|
Introduction: Similar biotherapeutic products (SBPs or biosimilars) include a rapidly expanding number of therapeutic products worldwide. However, agreement about the best practices for their regulation are lacking in many countries including in the Middle East and North Africa (MENA). |
Submitted: 12 September 2015; Revised: 10 October 2015; Accepted: 12 October 2015; Published online first: 26 October 2015
After pharmaceutical products lose their patent protection, follow-on medications have the potential to provide lower cost products with equal or even improved efficacy and/or safety. However, the actual savings generated by the marketing of follow-on products differs greatly depending on country specific differences in how they are regulated, marketed and used. The costs of bringing follow-on drug products to market, and therefore, how much companies can afford to discount their price, are largely determined by how much it costs to provide regulators with acceptable proof of chemical quality as well as clinically equivalent efficacy and safety.
For small-molecule drug products that can be relatively easily characterized and synthesized, and it is relatively inexpensive to obtain the proof of similar composition and bioavailability that allows regulators to conclude that they have equal therapeutic and toxic effects (the generics paradigm). This is not true for biological products. It is much more difficult to demonstrate the similarity of quality, efficacy and safety of follow-on biological products, because they are difficult or even impossible to completely characterize by their physio chemical properties and even minor changes in their manufacturing processes can result in clinically meaningful differences in efficacy or safety. For these reasons, such biological products, similar biotherapeutic products (SBPs), or biosimilars; are often regulated using a much more complicated similarity approach [1].
There are a growing number of follow-on SBPs either being used, or proposed for use, in MENA (Middle East, North Africa) countries. However, there are large differences in how SBPs are regulated in different MENA countries and also a lack of consensus as to the best practices for their evaluation, approval, clinical use and post-marketing surveillance.
As part of its educational commitment, the Generics and Biosimilars Initiative (GaBI) has conducted workshops to try to identify both such best practices as well as educational needs of various stakeholders to increase the patients’ access to biological products with adequate quality, safety and clinical responses. The workshop described was the first of such workshops to be held in the MENA region.
On 1 September 2015, GaBI held the First MENA Educational Workshop on Similar Biotherapeutic Products/Biosimilars in Dubai, UAE. As with prior workshops [2], the objective of the workshop was to review and explain the importance of biological product structure–function relationships in the regulatory assessment of SBPs, including best practice methods for their evaluation, clinical testing and post- marketing surveillance. After a series of didactic presentations, all workshop attendees participated in small breakout groups that evaluated fictitious data from the preclinical and clinical evaluation of either a follow-on erythropoietin or an anti-tumour necrosis factor (anti-TNF) monoclonal antibody.
Speakers included experts from academia, regulatory agencies and a biological manufacturer. Participants included regulators, academicians and practicing physicians from MENA countries as well as speakers from Germany, Saudi Arabia, Switzerland, Thailand, UAE, the UK and the US.
The results of these discussions were summarized verbally and in writing by moderators who were experts in the regulatory and/or clinical use of SBPs in MENA countries (Egypt, Jordan, Kingdom of Bahrain, Kuwait, Lebanon, Oman, Saudi Arabia and UAE).
The structure of the workshop was very similar to that reported for a previous workshop held in Mexico [2]. The Workshop Co-Chair and GaBI Journal Editorin- Chief, Professor Philip D Walson, welcomed the participants and presented the workshop goals and objectives.
Assistant Undersecretary Dr Amin Hussain Al Almiri of the UAE Ministry of Health gave an official welcome and described the current state of the regulatory approval of SBPs in the UAE.
Professor Walson gave an overview of the differences between small-molecule and biological products and the general approaches used to evaluate follow-on generics versus SBPs.
The Workshop Chair and GaBI Journal Deputy Editor-in-Chief, Dr Robin Thorpe, who is the former head of the Biotherapeutics Group at UK’s National Institute for Biological Standards and Control (NIBSC) presented an overview of ‘the global view of biologicals, biosimilars and nonoriginator (non-comparable) biologicals’ in which he clarified the differences between both follow-on biologicals and true biosimilars/SBPs. He also explained the differences in definitions, as well as the distinct appropriate and inappropriate uses of, Reference Products and International Standards/Reference Reagents used in biosimilar product development and regulation.
Dr Ivana Knezevic from the World Health Organization (WHO), Geneva, Switzerland, discussed both WHO standards for the evaluation of biotherapeutic products (including biosimilars) as well as many of WHO initiatives directed at developing best practices for the regulation approval, use and surveillance of SBPs throughout the world.
Dr Sundar Ramanan from Amgen Inc, USA, gave a review of the complexities of the relationships between structure and function of biological products, including biosimilars, and the various quality attributes that have been used to examine the complex relationships between structure and function during development of such biological products. He illustrated the critical role of structure–function in biosimilar development using some real-life monoclonal antibody examples.
Dr Meenu Wadhwa from UK’s NIBSC described how appropriate and informative immunogenicity testing of biotherapeutic products is done using carefully planned prospective studies in a suitable indication, a well-considered strategy and a panel of appropriately validated (or ‘fit for purpose’) assays for antibody detection and characterization in clinical samples.
Dr Ali M Al Homaidan from Saudi Food and Drug Authority (Saudi FDA) described the processes used by Saudi FDA to evaluate, approve and monitor SBPs as well as the current state of SBP marketing and use.
Mr Morakot Papassiripan from the Bureau of Drug Control of Food and Drug Administration (Thai FDA), Mini stry of Public Health, Thailand, described Thai FDA’s approach to the approval and post-marketing surveillance and relabelling of SBPs. He also gave an update on the incidence of pure red cell aplasia (PRCA) in Thailand and described attempts by Thai FDA to decrease the subcutaneous use of the 16 different follow-on erythropoietin products currently available in Thailand (not all of which qualify as biosimilars). He also discussed the effects of patient needs and physician practices, as well as the limited staff and resources available at Thai FDA, in addition to the lack of a mechanism to revoke marketing authorization on the success of these efforts.
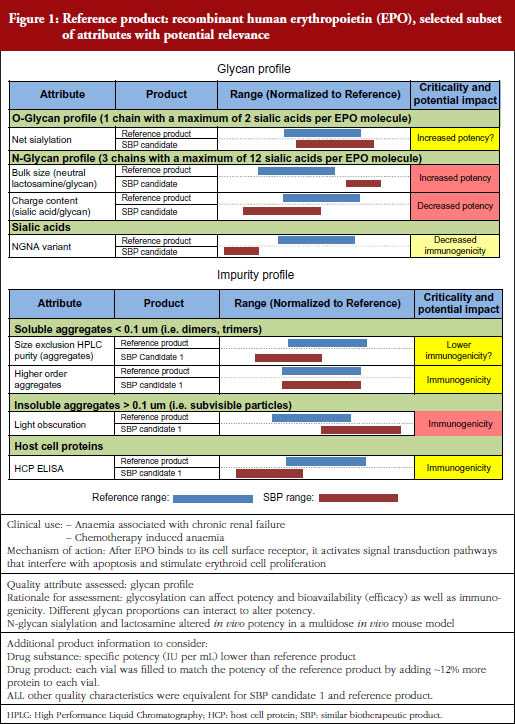
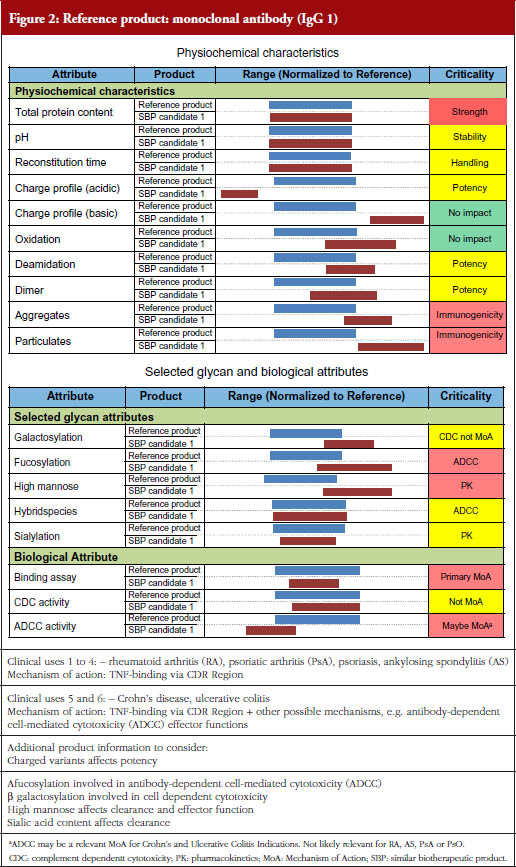
After these didactic presentations (available at the GaBI website: http://gabi-journal-net.server1.ech.be/about-gabi/current-activities), Professor Walson explained the format and objectives of the two case study discussions, followed by Dr Sundar Ramanan who presented in detail the data of the two fictional cases of biological products to be evaluated in the breakout sessions. The two fictional case studies – one an erythropoietin and the other an anti-TNF monoclonal antibody – contained comparisons to data from their respective originator proteins. The specific data presented, the putative effects of each characteristic on the performance of the biological, and the questions participants were asked to answer are presented in Figures 1 and 2.
Participants were divided into four groups. Each of the two biologicals was discussed by two different groups headed by experts from MENA countries, see list in Table 1. Results of the discussions were summarized and then presented by the moderators at the workshop for discussion.
Participants were provided time, and encouraged, to question the speakers during and after the presentations as well as during breaks.
There were differences of opinion expressed by both individuals as well as by the groups as to the acceptability of both the two fictitious follow-on biologicals described, see Figures 1 and 2, assuming that both products had failed to show any statistically or clinically important differences in efficacy or safety in a phase III human trial.
A brief overview of the responses of the four breakout groups, see Table 1 for listing of the group division and moderators, to some specific questions is provided below (detailed summaries of the responses are available upon request).
Do the data of the SBP candidate EPO qualify for biosimilarity with the reference product from a quality perspective with respect to efficacy?
Some but not all members of Group 1 felt that the differences in lactosamine and sialic acid content had potential to impact the efficacy of the product but stated that it was not possible to be sure without additional studies. Some felt that Kd calculations (based on in vitro binding studies) would be requested while others felt that additional studies would not be necessary since the phase III study failed to show nonequivalence. There was lack of agreement as to the impact of the differences on potency in terms of additional studies that might be requested before approval in order to deal with residual uncertainty. Some felt that a 20% difference in potency was unacceptable because biosimilars require that similar doses be used. Others felt that the differences might require the sponsor to develop and test a different formulation.
Group 2 felt that it was not possible to make an assessment of biosimilarity based on the limited data presented. They questioned whether it was possible to predict that the observed differences in post-translational modifications would have minimal impact to in vivo potency and pharmacokinetics. They also questioned whether it was possible to predict the impact of two offsetting differences in glycosylation.
Do the data of the SBP candidate EPO qualify for biosimilarity with the reference product from a quality perspective with respect to safety/immunogenicity?
Group 1 differed in their assessment of the ability of the data presented to predict similar safety/immunogenicity. The specificity of the test data presented was questioned by some who felt that additional data, e.g. from Surface Plasma Resonance testing and antibody measurements, were needed.
Group 2 felt that the observed post-translational differences could potentially impact in vivo potency and pharmacokinetics but that it was possible to predict this impact based on only the non- clinical in vivo and in vitro study data provided. They felt that it was not possible to decide on biosimilarity based on the data provided, and that additional in vivo studies in animals would be required before approval.
How could ‘residual uncertainty’ be addressed in the preclinical and/or clinical studies with respect to potency?
Different approaches were suggested by Group 1 members as to how residual uncertainty concerning efficacy should be addressed but this group felt that differences in haemoglobin responses could be used to look for efficacy differences.
Group 2 felt that additional preclinical in vivo animal studies were required.
How could ‘residual uncertainty’ with respect to immunogenicity be addressed in preclinical and/or clinical studies? Would it be possible to study rule out a 10-fold or 100-fold difference in immunogenicity based on these data?
Based on the rarity of clinically important antibodies and even rarer incidence of PRCA Group 1 agreed that a 10-fold or even a 100-fold difference in immunogenicity could not be identified by preclinical or clinical studies. They felt that such differences could only be identified through a large post- marketing pharmacovigilance programme.
Group 2 agreed that it would be difficult to detect a 10-fold increase but felt that it might be possible to detect a 100-fold increase. They suggested that to increase the chances of detecting such differences a specific protocol must be applied to all clinical study patients as well as patients in postmarketing EPO studies who show a lack of response. This protocol should include both ruling out all causes other than PRCA and testing for neutralizing antibodies.
Do the data of the SBP candidate monoclonal antibody qualify for biosimilarity with the reference product from a quality perspective?
Some members of Group 3 felt that the data provided justified giving conditional approval to the product if there was a commitment to perform and submit the results of a one-year post-marketing surveillance programme. Others felt that the differences, especially for charge, aggregates and particulates, were too great to allow for approval as an SBP.
Group 4 also felt that the differences were too great to approve the product as a biosimilar, especially with respect to aggregates and particulates. They also noted the potential for the differences in charge to alter potency, but felt that these concerns were lessened by the fact that the TNF binding tests showed equivalency.
What steps would you recommend to remediate the differences?
Group 3 felt that in order to decide they needed to know what type of antibodydependent cell-mediated cytotoxicity (ADCC) testing had been used to assess the products, but that additional confirmatory bioassays as well as a detailed risk management plan were needed for approval.
Group 4 felt that the binding/potency uncertainty needed to be addressed before performing an additional clinical trial in a sensitive indication. They felt that the causes of the change in glycosylation pattern would require additional animal testing to confirm the ADCC results and questioned, based on the ADCC results, whether a clinical trial of this product in a non-inflammatory bowel disease (IBD) population could be justified.
How could ‘residual uncertainty‘ be addressed in the preclinical and/or clinical studies?
Additional steps recommended by Group 3 included detailed stability studies (to establish when the aggregates form), requests for information on what steps had been taken to improve the quality of the product, additional pharmacokinetics data, and detailed safety data from their clinical efficacy study, especially for patients with ankylosing spondylitis.
Group 4 stated that fixing the aggregates and particulates issue was a mandatory quality step before even contemplating a trial in a sensitive indication but that fixing the differences in charges was not as much a concern. They also noted that while the effect of glycan and biological attributes differences (ADCC) may not be remediated as easily as the quality issue, this would limit only the extrapolation to other indications and not prevent use in the single indication already studied.
Would you recommend extrapolation of approval to all indications?
All members of Group 3 felt that extrapolation was not possible without additional data showing a similar mechanism of action using ADCC as part of the preclinical assessment as well as a clinical study that included at least some patients with IBD indications.
Group 4 noted that neither approval for IBD alone nor consideration of extrapolation of other indications was justified until all quality issues were addressed. If such quality issues were adequately addressed they felt that extrapolation could be done but only for psoriatic arthritis and psoriasis.
While there was some areas of general agreement, there were both individual and group differences of opinion as to how the two products should/would be handled. Some participants felt that the information/data provided was adequate to approve the products but others felt it was incomplete, and therefore that additional studies were needed before the products could be approved.
The lack of consensus as well as the questions and concerns raised suggested that there is a need for clear regulatory pathways to be developed and that in order to develop such pathways, basic scientific and clinical data are needed, as well as training and education of regulators who implement these pathways. It was not clear what methods would be the most effective to identify pathways that would be suitable in MENA countries. However, during the presentations all participants were given links to a number of applicable documents, such as from WHO, European Medicines Agency and US Food and Drug Administration, and that publishing a report of this workshop will lead to greater local or international regulatory cooperation and more effective approval, use and monitoring of these products.
The GaBI Journal will continue to focus on SBPs in future publications and educational workshops both in MENA and globally.
A growing number of follow-on biotherapeutic products are either being evaluated by regulators for use or are already being used in MENA countries. Approval of such products is difficult because even minor changes in production methods may have major but unpredictable impacts on their therapeutic and toxicological profiles when used in patients. In addition, products can have varying performance characteristics in a number of currently available preclinical testing methods but it is not always clear how important these differences are in predicting clinical performance. Financial limitations on public health costs and the need to provide patients with access to biologicals demand that effective methods be found to make SBPs available in MENA countries and worldwide.
The many challenges to the rational, effective approval of SBPs may explain the differences in opinions and approaches among MENA country stakeholders who attended this workshop. In addition, there appear to be gaps in the understanding of the properties of and the preclinical testing of these agents, especially amongst practitioners, that need to be addressed before harmonized, uniform approaches to their approval and use can be developed. These gaps might be at least partially addressed by educational programmes such as the workshop described herein as well as by current WHO initiatives.
Speakers
Assistant Undersecretary Amin Hussain Al Amiri, PhD, UAE
Ali M Al Homaidan, PhD, Saudi Arabia
Ivana Knezevic, MD, PhD, Switzerland
Morakot Papassiripan, MSc, Thailand
Sundar Ramanan, PhD, USA
Robin Thorpe, PhD, FRCPath, UK (Chair)
Meenu Wadhwa, PhD, UK
Professor Philip D Walson, MD, USA/Germany (Co-Chair)
Moderators
Associate Professor Ahmed Al-Jedai, PharmD, MBA, BCPS, FCCP, Saudi Arabia
Professor Ali K Abu-Alfa, MD, FASN, Lebanon
Professor Aws Alshamsan, PhD, Saudi Arabia
Professor Mazen Kurdi, PhD, Lebanon
Professor Mohammad Ghnaimat, MD, FRCP, FACP, Jordan
The Generics and Biosimilars Initiative (GaBI) wishes to thank Assistant Undersecretary Amin Hussein Al Amiri, PhD, UAE Ministry of Health, and his colleagues (specifically Dr Rasha Sayed Salama) for their strong support through the offering of advice and information during the preparation of this educational workshop.
The authors would like to acknowledge the help of all the workshop speaker faculty and participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators: Associate Professor Ahmed Al-Jedai, Professor Ali K Abu-Alfa, Professor Aws Alshamsan, Professor Mazen Kurdi and Professor Mohammad Ghnaimat, in facilitating meaningful discussion during the parallel case study working session, and presented the discussion findings at the workshop.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; internally peer reviewed.
Robin Thorpe, PhD, FRCPath
Deputy Editor-in-Chief, GaBI Journal
References
1. Alvarez AA, Mysler E, Ruiz de Castilla EM, Flores-Murrieta FJ, Hughes J, Azevedo VF. Recommendations for the regulation of biosimilars and their implementation in Latin American. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):143-8. doi:10.5639/gabij.2014.0303.032
2. Walson PD, Thorpe R. First Latin American educational workshop on similar biotherapeutic products, Mexico City, Mexico, 20 January 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):143-8. doi:10.5639/gabij.2015.0403.032
|
Author for correspondence: Professor Philip D Walson, MD, Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/first-mena-educational-workshop-on-regulation-and-approval-of-similar-biotherapeutic-products-biosimilars-dubai-united-arab-emirates-1-september-2015.html
Copyright ©2024 GaBI Journal unless otherwise noted.

2 Responses to First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars, Dubai, United Arab Emirates, 1 September 2015