessment
Mr Castillo noted that biosimilarity has not yet been officially defined in Colombia. This is an issue when choosing the exact pathway to follow for similarity assessment. It can also lead to redundant studies being done that have already been carried out in other countries.
In response, Professor Borg highlighted that WHO has a reliance assessment that helps regulators outside the EU assess biological products. As part of this, WHO has a formal agreement with EMA that allows EMA’s CHMP assessment report to be shared so that product assessment can be targeted on areas which are relevant to the country. As such, they do not need to waste time or money on reassessment and revaluation. This aims to allow for faster access to medicines for patients and improved healthcare systems in the non-EU countries. He suggested that Colombia could take advantage of this WHO reliance assessment.
Biosimilar education of the medical and patient community/Improving education about biosimilars
Professor García Cortes noted that the implementation of Decree 1782 is a big challenge for Colombia. Medical doctor education is a key part of ensuring its success. Colombia has done a lot of work to ensure correct evaluation of biosimilars from a qualitative, safety and efficacy point of view. It is now important that doctors have clarity about and trust in the medicines approved. As such, INVIMA’s next goal is to ensure that doctors and healthcare personnel are well educated when it comes to biosimilars. They should understand what they are and should trust the information available. She believes that, through this, INVIMA can create confident prescribers and good therapeutic results.
Mr Castillo stressed the importance of having a medical community that has knowledge and trust in the medicinal products it prescribes. INVIMA is working hard to ensure products receive a comprehensive evaluation and that there are channels of communication open to prevent any issues from causing problems or harm. With these mechanisms, and any other means, INVIMA will reduce the impact on the public health of Colombians.
Professor Laslop concluded that education of the medical community and the patient community is extremely important. In Europe, it became quickly apparent that it is not always efficient to simply publish something on a website. Instead, proactively approaching all stakeholders (the pharmaceutical manufacturers, or the patients, or the medical community) to educate, explain and communicate, has had far greater impact.
Dr Néstor Álvarez Lara noted that, when it comes to the communication of information, it is important to pay close attention to pharmaceutical marketing in Colombia. Advertising must be kept in check to ensure it does not negatively affect government spending or patient access to medicines.
Regulatory panel discussion conclusion and future actions
Dr Gramajo stated that the subjects discussed are very controversial, and there are a lot of things to consider which require more communication, more information, more collaboration between different agencies. Colombia is unique in that it is going to determine its own regulations and elect medical authorities in accordance to public opinion at a national and an economic level.
Discussion groups were provided with data on two semi-fictional trastuzumab biosimilar candidates. They were provided with physicochemical characteristics, selected glycan and biological attributes, and the results of a phase I study for Candidates 1 and 2.
Following the same format as previous GaBI meetings, each discussion group was asked whether the data for the candidates qualified for biosimilarity with a reference product from a quality (CMC) perspective. If not, they were asked what steps they would recommend fixing this. Discussion groups were also asked how ‘residual uncertainty’ could be addressed in preclinical or clinical studies. They were then asked, given that Candidates 1 and 2 had both the CDR (complementarity-determining region) and Fc (fragment crystallizable) region involved in their MoA for some of the indications, whether they would recommend extrapolation to all indications.
There were six discussion groups that evaluated the case studies. Overall, they all concluded that Candidate 1 is a biosimilar, from a quality perspective. However, there are differences between the candidate and the reference and some elements of residual uncertainty which require further investigation. If results of these investigations are as expected, then extrapolation is acceptable. When considering Candidate 2, all groups concluded that this is not a biosimilar, from a quality perspective. The differences between it and the reference cannot be solved and residual uncertainty is too large to warrant further study or clinical trial. Extrapolation is not acceptable.
Group 1 was moderated by Professor Fabio Ancizar Aristizábal Gutiérrez, and co-moderated by Judy Hasleidy Martínez Martínez. Based on the data presented for Candidate 1 this group suggested that further physicochemical or in vitro tests should be carried out to remediate differences. In addition, potency studies should be carried out. To address residual uncertainty, preclinical models could be used in vitro or in a parallel animal model. Here, clinical evaluation may be required for PK/PD data. Group 1 stated that Candidate 1 did qualify for extrapolation if the results obtained from the required physicochemical studies and in vitro studies resolve the uncertainties related to the quality attributes mentioned (fucosylation – antibody-dependent cellular cytotoxicity (ADCC); glycosylation – complement-dependent cytotoxicity (CDC) given that the therapeutic indication studied was relevant. Regarding Candidate 2, there are differences in some quality attributes, a larger sample of batches must be available to reduce uncertainty. Clinical studies do not allow the differences to be resolved, so do not recommend as biosimilar.
Professor Gutiérrez noted that having open communication channels and strong teamwork are very important when assessing biosimilars. This ensures everyone analyses and understands critical variables rather than criticizes information. Analysis methodology is important to optimise time use and to reduce questions following analysis. These case studies showed that the manner in which information is exchanged between different levels of experience in technical areas is critical. In addition, another important aspect is quality. The case studies put forward two quality parameters in the form of clinical trials. If preliminary quality results are obtained that raise doubt about the efficacy in the clinical trial, these can be used to confirm biosimilarity of a candidate product. In addition, Group 1 felt that the extrapolation of uses should only be allowed with better information regarding MoAs for each indication to know if they are complimentary. This is what was shown in the clinical trial provided.
Group 2 was moderated by Associate Professor Claudia Patricia Vaca González and co-moderated by Joseph Sebastián Cepeda Santamaría. The group confirmed similar outcomes to Group 1. They discussed that it is very important to compare the primary structures, yet in these cases there was no data with respect to that. It was also noted that there was no comparability data or immunogenicity data. For Candidate 2, they observed that some parameters recorded were outside the ranges expected to directly impact the immunogenicity and as such, the exercise did not enable the evaluation of immunogenicity.
Group 3 was moderated by Arley Gómez López and co-moderated by Gloria Cecilia Peñuela Sánchez. During their discussions, Group 3 noted that for Candidate 1, from a quality perspective, there are differences in the acid charge profile and the profile of deamidation which would affect the potency of the product. To remedy the differences, they should be evaluated in terms of their impact on the potency with clinical studies. They suggested that the residual uncertainty should be evaluated with the use of a small clinical study which is sufficiently sensitive in early stages of the disease. If this can be carried out, then extrapolation is possible. However, the group also noted that extrapolation cannot be done because the bioequivalence study was carried out on a single dose in healthy subjects and a more complete PK study would be required. When evaluating Candidate 2, Group 3 noted that it does not meet criteria to be a biosimilar from a quality perspective. The differences cannot be solved, and a clinical study and extrapolation should not be carried out.
Group 4 was moderated by Dr Néstor álvarez Lara and co-moderated by Giovanny de Jesús Otálvaro Rojas. This group’s opinion of Candidate 1 was aligned with that of Groups 1–3. In discussion about Candidate 2, it was noted that the clinical study design was not of good quality and the efficacy of the candidate was low.
Group 5 was moderated by Mr Castillo and co-moderated by Deyanira Duque Ortíz. With regards to Candidate 1, Group 5 noted that they had some concerns about the differences in the acid charge profile and ADCC binding test which could directly impact the PK and lead to inferior results. They suggested that information is needed to support the differences in acid charge profile. With regards to residual uncertainty, they noted that they would need the secondary evaluation results of overall survival (OS) and progression-free survival (PFS). In addition, they wanted clarification as to why the PCR evaluation variable was the primary evaluation variable. Overall, Group 5 said the candidate could be extrapolated but they would want to address the specific implications for gastric cancer.
Other observations the group made included:
Mr Castillo highlighted some brief adjustments in examinations that influenced the part of the bond strength, the power. The differences in glycosylation also affected these factors. In the PK table, with reference to the higher value, which is the expected value, the comparator result was lower than the expected value but not by much whilst the reference value was within or even higher. From an efficacy level, it caught the attention that even with these differences, the biosimilar presented a slightly higher efficacy compared to the reference, about 0.2% higher than the reference.
Group 6 was moderated by Johanna Andrea Garcia Cortes, and co-moderated by Ivan David Fonseca García. With regards to Candidate 1, the group noted that charge profile and deamidation were observed in the fucosylation particles. It was explained that the former affects ADCC and the latter, potency. However, when reviewing the data of ADCC, CDC and binding tests, they all fall within the required parameters. In addition, the group found it striking that there was nothing about comparison of comparative primary structure or comparative immunogenicity. For Candidate 2, the PK data endpoints do not fall in the correct regions and thus it is not a biosimilar.
In addition to the points made by the group moderators, Dr Wang noted that interestingly in the case of Candidate 1, the PK data is very similar to the reference. If this case had a true PD surrogate you might be able to determine a difference, but here you have no PD data so clinical studies are needed. However, looking at this case, you will need a sensitive study population as the PK values for the two products are comparable; if you had a non-sensitive study population you may reach a wrong conclusion. At each step, comparison must be sensitive to detect potential differences.
Ms Sánchez noted that the case studies were useful as it allowed workshop participants to have a complete discussion about both quality evaluation and clinical studies and the different parameters that can affect quality. Quality is very important when it comes to a products efficacy and safety and it allows for faster conclusion about whether a product is a biosimilar.
Professor Borg concluded that there was an issue with the clinical studies. The studies for the two candidates have two different designs and this could have been further discussed during the workshop. In addition, the primary endpoints warranted further discussion. For one candidate this was the overall response rate (ORR), which he believes is not the best endpoint for a study. He noted that the results clearly show that, even with an insensitive endpoint, there are huge differences in the efficacy of the product. This clearly demonstrates an inferior product. With this in mind, he added that the take home message is to consider the totality of evidence when in decision-making.
The 3rd Colombian educational workshop was successful in bringing together those involved with biological/biosimilar regulation in Colombia with experts from Canada, Europe and the US. It highlighted the progress that has been made in Colombia in terms of biosimilars regulation since the 2018 implementation of Decree 1794 and the future steps that are needed to improve biosimilars uptake and access to medicines. The workshop also highlighted many important issues surrounding biosimilars regulation and regulatory assessment and helped to clarify many regulatory concepts and concerns. The attendees shared ideas with the speakers and received clarification on issues of interest and concern.
The Generics and Biosimilars Initiative (GaBI) wishes to thank Professor Andrea Laslop and Dr Robin Thorpe, Chair and Co-chair of this workshop; the moderators and co-moderators in implementing the parallel discussions and clarifying information of the parallel discussion when finalizing the meeting report; as well as Mr Francisco Javier Sierra Esteban of INVIMA for his strong support through the offering of advice and information during the preparation of the workshop.The authors would like to acknowledge the help of the workshop speaker faculty and all participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators and co-moderators in facilitating meaningful discussion during the parallel discussions and case study working sessions, presenting the discussion findings at the meeting, and contributing in the finalization of this meeting report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Speakers
Professor John-Joseph Borg, PhD, Malta
Yolanda Elias Gramajo, MD, Canada
Thomas Felix, MD, USA
Johanna Andrea García Cortes, MSc, Colombia
Professor Andrea Laslop, MD, Austria
Jennifer Liu, PhD, USA
Aurelio Enrique Mejía Mejía, MSc, Colombia
Robin Thorpe, PhD, FRCPath, UK
Jian Wang, MD, PhD, Canada
Moderators
Néstor Álvarez Lara, PharmD
Professor Fabio Ancizar Aristizábal Gutiérrez, PhD
Joseph Sebastián Cepeda Santamaría
Deyanira Duque Ortíz, MSc
Ivan David Fonseca García
Johanna Andrea Garcia Cortes, MSc
Arley Gómez López, MD, PhD
Juan Fernando Juez Castillo
Judy Hasleidy Martínez Martínez
Giovanny de Jesús Otálvaro Rojas, PharmD
Gloria Cecilia Peñuela Sánchez, PharmD
Associate Professor Claudia Patricia Vaca González, MSc
Speakers and moderators had provided feedback on the regulatory panel discussion and the case study group discussion, respectively; read and commented the revised content of the manuscript, and approved the final report for publication.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor John-Joseph Borg, PhD, Malta
Yolanda Elias Gramajo, MD, Canada
Professor Andrea Laslop, MD, Austria
Robin Thorpe, PhD, FRCPath, UK
Jian Wang, MD, PhD, Canada
References
1. Generics and Biosimilars Initiative. First INVIMA Educational Workshop on Assessment of Similar Biotherapeutic Products 2016; 14 June 2016; Bogotá, Colombia. Available from: www.gabi-journal.net/about-gabi/educational-workshops/first-invima-educational-workshop-on-assessment-of-similar-biotherapeutic-products-2016
2. Generics and Biosimilars Initiative. Second Colombian Scientific Meeting on Quality Assessment of Biosimilars/Similar Biotherapeutic Products 2017; 15 August 2017; Bogotá, Colombia. Available from: www.gabi-journal.net/second-colombian-scientific-meeting-on-quality-assessment-of-biosimilarssimilar-biotherapeutic-products-2017
3. Gray E, Matejtschuk P, Thorpe R. Quality assessment of biosimilars in Colombia – reducing knowledge gaps. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):79-83. doi:10.5639/gabij.2018.0702.017
4. Generics and Biosimilars Initiative. First Latin American educational workshop on similar biotherapeutic products, Mexico; 20 January 2015; Mexico City, Mexico. Available from: www.gabi-journal.net/first-latin-american-educational-workshop-on-similar-biotherapeutic-products-mexico-city-mexico-20-january-2015.html
5. Walson PD, Thorpe R. First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars, Dubai, United Arab Emirates, 1 September 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):173–7. doi:10.5639/gabij.2015.0404.039
6. Laslop A, Wang J, Thorpe R. 2nd MENA Stakeholder Meeting on Biosimilars 2018 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2019;8(2):76-87. doi:10.5639/gabij.2019.0802.009
7. 3rd Colombian educational workshop on regulatory assessment of biosimilars 2019. 30 April 2019, Bogotá, Colombia. Available from: www.gabi-journal.net/about-gabi/educational-workshops/3rd-colombian-educational-workshop-on-regulatory-assessment-of-biosimilars-2019
8. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 22 May 2014 [homepage on the Internet]. [cited 2020 Jun 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf
9. European Medicines Agency. Guideline on similar biological medicinal products. 23 October 2014 [homepage on the Internet]. [cited 2020 Jun 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
10. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 18 December 2014 [homepage on the Internet]. [cited 2020 Jun 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/3rd-colombian-educational-workshop-on-regulatory-assessment-of-biosimilars-2019-report.html
Copyright ©2024 GaBI Journal unless otherwise noted.