|
Abstract: |
Submitted: 21 November 2012; Revised: 11 February 2013; Accepted: 13 February 2012; Published online first: 19 February 2013
The population of Iran is now over 74 million. The country’s gross domestic product (GDP) per capita in 2011 was reported to be over US$12,000 and the country spends about 6% of its GDP on health. In the past three decades the government of Iran has devoted substantial resources for improving the national health system. Through implementation of many long-term initiatives Iran has achieved substantial success in extending health care to every corner of the country and most of the major healthcare indicators have significantly improved during the past decades [1–2]. The availability of World Health Organization (WHO) designated essential medicines in Iranian health centre pharmacies is reported to be over 92% [3]. The healthcare system in Iran was established in response to its primary healthcare needs. However, recent changes in the Iranian lifestyle have caused a shift in mortality and morbidity patterns towards a predominance of the non-communicable diseases of an ageing population [1].
After the 1979 Islamic revolution, Iran adopted a full generic-based medicine policy and prioritized local production of medicines. A formulation based national industry focused on self-sufficiency is one of the main goals of Iran’s medicine policy. The policy is mainly based on generic medicines, in which all medicines are only registered by their generic identity. In 2002, however, Iranian policymakers introduced the concept of branded generics in order to promote competition within the national pharmaceutical industry [4–5].
Although local Iranian pharmaceutical companies are able to produce small molecule medicines for the domestic market, the national health system depends on certain imported life-saving or disease-modifying patented medicines including biopharmaceuticals [6]. Although modern Iranian pharmaceutical companies were established 80 years ago, departure of international pharmaceutical companies following the 1979 Islamic revolution limited the access of the local pharmaceutical industry to both new technology and needed raw materials [4–6]. Although due to its population size, Iran has one of the largest pharmaceutical markets in the Middle East, its per capita spending on medicines is low. However, the Iranian pharmaceutical market has experienced rapid growth in recent years, increasing on average more than 30% annually in the period 1993–2003. Most of this increase is attributed to the cost of imported medicines and devaluation of the national currency. The cost is mostly because local manufacturers do not produce the new, hi-tech medicines being demanded from prescribers by consumers. Currently locally produced medicines provide only about 65% of the Iran pharmaceutical market needs and this percentage has been declining substantially in recent years. Despite a substantial decrease in the country’s population growth rate in the past decade, drug expenditures have drastically increased to over US$4 billion [4–6].
The increasing costs of health care, including the costs of pharmaceuticals, pose challenges for all nations. In developing countries including Iran, pharmaceutical expenditures account for 25–65% of total public and private health expenditures, and for 60–90% of household out-of-pocket expenditures on health. In order to improve affordability of the medicines in Iran, the government has implemented a direct and not targeted subsidy mechanism for medicines. The main goal of this strategy is to reduce the price of medicines on the market. Obviously this universal subsidy mechanism can cause corruption. Direct subsidization of some very expensive imported medicines by the government has promoted smuggling of these medicines into the neighbouring countries, where they can be sold with a very high profit [7]. Therefore, this strategy was stopped and now only a few, mainly hi-tech medicines are receiving direct subsidy from the government. Currently the national health insurance system is the main tool in Iran for improving the affordability of medicines. Local production of copies of biopharmaceuticals is a new development in the Iranian pharmaceutical sector. In recent years some small science-based pharmaceutical companies have started to manufacture biological pharmaceuticals using modern biotechnology methods [5].
Biopharmaceuticals are large and complex biological molecules. Their final biological activities very much depend on the methods used for their production. Recombinant proteins such as blood coagulating factors, erythropoietins, gonadotrophins, granulocyte colony-stimulating factors (G-CSF), human growth hormones, interferons, interleukins and monoclonal antibodies are among the most important biopharmaceuticals marketed in past decades [8].
Biopharmaceuticals represent a rapidly growing market. It is reported that 32% of the products in development pipelines and 7.5% of marketed medicines are biopharmaceuticals. By 2020, biopharmaceuticals are forecasted to sell for around US$23 billion in the EU and US$29 billion in the US [9]. Although currently many original brand-name biopharmaceuticals are protected by intellectual property laws and regulations, with expiration of their patent protection in the next few years the pharmaceutical industry faces a unique business challenge from both well regulated rival biosimilars as well as less regulated copied biopharmaceutical. Despite the increasing clinical significance of biopharmaceutical medicines, economic barriers limit their use for many patients, especially in developing countries. In the World Trade Organization (WTO) member countries, failure of the pharmaceutical industry to meet the needs of national health services may result in compulsory licensing regulation changes in order to permit local industries to produce and market copies of such patented medicines to be sold at more affordable prices.
‘Biosimilars’ are biopharmaceuticals that are manufactured and marketed, usually by non-originator pharmaceutical companies, following expiration of the originator patents. Currently, marketing authorization of such biosimilars for highly regulated markets depends on demonstration of ‘similarity’ between a biosimilar and its corresponding originator biopharmaceutical. In the next few years substantial numbers of biopharmaceuticals will lose their patent protection and therefore will be open to the production and competitive marketing of such medicines. However, in contrast to small molecule medicines the replication of the biopharmaceuticals is not an easy task. In many cases even small changes in the structure of the final molecule can create a different safety and efficacy profile. This is why adequate evaluation of biosimilars has become such a challenge for both the scientific community and regulatory agencies.
Generic small molecule medicines are usually approved on the basis of their established bioavailability compared to the originator brand comparator. In most cases, generic medicines are also considered as interchangeable with their corresponding original brand medicines. In contrast to small molecule medicines, biopharmaceuticals are manufactured in living systems, e.g. plant or animal cells. The methods used to produce the biopharmaceuticals; including cloning, expression system, expansion, recovery, purification and formulation of the final product, determine the ultimate biological activity of these products. Therefore, any change in any of these steps could create substantial changes in the efficacy or safety profile of the medicine, including its immunogenicity. Despite the introduction of biosimilars into world pharmaceutical markets many years ago some countries still lack regulations for registration of these medicines. The European Medicines Agency (EMA) was the first well-established regulatory authority to develop comprehensive guidelines in 2004 [10]. WHO has also published its guidelines for the evaluation of biosimilars in 2010 [11]. These guidelines are mainly focused on a head-to-head demonstration of biosimilarity with a registered original brand biopharmaceutical through both preclinical and clinical trials. The clinical studies must be designed in a way that can demonstrate comparable safety and efficacy between biosimilar and the reference biopharmaceutical.
With the increasing importance of biopharmaceuticals in disease therapy and their high costs, the lower costs of their alternatives, biosimilars, is a driving force for their marketing. Due to very high costs both for each treatment, and over a full treatment time period, even a slight cost reduction through the marketing of biosimilars could facilitate access to biopharmaceuticals; especially in resource limited healthcare systems [12–14]. As has happened with generic versions of small molecule medicines, the introduction of biosimilars is expected to create substantial reductions in national healthcare expenditures. Published data show that these medicines will result in savings of about US$9–12 billion for the US Medicare program in a decade [15]. It is also reported that biosimilars could create savings of between Euros 11.8 and Euros 33.4 billion between 2007 and 2020 for European healthcare systems. The savings for erythropoietins alone could be between Euros 9.4 and Euros 11.2 billion. However, most of these savings will come from administration of biosimilar monoclonal antibodies. [16].
It is estimated that developing a biosimilar for highly regulated markets such as the EU and the US will cost between US$75 and US$250 million [17]. Since many pharmaceutical companies in developing countries cannot afford such costs, national companies try to meet local demand by manufacturing copied biopharmaceutical medicines without actual demonstration of biosimilarity. Obviously, these copied biopharmaceuticals cannot be compared to the original brand biopharmaceuticals approved in highly regulated markets. These countries instead must perform close pharmacovigilance of copied biopharmaceuticals for both patient safety and proving efficacy of these medicines [18].
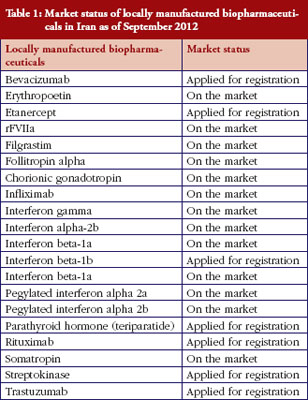
Iran’s national pharmaceutical industry is mostly a generic-based industry that produces small molecule chemical medicines. Since a decade ago some newly established science-based Iranian pharmaceutical companies began developing biopharmaceuticals. Iran’s Government has also allocated substantial resources including financial and administrative support for improving the capability of local pharmaceutical companies to manufacture biopharmaceuticals. Because they do not have access to the production procedures of originators, including cell type, fermentation and purification procedures, they cannot claim ‘similarity’ to originator brands. These copied biopharmaceutical including IFNs, G-CSF and GH have received marketing authorization for the local Iranian market, but none have received evaluation according to internationally recognized guidelines for biosimilars. Registration has mainly followed the path for ‘biogeneric’ medicines, and their application for marketing authorization has been handled on a case by case basis. Since 2003, about 20 locally manufactured biopharmaceuticals either entered the market or are awaiting marketing authorization, see Table 1; from Iran’s national regulatory authority (NRA). This table shows that the most clinically important biopharmaceuticals, especially recombinant proteins, are manufactured locally by Iran’s national industry. Some additional monoclonal antibodies have already been filed for registration and are expected to enter the market in 2013–2014. All medicines, including biopharmaceuticals in Iran, have their prices set by national authorities, and have to be sold at a fixed price all over the country. The prices of locally manufactured biopharmaceuticals are between 27–72% lower than their corresponding imported original brands. Obviously, this could significantly improve affordability and accessibility of the biopharmaceuticals for both patients and the national health service.
In 2001, Iran became a member of the World Intellectual Property Organization and registers trademarks for medicines. However, there is no patent protection for imported medicines in Iran [5]. As of mid 2005, Iran became an observer member of WTO. However, due to the current international political situation full WTO membership is unlikely to happen in the near future. Iran’s local industry does not consider WTO membership to be an eminent concern with regard to producing copies of patented biopharmaceuticals. Therefore, the local pharmaceutical companies would be able to manufacture both patented and off-patent biopharmaceuticals provided that they gain access to their production procedures.
In order to support local manufacturers, Iranian officials impose a tariff on imported original brand medicines as soon as a local version enters the market. This tariff can be as high as 65% and therefore substantially increases the price of imported medicines, enabling locally made products to dominate the market. However, if either local manufacturers or authorities overestimate production capability, there may be a shortage of cheaper, locally manufactured biopharmaceuticals, thus causing a huge economic burden on patients who then have to buy more expensive original brand medicines.
The process of registration of biopharmaceuticals in Iran
Iran established its NRA in the 1950s, and passed the first laws for the regulation of the national pharmaceutical market in 1955. The Ministry of Health and Education of Iran is responsible for regulation of the market. Although the registration of small molecule medicines is a well-developed system it is not always as transparent as it should be according to NRA’s guidelines for the marketing of locally produced copied biopharmaceuticals [5]. In 2006, the Iranian NRA announced national guidelines for the marketing of biosimilars. These guidelines, which are mainly adapted from the WHO guidelines, specify the need for a clinical trial with a small sample size comparing locally manufactured biopharmaceuticals with the original brand. However, there are clear differences between the WHO guidelines and current Iranian national guidelines for the registration of locally produced biopharmaceuticals [19–20]. Currently, the Iranian NRA does not ask for comprehensive new preclinical or clinical data for proving similarity between locally produced biopharmaceuticals and original brand medicines.
The Iranian NRA has so far relied on national post-marketing surveillance to produce data on the ‘safety’ of all marketed, copied biopharmaceuticals. Iran has a fairly well-established national adverse drug reaction (ADR) reporting system and so far no serious or unexpected ADRs related to administration of locally manufactured biopharmaceuticals have been reported to the national health authorities [20–21].
Healthcare expenditures in Iran have risen dramatically in recent years, due mainly to an increase in expenditure on biopharmaceuticals. Iran’s policymakers believe that although biosimilars are a cost-effective intervention for the treatment of patients, the regulatory approaches for their marketing used by organizations such as EMA, US Food and Drug Administration (FDA) or even WHO may not suit the needs of the Iranian market [20–21].
Although highly regulated markets such as the EU and the US have specific requirements for marketing of biosimilars, these might not be appropriate for the marketing of biopharmaceuticals in less regulated markets. Biosimilars in these markets therefore have different definitions. While they may have acceptable safety and efficacy profiles based on local requirements these ‘copied biopharmaceuticals’ manufactured in these countries are not ‘biosimilars’ as defined by EMA, FDA and WHO guidelines.
In order to create a balance between the regulation and affordability of locally produced biopharmaceuticals, Iran’s NRA tries to use its national pharmacovigilance system as a tool for assessing the safety of the marketed locally manufactured biopharmaceuticals, including the incidence of ADRs associated with their use. There is a general consensus that patient’s safety should not be compromised in exchange for access to the less expensive biosimilars [22]. Iran’s NRA mainly accepts evidence of pharmacokinetic and pharmacodynamic equivalence between the originator and locally manufactured biopharmaceuticals and a small size clinical trial as a measure of clinical comparability for copied biopharmaceuticals. However, biopharmaceuticals have varying potential for immunogenicity that can change based on a number of factors, including manufacturing processes. Although small differences between copied biopharmaceuticals and original brand medicines might not cause significant differences in clinical efficacy, in rare cases these differences might lead to safety problems. Therefore, post-marketing surveillance could be used as an effective tool to detect any adverse reactions which could be considered as a breach of safety profile of these biopharmaceuticals. Since biological medicines have unique characteristics the surveillance system must be both very sensitive and reliable if it is to detect and assess any adverse event in a timely manner [22].
The Iranian pharmaceutical market is susceptible to the use of counterfeit medicines. Iran is one of the major transit routes for illicit narcotics produced in Afghanistan. It is assumed that the same transit pathways could also be used for counterfeit medicines including biopharmaceuticals. Indeed, illegal medicines and supplements now comprise up to 10% of the total pharmaceutical market [7]. There is a danger that the ambiguity and uncertainty in the regulatory requirements for the marketing of biopharmaceuticals in Iran will encourage manufacturers to avoid doing the clinical trials necessary for proving the comparable efficacy and safety of their products. The use of clinically not comparable biopharmaceuticals could then impose extra medical and financial burdens on patients and the national health system if this leads to treatment failure, toxicity, and the need for corrective interventions. This also raises the possibility of yet unidentified short- and long-term safety concerns. The government of Iran currently supports the local pharmaceutical industry by imposing high tariffs on imported medicines. Therefore, it is expected that patients should benefit from the availability of copied biopharmaceuticals through improved access for effective treatment of chronic and lifelong diseases.
In the past few years, the Iranian national industry has manufactured copies of biopharmaceuticals. Although granted marketing authorization in Iran, none of these medicines has had comprehensive evaluation according to FDA or EMA guidelines. The lower cost of these copied biopharmaceuticals could improve the affordability of these clinically important medicines. However, authorities need to perform close vigilance of these biopharmaceuticals in order to evaluate their safety and efficacy.
The manuscript by Professor Majid Cheraghali in this edition of the GaBI Journal raises two important issues that need to be considered. First, economic pressures exist that will encourage some countries to pursue copied biologics which will not be adequately tested for ‘biosimilarity’. This may or may not result in additional risks or cost savings. Second, the article illustrates how politically motivated sanctions, whether or not justified, can have major (hopefully unintended) negative effects on public health even in a resource rich, developed country like Iran. Finally, the article mentions the impact of drug smuggling and counterfeit medicines, both of which have major public health implications throughout the developing and developed world.
Our readers are encouraged to submit manuscripts that explore these important aspects of the use of generics and biosimilars.
The author wishes to thank Dr Shekoufeh Nikfar for her valuable assistance on providing some data.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Asadi-Lari M, Sayyari AA, Akbari ME, Gray D. Public health improvement in Iran–lessons from the last 20 years. Public Health. 2004 Sep;118(6):395-402.
2. Goudarzi S, Kameli ME, Hatami H. Improvement in health indicators of Islamic Republic of Iran in the years 2004 and 2008. Iran Red Crescent Med J. 2011;13(8):574-7.
3. Cheraghali AM, Nikfar S, Behmanesh Y, Rahimi V, Habibipour F, Tirdad R, et al. Evaluation of availability, accessibility and prescribing pattern of medicines in the Islamic Republic of Iran. East Mediterr Health J. 2004 May;10(3):406-15.
4. Basmenji K. Pharmaceuticals in Iran: an overview. Arch Iranian Med. 2004;7(2):158-64.
5. Cheraghali AM. Iran Pharmaceutical Market. Iran J Pharm Res. 2006;1:1-7.
6. Dinarvand R. New national drug policy in Iran leading to expanded pharmaceutical market and extended access of public to medicines. Iranian J Publ Health. 2009;38 Suppl 1:158-61.
7. Hosseini SA, Darbooy Sh, Tehrani Banihashemi SA, Naseri SM, Dinarvand R. Counterfeit medicines: report of a cross-sectional retrospective study in Iran. Public Health. 2011;125(3):165-71.
8. Brockmeyer C, Seidl A. Binocrit: assemssment of quality, safety and efficacy of biopharmaceuticals. Eur J Hosp Pharm Prac. 2009;15(2):34-40.
9. GaBI Online – Generics and Biosimilars Initiative. Biosimilars: barriers to entry and profitability in the EU and US [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 February 11]. Available from: www.gabionline.net/Biosimilars/Research/Biosimilars-barriers-to-entry-and-profitability-in-the-EU-and-US
10. World Health Organization [homepage on the Internet]. Guidelines on evaluation of similar biotherapeutic products (SBPs). 2009 [cited 2013 Feb 11]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
11. European Medicines Agency [homepage on the Internet]. Annex guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing somatropin. 2006 [cited 2013 Feb 11]. Available from: http://www.ema.europa.eu/pdfs/human/biosimilar/9452805en.pdf
12. Stewart A, Aubrey P, Belsey J. Addressing the health technology assessment of biosimilar pharmaceuticals. Curr Med Res Opin. 2010;26(9):2119-26.
13. Bruce ES, Armour K, Romiti R, Smith C, Tebbey PW, Menter A, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66(2):317-22.
14. Simoens S. Biosimilar medicines and cost-effectiveness. Clinicoecon Outcomes Res. 2011;3:29-36.
15. Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program, Medicare Payment Advisory Commission, Washington (DC) [homepage on the Internet]. 2009 [cited 2013 Feb 11]. Available from: http://www.medpac.gov/documents/jun09_entirereport.pdf
16. Höer A, de Millas C, Häussler B, Haustein R. Saving money in the European healthcare systems with biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):120-6. doi:10.5639/gabij.2012.0103-4.036
17. McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3(2):209-17.
18. Joshi SR. Biosimilar peptides: need for pharmacovigilance. J Assoc Physicians India. 2011 Apr;59 Suppl:44-7.
19. Hadavand N, Valadkhani M, Zarbakhsh A. Current regulatory and scientific considerations for approving biosimilars in Iran. Biologicals. 2011;39(5):325-7.
20. Cheraghali AM. Biosimilars; a unique opportunity for Iran national health sector and national pharmaceutical industry. Daru. 2012 Sep 10;20(1):35.
21. GaBI Online – Generics and Biosimilars Initiative. Significance of locally produced biosimilars in Iran [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 February 11]. Available from: www.gabionline.net/Biosimilars/Research/Significance-of-locally-produced-biosimilars-in-Iran
22. Straus SMJM, Giezen TJ. Pharmacovigilance of biosimilars: challenges and possible solutions. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):118-9. doi:10.5639/gabij.2012.0103-4.033
|
Author: Professor Abdol Majid Cheraghali, PharmD, PhD, Department of Pharmacology, University of Baqiyatallah Medical Sciences, Tehran, Iran |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
Biosimilars in the European market
Source URL: https://gabi-journal.net/current-status-of-biopharmaceuticals-in-iran-s-pharmaceutical-market.html
Copyright ©2025 GaBI Journal unless otherwise noted.