Author byline as per print journal: Mihaela Buda, PhD; Olga Kolaj-Robin, PhD; Emmanuelle Charton, PhD
|
Abstract: |
Submitted: 7 January 2022; Revised: 26 January 2022; Accepted: 31 January 2022; Published online first: 14 February 2022
The monographs and associated physical reference standards of the European Pharmacopoeia (Ph. Eur.) are legally binding public standards in the Member States of the European Pharmacopoeia Convention and thus play a major role in ensuring the quality of medicines in Europe – and beyond [1]. While Ph. Eur. monographs on biotherapeutics (covering active substances as well as medicinal products) have existed for nearly three decades, their elaboration has faced challenges in recent years. The difficulties encountered relate mostly to the advent of biosimilars and a misunderstanding of the role of monographs in this context. These challenges were discussed at the international conference entitled European Pharmacopoeia: tackling future challenges of the quality of medicines together in September 2016 in Tallinn, Estonia (hereinafter ‘the Tallinn conference’). Following the event, ways were proposed to identify and elucidate these challenges and determine how they can be overcome [2]. The dialogue with stakeholders on the development of public standards for biotherapeutics continued in June 2019 at the dedicated workshop of the International Conference entitled EDQM & European Pharmacopoeia: state-of-the-art science for tomorrow’s medicines held to mark the publication of the 10th Edition of the Ph. Eur. (hereinafter ‘the Strasbourg conference’) [3]. On the eve of the launch of the 11th Edition of the Ph. Eur. in 2022, this manuscript provides an overview of the key points that emerged from the discussions held with Ph. Eur. users, such as representatives from innovator and biosimilar companies, National Control Laboratories and licensing authorities. It also offers a status update on Ph. Eur. monographs in the field of biotherapeutics.
EDQM perspective – case studies
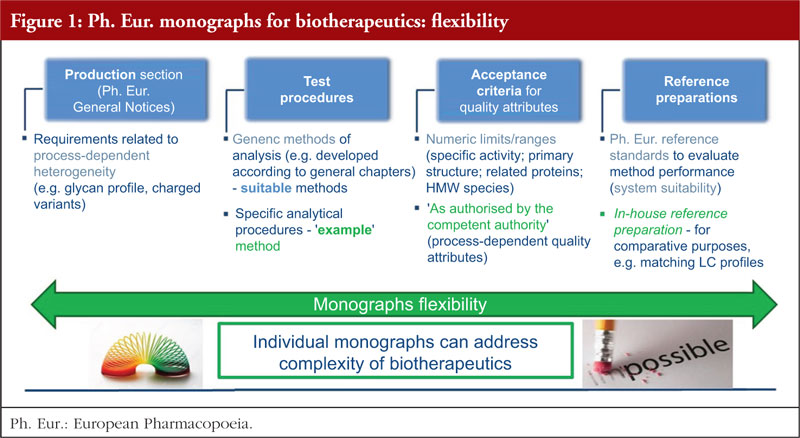
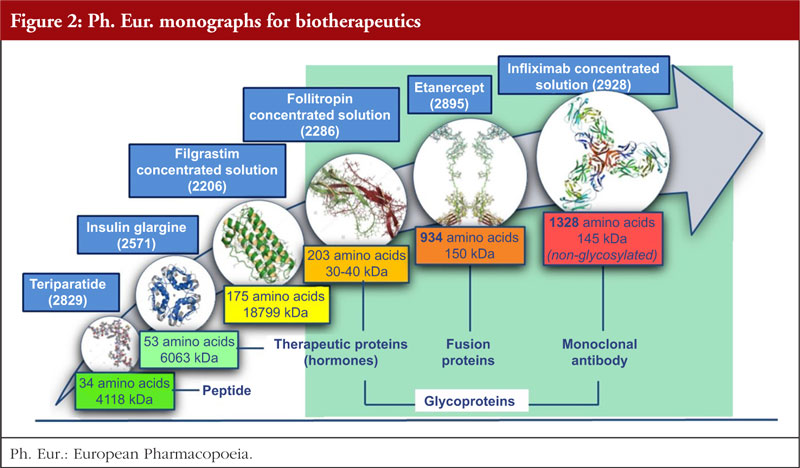
Previous discussions revealed that opposition to individual monographs was often a result of their misuse and misinterpretation, for example, their use to assess biosimilarity outside Europe. The European Directorate for the Quality of Medicines & HealthCare (EDQM) has taken action – including wide and regular communication in international conferences, providing a dedicated webpage on the EDQM website [4] and publishing scientific articles [2, 5] – to help ensure that the role played by monographs in defining quality standards for biotherapeutics is correctly understood. Another important milestone in this action plan has been the publication of a revised version of the Technical guide for the elaboration of monographs on synthetic peptides and recombinant DNA proteins [6] with the introduction of a new section on flexibility, see Figure 1 and section 3 for more details. The biotherapeutics covered by monographs of the Ph. Eur. range in size from a 30 amino acid polypeptide (teriparatide) to a 145 kDa glycoprotein (infliximab), see Figure 2. The contents of the monographs for these substances vary significantly, with monographs covering large, complex structures having built-in flexibility in addition to that already allowed by the Ph. Eur. The monograph elaboration or revision process relies on stakeholder participation; the approach is dynamic, collaborative and transparent, with the involvement of interested parties actively sought and encouraged from the early stages of the process. The consultation phase (publication in Pharmeuropa), during which manufacturers should perform testing to verify their substances against the new or revised standard, is of key importance. As compliance with the Ph. Eur. monograph is mandatory, interested parties must be able to resolve any issues before the implementation date of the text: working closely with the EDQM at an early stage and up to the public consultation phase is therefore strongly encouraged. This topic is further explained in section 4.
In sum, Ph. Eur. monographs provide a common framework for setting the quality and for standardisation of the medicinal products available on the market and under development and thus help ensure a better understanding of potential drift and evolution in product quality. This helps ensure that consistently high product quality is maintained.
Ph. Eur. monographs prescribe the use of reference standards, e.g. biological reference preparation (BRP) and chemical reference substance (CRS): these contribute to defining the quality requirements. The first Ph. Eur. BRP established by the EDQM for a monoclonal antibody, Infliximab BRP for cell-based assay calibration, was the result of a collaborative study involving laboratories from regulatory authorities, official medicine control laboratories and the pharmaceutical sector [7]. The study also shed light on critical parameters and possible contributors to assay variability and helped ensure that the level of method detail and assay conditions were suitably reflected in the monograph. Consequently, the Infliximab concentrated solution (2928) monograph [8] prescribes the use of a ‘suitable cell-based assay based on the inhibitory action of infliximab on the biological activity of TNF-alpha’. The specific assay used in the collaborative study measures the cytotoxic effect of antitumour necrosis factor-alpha (TNF-a) on WEHI-164 cells, and is provided as an example procedure (described in detail) in the monograph, using Infliximab BRP as a standard. It is important to note that Ph. Eur. BRPs are qualified for the use(s) described in Ph. Eur. monographs; they are established as a result of international collaborative studies coordinated through the Biological Standardisation Programme and the full reports are published in Pharmeuropa Bio & Scientific Notes (https://pharmeuropa.edqm.eu/app/BioSN/search/), thereby giving users access to important information on assay procedure, reagents, reference standards and products [9, 10]. There have been significant developments in Ph. Eur. reference standards established to support the physicochemical testing of recombinant glycoproteins in recent years. Reference standards for glycan analysis may serve two functions: 1) to support controlling the performance of a multi-step procedure, and thus serve the purpose of system suitability testing, described in monographs; 2) to assess compliance with both qualitative and quantitative acceptance criteria, therefore acting as a quality benchmark. These functions can be illustrated using two recently established Ph. Eur. CRSs, Infliximab CRS and Etanercept CRS, both of which have been used to support analytical procedure transfer and independent testing.
For the assessors present at the Strasbourg conference, among the challenges identified during monograph elaboration is the question of maturity: is a substance/subject sufficiently well developed for standardisation? A pragmatic approach to the issue would be appropriate: each case will be different and will need to be addressed based on experience. In their views, a monograph should not necessarily be ‘exhaustive’, but should aim at comprising a number of key quality attributes that are both critical and amenable to standardisation. The identification of appropriate, key quality attributes to be covered by the future standard is seen as a major challenge. The recently published monographs for complex recombinant proteins such as etanercept and infliximab demonstrate that this exercise is possible.
On a different topic, from an innovator standpoint, a biopharmaceutical manufacturer must be able to absorb a substantial and challenging workload following implementation of a Ph. Eur. monograph. The following question was raised: can a biotherapeutic product monograph sufficiently describe acceptable quality for market use and be a reliable predictive model of this acceptable quality? From an industry perspective, a monograph can only fulfil this role if it goes through multiple revision cycles, but the regulatory burden these generate would be challenging. In the view of industry representatives present at the Strasbourg conference, a forward-looking strategy should be developed at the level of the Ph. Eur. to move from a ‘product-specific’ toward a ‘modular adaptative’ approach involving class- and performance-based monographs and general texts. In the opinion of biosimilar manufacturers present at the Strasbourg conference, monographs should champion high quality products and analytical procedures but provide sufficient flexibility to address complexity of large biologicals and to facilitate the use of novel technologies.
The following sections describe the approaches taken by the EDQM to address these challenges.
The number of Ph. Eur. public standards for biotherapeutics has grown continuously over the last three decades and now covers a broad selection of substances ranging from peptide hormones, interferons and interleukins to growth factors, blood coagulation factors and monoclonal antibodies. In an increasingly evolving multi-product market, the development and evolution of monographs for highly complex biotherapeutics – where the process defines the product – has faced significant challenges. Unlike many other proteins, glycoproteins (including monoclonal antibodies) have complex structures and intrinsic heterogeneities; they display a large diversity of quality attributes, which put high demands on the techniques required for their analysis, far beyond traditional physicochemical procedures. These aspects make it impossible to fully standardise complex biotherapeutics and raise the fundamental question of how to reflect key quality attributes (QAs) and associated testing strategies, and how to establish suitable common expectations in a monograph.
Work on monographs for complex biotherapeutics has shown that additional flexibility (other than that defined in the Ph. Eur. General Notices) is needed to address the structural complexity and process-dependent product heterogeneity of these substances, the complexity and specifics of (often multi-step) procedures for analysis, as well as the potential diversity of the product resulting from different manufacturing processes. A key challenge is how to build this flexibility into a public standard, so that it still provides sufficiently prescriptive requirements (including tools to support analytical procedure control strategy and facilitate successful independent testing, and defined acceptance criteria for key QAs to enable standardisation of functionality), while remaining compatible with the development of follow-on versions.
As a result, the following elements of additional flexibility have been built into monographs for complex biotherapeutic proteins, pioneering a unique approach across pharmacopoeias:
In combination, these elements provide a means of enhancing monograph flexibility under well-defined conditions, an approach that addresses structural complexity of biotherapeutics and is compatible with development of biosimilars. This approach has been established based on extensive input from the Ph. Eur. experts and the EDQM and relies on scientific and experimental evidence generated through dedicated laboratory studies. Combined with close collaboration with industry parties, these factors have proved essential in finding the best way forward for public standard setting. This concept was also described in detail in the Technical guide for the elaboration of monographs on synthetic peptides and recombinant DNA proteins [6], which was revised in 2018 to take into account these developments.
At the Strasbourg conference, the huge progress in the way monographs address the glycosylation issue was acknowledged and the participants commended both the flexibility offered by the Ph. Eur. and the communication surrounding it. However, this approach was not created overnight – monographs on biotherapeutics are not new: the first texts were published nearly three decades ago and new texts continue to be developed, even for more complex molecules such as monoclonal antibodies. Development of the latest monographs for etanercept or infliximab illustrated the fact that there is an added value for certain standardisation activities subsequent to development of individual monographs: this ‘bottom-up approach’ has allowed the Ph. Eur. to base its approaches and policies on facts and put an end to earlier sterile theoretical debates about whether or not monographs can cover large molecules. Much was learned from these individual cases and it became apparent that monographs should not be expected to cover all quality attributes that are used to characterize the product and should focus on issues that can be standardised. This specific idea is the starting point for standardisation of a particular quality attribute in a multi-product setting. The potency assay is an illustrative example: the bioassay standardisation efforts and establishment of Ph. Eur. BRPs for etanercept and infliximab potency assays enhanced our understanding of the anti-TNF-a neutralization assay that could serve as a multi-product procedure for monoclonal antibodies, exercising the same function. Most importantly, it helped establish a robust assay procedure, with confirmed applicability and transferability, that became part of the infliximab monograph. On the one hand, both regulators and representatives of Official Medicines Control Laboratories considered this to be a very important feature: the monograph sets up the minimum requirements and allows alignment with current expectations. It is important to have a common public standard for all products and robust test procedures. However, on the other hand, some industry representatives claim that individual monographs are underutilized. The point was well-taken and the example described above shows that the knowledge gained in the elaboration of individual monographs can be the basis for future standardisation of general matters and, by extension, to explore flexible concepts of standardisation.
The same bottom-up approach for biological assays could be applied to classes other than TNF-a antagonists, of different complexities, e.g. ErbB/HER, CD20, vascular endothelial growth factor (VEGF) antagonists. As a first step, specific monoclonal antibodies of a given class are to be considered individually in view of their specific quality attributes related to the mechanism of action, e.g. Fc-effector functions; the outcome of such exercise would help better understand how to address specific challenges to bioassay standardisation within a class of monoclonal antibodies, and thus drive forward the development of class-based, general Ph. Eur. texts. Further reflection on the application of this model to the development of monographs for trastuzumab or rituximab, for example, is necessary.
Consultation of stakeholders
Ph. Eur. texts are public standards legally binding in the 39 states parties to the Convention on the Elaboration of a European Pharmacopoeia and applied in more than 120 countries worldwide.
They become mandatory at the implementation date, six months after their publication. Therefore, users have six months to take appropriate measures to assure compliance of the substances or products covered by the Ph. Eur. Seen from the outside, this period may seem to be short, but it should be noted that all the texts published in the Ph. Eur., whether monographs or general chapters, whether revised or newly elaborated, undergo publication consultation before their official publication and implementation. The forum used for consultation, Pharmeuropa, is freely available online (https://www.edqm.eu/en/How-to-register-for-Pharmeuropa-Online-1457.html). It is essential that the users of the Ph. Eur. examine closely the texts published in Pharmeuropa; they are also strongly encouraged to carry out experimental verification of the described analytical procedures on their own products, after which they might see the need to comment on the texts. Commenting is done either through their national pharmacopoeia authority if they belong to one of the 39 signatory countries to the Ph. Eur. Convention, or directly to the EDQM if they are commenting from outside Europe. The comments are carefully reviewed by the experts of the Ph. Eur., who then – when necessary – take appropriate action. It is important to note that comments submitted after official publication cannot be addressed. Or rather, they will be addressed in a subsequent round of revision.
In this context, the importance of the public consultation period must not be underestimated: the worst-case scenario would be for a user to discover that their product fails to meet all the requirements of the newly implemented text. It can also happen that a user will only perform a paper evaluation of the pharmacopoeia text during the public enquiry, comparing the elements of the new analytical procedure against the in-house procedure without trying out the new procedure experimentally.
During past events, notably at the Strasbourg conference, industry representatives commented that the main issue with biotherapeutic monographs was not the monographs themselves, but the timing at which comments were requested: for example, the commenting period might coincide with the final phase of a dossier submission, or because of the time needed to approach competent authorities, the slot for commenting on a monograph might be missed. The EDQM also received comments from stakeholders who had not yet received a marketing authorization. This grey area during which a product is still the subject of confidential discussions between industry and authorities prevented any contribution to pharmacopoeia efforts.
Implementation of Ph. Eur. monographs
Implementation of monographs has an impact on already approved products. During the Strasbourg conference, some industry representatives expressed concerns about the regulatory burden linked with the implementation of new or revised pharmacopoeia texts, especially when a text is revised repeatedly over the years. The Ph. Eur. is keen to reduce the number of revisions rounds for a given text, but this is only possible if contributions from all stakeholders are submitted within the given timelines (for example, public enquiries). If marketing authorization holders reacted too late, then yes, multiple revisions are inevitable.
As regards regulatory updates linked with revisions of Ph. Eur. texts, the EU has introduced the following statement in its guideline on how to deal with variations: ‘There is no need to notify the competent authorities of an updated monograph of the European Pharmacopoeia or a national pharmacopoeia of a Member State in the case that compliance with the updated monograph is implemented within six months of its publication and reference is made to the ‘current edition’ in the dossier of an authorised medicinal product’ [11]. This implies that, in the event that the user does not need to notify the authority, they must ensure compliance with the text. To implement a new or revised text, the user may choose between two options: 1) to implement the text as published; 2) to keep their in-house procedure; in the latter case, to assure compliance with the Ph. Eur., the method will be considered as an alternative procedure, as defined in the General Notices and the user would have to be able to demonstrate, to the satisfaction of the regulatory authority, that the substance or product would comply with the Ph. Eur. if tested. This will most probably require experimental testing and undertaking the necessary regulatory steps with the competent authorities for the respective changes. Industry representatives reported that such changes might be easy to handle in Europe, but 3–5 years may be required to complete a change in test procedure worldwide – a huge burden. At the Strasbourg conference, regulators confirmed that there is regulatory obligation to update the dossier: ‘your analytics have to be up to date’. However, the difficulty of implementation worldwide was unanimously recognized. Reference was made to ICH Q12, which strives for a globally harmonized approach to technical and regulatory considerations. A plea was made for a regulatory pathway that would facilitate the process for biotherapeutic monograph implementation. For a biosimilar manufacturer, life-cycle management is particularly difficult since the appearance of a new standard may call for a change in the biosimilar development.
One of the main recommendations that emerged from the Tallinn conference was to enhance communication with stakeholders on the activities of the Ph. Eur. Commission in the field of biotherapeutics. Enhanced communication was also viewed necessary to emphasize that, although compliance with the Ph. Eur. monographs is required, it must not be considered as sufficient for proving biosimilarity. Moreover, further feedback from stakeholders on the concept of additional flexibility in monographs for biotherapeutics as proposed in the Infliximab concentrated solution (2928) monograph [8], published at that time for consultation in Pharmeuropa, was to be gathered.
To address these requests, two articles were published shortly after the Tallinn conference, one on the role of Ph. Eur. monographs in setting quality standards for biotherapeutics [2] and another on the process of elaboration of monographs for biotherapeutics using substances and medicinal products from a single source, together with the lessons learnt during this exercise [5]. In addition, in early 2017, the EDQM and the European Medicines Agency (EMA) organized a joint workshop on biosimilars [12]. It was broadcast live to increase its outreach and was highly appreciated by the participants. Two further Ph. Eur. training sessions on biologicals with modules and workshops dedicated to biotherapeutics were also organized in 2017 and 2020 [13, 14]. Finally, a page devoted exclusively to biotherapeutics was created on the EDQM website [4]. In addition to procedural explanations, it gathers all the related news, articles and information on events as well as the regularly updated Ph. Eur. biotherapeutics monograph portfolio.
The participants at the Strasbourg conference were unanimous: the EDQM had done a good job documenting the challenges and answering them. The actions devoted to communication with its stakeholders had contributed to focusing the debate on real, concrete issues. The EDQM has created a safe space for Ph. Eur. experts to debate and discuss different views. The examples given in the presentations shared at the Strasbourg conference support these findings [3].
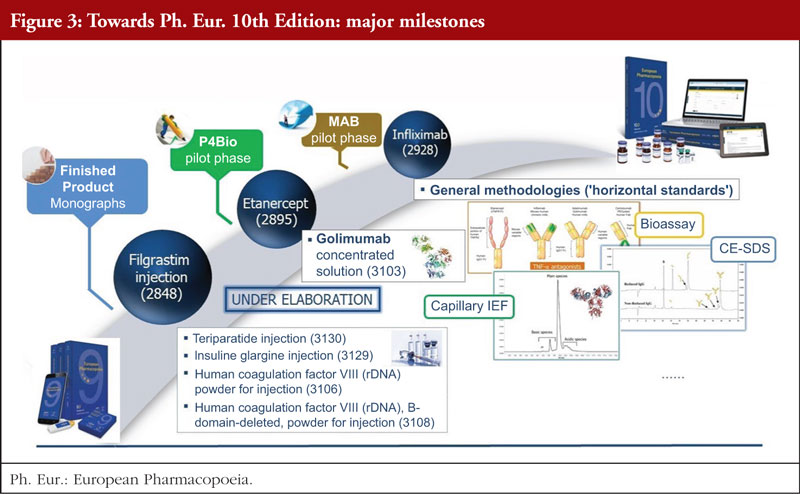
The work of the recent years and its many positive outcomes have demonstrated that elaboration of monographs on complex biotherapeutics is not only possible but also very useful. In 2016, with the adoption of Etanercept (2895) and Human coagulation factor IX (rDNA) powder for solution for injection (2994), the Ph. Eur. Commission concluded the so-called P4-Bio pilot phase, thus establishing a routine framework for setting public standards for biotherapeutics using substances and medicinal products from a single source and working in close collaboration with an innovator company. A year later, the adoption of the first monograph on a monoclonal antibody, Infliximab concentrated solution (2928), marked a significant milestone in setting standards for complex biotherapeutics.
Recently, Erythropoietin concentrated solution (1316), the first monograph on a complex glycosylated molecule, first published in Supplement 1999 to the 3rd Edition of the Ph. Eur. in 1999, was revised following this approach (Supplement 10.4).
In addition to regular revision of monographs on biotherapeutics, e.g. Insulin glargine (2571) in Supplement 9.5 or Erythropoietin concentrated solution (1316) in Supplement 9.6, a new monograph on Filgrastim injection (2848), elaborated using the multisource procedure, was published in Supplement 9.8. The commitment of the Ph. Eur. in the field of biotherapeutics is also reflected in the number of items recently added to the work programme. Elaboration of several new monographs via the single source procedure (Golimumab concentrated solution (3103) and Ustekinumab (3165)) as well as via the multisource procedure (Insulin glargine injection (3129), Teriparatide injection (3130), Human coagulation factor VIII (rDNA) powder for injection (3106) and Human coagulation factor VIII (rDNA), B-domain-deleted, powder for injection (3108)) is currently ongoing. Moreover, another individual monograph on a monoclonal antibody, Adalimumab concentrated solution (3147), is being prepared within the framework of the ongoing monoclonal antibodies (mAb) pilot phase. Finally, the mAb WP is also working on the development of horizontal standards applicable to monoclonal antibodies. Three general chapters on methodologies for potency determination for anti-TNF-a antagonists
(Cell-based assays for potency determination of TNF-alpha antagonists (2.7.26)) and on physicochemical testing applicable to various monoclonal antibodies (Capillary isoelectric focusing for recombinant therapeutic monoclonal antibodies (2.5.44), Size-exclusion chromatography for recombinant therapeutic monoclonal antibodies (2.5.43)) are currently in preparation, see Figure 3.
The experts of the Ph. Eur. present at the Strasbourg conference considered that the monograph development exercise enhances product understanding. Monographs based on a single product bring a lot of insight; a monograph based on many products helps build a bigger picture, a robust standard and thus a foundation for the rationalization of test procedures in a multi-product setting. Overall, it is beneficial for the field to work on and add to this understanding together. The resulting standards are more robust and of greatest benefit to patients. The Ph. Eur. portfolio of quality requirements for biotherapeutics will continue to rely on the experience gathered from product-specific cases and use it as a basis for driving general, transversal matters. A number of studies are underway, and the Ph. Eur. looks forward to making further progress in the field in the years to come.
Competing interests: Mihaela Buda, Olga Kolaj-Robin and Emmanuelle Charton are employees of the EDMQ, Council of Europe. All authors declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
Provenance and peer review: Not commissioned; externally peer reviewed.
Mihaela Buda, PhD
Olga Kolaj-Robin, PhD
Emmanuelle Charton, PhD
European Pharmacopoeia Department, European Directorate for the Quality of Medicines & HealthCare (EDQM), Council of Europe, Strasbourg, France
References
1. Council of Europe. European Directorate for the Quality of Medicines & HealthCare. The European Pharmacopoeia (Ph. Eur.). Background Mission [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://www.edqm.eu/en/European-Pharmacopoeia-Background-Mission
2. Charton E. The role of European Pharmacopoeia monographs in setting quality standards for biotherapeutic products. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(4):174-9. doi: 10.5639/gabij.2016.0504.045
3. Council of Europe. European Directorate for the Quality of Medicines & HealthCare, EDQM & European Pharmacopoeia: State-of-the-art science for tomorrow’s medicines, June 2019, Strasbourg, France. Workshop on biotherapeutics [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://www.edqm.eu/sites/default/files/medias/fichiers/Events/edqm_european_pharmacopoeia_state-of-the-art_science_for_tomorrows_medicines_-_workshop_on_biotherapeutics.pdf
4. Council of Europe. European Directorate for the Quality of Medicines & HealthCare. Biotherapeutics [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://www.edqm.eu/en/biotherapeutics
5. Buda M, Wicks S, Charton E. Elaborating European Pharmacopoeia monographs for biotherapeutic proteins using substances from a single source. Pharmeur Bio Sci Notes. 2016:129-34.
6. Council of Europe. European Directorate for the Quality of Medicines & HealthCare. Technical guide for the elaboration of monographs on synthetic peptides and recombinant DNA proteins. 2018 [homepage on the Internet]. [cited 2022 Jan 26]. Available from https://www.edqm.eu/sites/default/files/medias/fichiers/European_Pharmacopoeia/Find_information_on/Technical_Guides/guide_ph_eur_synthetic_peptides_and_rdna_proteins_2018.pdf
7. Metcalfe C, Dougall T, Bird C, Rigsby P, Behr-Gross M-E, Wadhwa M, et al. The first World Health Organization International Standard for infliximab products: a step towards maintaining harmonized biological activity. MAbs. 2019;11(1):13-25.
8. Infliximab concentrated solution, monograph 2928. Ph. Eur. 10th Edition. Strasbourg, France: Council of Europe 2019.
9. Wadhwa M, Rigsby P, Behr-Gross M-E. Collaborative study for the establishment of infliximab biological reference preparation batch 1. Pharmeuropa Bio Sci Notes. 2020;2020:49-52.
10. Wadhwa M, Rigsby P, Behr-Gross M-E. Collaborative study for the establishment of etanercept biological reference preparation batch 1. Pharmeuropa Bio Sci Notes. 2020;2020:203-5.
11. Official Journal of the European Union. C 223. Guidelines on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products and on the documentation to be submitted pursuant to those procedures [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C:2013:223:FULL:EN:PDF
12. Council of Europe. European Directorate for the Quality of Medicines & HealthCare. Joint EDQM-EMA Session on Biosimilars. February 2017, Strasbourg, France [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://www.edqm.eu/en/european-pharmacopoeia-training-resources#Biologicals
13. Council of Europe. European Directorate for the Quality of Medicines & HealthCare. European Pharmacopoeia training session on Biologicals. February 2017, Strasbourg, France [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://www.edqm.eu/en/european-pharmacopoeia-training-resources#Biologicals
14. Council of Europe, European Directorate for the Quality of Medicines & HealthCare. EDQM Training Session – Biologicals. February 2020, Strasbourg, France. [homepage on the Internet]. [cited 2022 Jan 26]. Available from: https://www.edqm.eu/en/european-pharmacopoeia-training-resources#Biologicals
|
Author for correspondence: Emmanuelle Charton, PhD, European Pharmacopoeia Department, European Directorate for the Quality of Medicines & HealthCare (EDQM), Council of Europe, 7 allée Kastner, CS 30026, FR-67081 Strasbourg, France |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2022 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/biotherapeutic-products-in-the-european-pharmacopoeia-have-all-challenges-been-tackled.html
Copyright ©2024 GaBI Journal unless otherwise noted.