|
Abstract: |
Submitted: 30 November 2011; Revised manuscript received: 2 March 2012; Accepted: 5 March 2012 Published online first: 2 May 2012
In the anticipated upcoming renewal of the Best Pharmaceuticals for Children Act (BPCA) and the Pediatric Research Equity Act (PREA), four changes will markedly improve these programmes. These changes concern expanded attention paid to neonatal studies, support for the off-patent programme, timing and submission of paediatric plans, and the permanence of the legislation.
Both BPCA and PREA have failed to encourage meaningful trials for American neonates, see Figure 1. For example, of the first 121 products studied under the exclusivity provisions of BPCA, only five trials enrolled 198 patients in studies conducted in American neonatal intensive care units, although several well-conducted trials enrolled HIV-exposed newborns globally. Of these five trials, the only drugs studied that were routinely used in neonates were ranitidine (the 16th most prescribed medicine in newborns) and linezolid (in the only trial that included premature infants). The dearth of neonatal clinical pharmacology data is due generally to issues involving the technical feasibility of conducting trials in very sick young infants, which often lead to the issuance of a waiver obviating the need for a sponsor to conduct such studies.
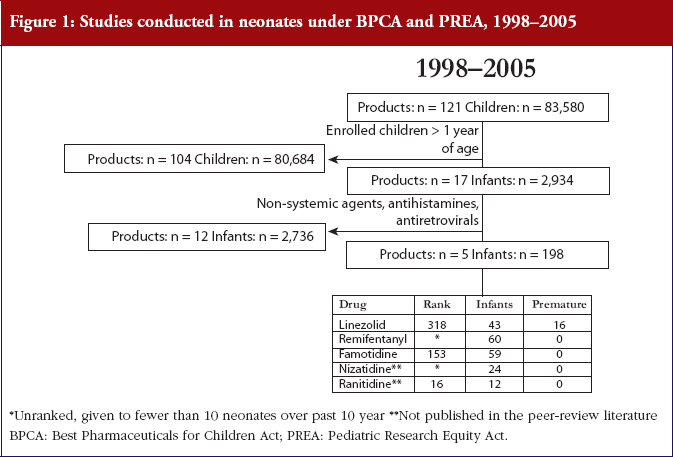
This lack of trials in neonates can be solved by mandating that FDA requires, at a minimum, exposure (pharmacokinetic), response (pharmacodynamic), and safety trials in children of all ages, even when a pivotal trial is not possible. Such a mandate will at least produce data that will better characterise the clinical pharmacology of a drug in neonates—information that can be translated into critical knowledge, such as information on exposure-response relationships and safety, that can be used by clinicians.
The general failure of the pharmaceutical industry to conduct critical trials in neonates is well described in the recent Institute of Medicine report titled ‘Safe and Effective Medicines for Children: Pediatric Studies Conducted Under the Best Pharmaceuticals for Children Act and the Pediatric Research Equity Act’ [1]. This deficiency and the information gap that it produces must be addressed so that neonatal therapeutics can advance. While part of the solution to this problem can be achieved through FDA mandate, especially for new drugs and those with existent patent life, the remaining challenge must be met by keeping a viable mechanism for the study of off-patent drugs firmly in place.
As outlined by Professor Walson [2], governmental funding for the study of off-patent therapeutics has never been robust. Although the effort of the National Institute of Child Health and Human Development in this regard has been substantial, annual funding for the programme has been less than what is typically required by a pharmaceutical company to conduct one large pivotal trial for labelling. If the current rate of National Institutes of Health (NIH) support and its approach to funding for the study of off-patent drugs is maintained, only one or two molecules will be able to be investigated per year, severely crippling efforts to provide meaningful safety and efficacy data for off-patent drugs. This situation stands in stark contrast to that seen in the private sector represented by the pharmaceutical industry. When a company is successful in addressing the requirements of a ‘written request’ from FDA, the reward of extended marketing exclusivity can produce hundreds of millions of dollars in incentives, some of which may not be used to leverage the further study of drugs in children.
Linking the ‘exclusivity programme’ to the off-patent programme can solve this problem. While the structure of the ‘exclusivity programme’ is fundamentally sound, a small fraction of these exclusivity dollars could be diverted to the NIH in the form of a ‘processing fee’. Once all of the trials for exclusivity have been conducted, the data reviewed, and decisions made, companies could rebate back a small portion of the profit emanating from the awarding of exclusivity, e.g. US$10,000,000, to support off-patent investigation. With approximately 10–15 molecules being granted exclusivity on an annual basis, the aforementioned approach would markedly contribute to the public good by: 1) enabling a renewable resource that provides dedicated support for two programmes charged with performing research critical to the safe and effective use of many drugs in paediatric patients and; 2) not requiring the NIH to carve out budget dollars from programmes dedicated to discovery and the fostering of new science and technology in order to meet the mandates of BPCA or PREA. Without question, such an approach would require extension of the already established partnership between the pharmaceutical industry, NIH, and FDA, all of whom want the most well-studied drugs available for paediatric use. The risk to industry with this proposal is small because the fee comes only if exclusivity is granted.
In the meantime, the US Congress can strengthen the off-patent programme by re-authorising the BPCA NIH programme at current levels and supporting the appropriation of funds to support this much-needed research.
Paediatric use is an afterthought in the current American drug development system. Typically, plans are put together at the very end of drug development. Late development results in products licensed for years without paediatric exposure or safety information.
Lessons learned from the EU experience can provide a solution to this problem. EMA requires submission of a paediatric plan at the end of phase I. One drawback to the European approach is that many molecules that complete phase I do not move beyond phase II. Putting together a paediatric plan for a large number of molecules that will not be licensed is not an optimal use of industry or regulatory resources. While requiring submission of a paediatric plan at the end of phase I is better than the current American system, a middle path is optimal. Paediatric plans, and testing, should begin at the end of phase II.
Despite its imperfections, paediatric legislation has proven remarkably successful. The programmes have finally resulted in clinical trials that are vital to children’s public health. The most important component of the forthcoming paediatric legislation is that the programmes should become permanent. We do not require that the principles of good drug development be renewed every five years for adults; the same should be true for children.
Competing interests: None.
Provenance and peer review: Commissioned; internally peer reviewed.
Professor Gregory L Kearns, PharmD, PhD, Marion Merrell Dow/Missouri Chair of Medical Research; Professor of Pediatrics and Pharmacology; University of Missouri-Kansas City; Chairman of the Department of Medical Research; Associate Chairman of the Department of Pediatrics; Director of the Pediatric Pharmacology Research Unit; Children’s Mercy Hospitals and Clinics; 2401 Gillham Road, Kansas City, MO 64108, USA
References
|
Author for correspondence: Professor Daniel Kelly Benjamin Jr, MD, MPH, PhD, Professor with Tenure, Pediatric Infectious Diseases, Duke University, Faculty Associate Director, Duke Clinical Research Institute; Chair, Pediatric Trials Network; 2400 Pratt Street, Durham, NC 27705, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
Generic and therapeutic orphans
Source URL: https://gabi-journal.net/equal-protection-under-the-law-children-and-the-best-pharmaceuticals-for-children-act.html
Copyright ©2024 GaBI Journal unless otherwise noted.