|
Abstract: |
Submitted: 16 August 2016; Revised: 1 September 2016; Accepted: 1 September 2016; Published online first: 14 September 2016
Biologicals are a class of drug that is derived from living organisms [1]. The majority of biologicals are utilized in the areas of oncology, inflammatory diseases and diabetes [2–4]. According to a recent report published by the University of British Columbia, the per capita spending on biologicals for inflammatory conditions alone in Canada have more than doubled from CAN$18 in 2007–2008 to CAN$45 in 2012–2013 [5], which contrasts with relatively stable overall per capita spending for all prescription drugs (CAN$661 and CAN$656 in 2007–2008 and 2012–2013, respectively) [5]. In 2010, spending on biologicals was approximately CAN$3 billion, which constituted 14% of the Canadian pharmaceutical market. It is expected to grow to about 20% by the end of the decade, creating substantial financial impact for public and private payers.
A large number of patents for high cost innovator biologicals are set to expire in Canada within the next 10 years, e.g. Avastin, Herceptin, Humira, Lucentis, Rituxan. This will lead to the rapid development of subsequent entry biologics (SEBs). For a review of the current list of innovator biologicals set to expire in Canadaand SEBs in development, please refer to a report published recently by the Canadian Agency for Drugs and Technologies in Health (CADTH) [6]. Introduction of SEBs may lead to increased access for many patients, although SEB developers face many challenges prior to having their products reach patients. This paper provides an overview of the current regulatory and health technology assessment (HTA) environments for SEBs in Canada,positions of various medical and disease associations, as well as lessons that can be learned from Europe to increase uptake of SEBs. Please note that the terms ‘biosimilar’ and ‘SEB’ will be used interchangeably in this paper.
Federal
Around the time of the approval of the first SEB in the European Union (EU) (Omnitrope) in 2006, Health Canada released an SEB Factsheet [1], which signalled the development of the first SEB regulatory guideline in Canada. In March 2010, the Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs) was released [7]. The term ‘subsequent entry biologic’ is defined as ‘a biologic[al] drug that enters the market subsequent to a version previously authorized in Canada, and with demonstrated similarity to a reference biologic[al] drug. An SEB relies in part on prior information regarding safety and efficacy that is deemed relevant due to the demonstration of similarity to the reference biologic[al] drug and which influences the amount and type of original data required’.
Because biologicals are produced in living organisms, and they are highly sensitive to process changes, it is virtually impossible to make identical copies of these protein-based therapeutics. Therefore, an important consideration for SEBs is that they are not ‘generic biologicals’. Many characteristics associated with the market authorization process use for generic pharmaceutical drugs do not apply. As a result, SEBs cannot be considered pharmaceutically or therapeutically equivalent to the reference biological products.
Under the Health Canada SEB pathway, the SEB manufacturer presents a reduced clinical and non-clinical package to show that the SEB is highly comparable to the reference biological via a series of comparability exercises, e.g. analytical testing and biological assays. This is particularly important for SEB manufacturers since additional clinical/non-clinical requirements would add significant time (7–8 years) and cost (US$100 million to US$250 million) to develop an SEB compared to small-molecule generics (US$1 million to US$4 million) [8]. As discussed later, the importance of demonstrating physicochemical and in vitro functional comparability between the SEB and the reference biological is also a key for the approval of indication extrapolation (the granting of indication approved for the reference biological to the SEB without conducting clinical trial for that indication).
In December 2015, Health Canada issued a consultation to stakeholders on a revised Information and Submission Requirements for Subsequent Entry Biologics (SEBs) guidance documents [9, 10]. Key revisions included addition of further guidance with respect to considerations in the selection of a reference biological drug; additional detail with respect to considerations when performing non-clinical and clinical studies for SEBs, including discussion with respect to immunogenicity, the use of the most sensitive population in clinical trial design and a new section on extrapolation; and a new section which promotes early consultation with Health Canada, as well as the launch of three-year pilot for SEB Scientific Advice Meetings to allow for discussion of an SEB with Health Canada early in the development process [9]. The latter addition is very much similar to the European Medicines Agency’s (EMA) scientific advice and protocol assistance process [11], suggesting Health Canada’s continuing effort to harmonize its SEB regulatory process with other international agencies.
The Canadian drug plan market is a mix of public and private systems. Public drug plans make extensive use of HTA as part of the reimbursement decision-making process. Eligible non-cancer prescription drugs approved by Health Canada are reviewed by the Common Drug Review (CDR) and the Institut national d’excellence en santé et en services sociaux (INESSS; province of Quebec only) [12, 13]. In contrast, eligible approved cancer drugs are reviewed by the pan-Canadian Oncology Drug Review (pCODR) [14]. INESSS also reviews eligible cancer drugs. Both the CDR and pCODR are part of the CADTH that provides reimbursement recommendations that are considered by participating public drug plans. The province of Quebec does not participate in the CDR and the pCODR process.
In the fall of 2009, CADTH initiated a pilot project specific to the submission and evaluation of SEBs due to the different regulatory data requirements (CDR Update – Issue 59, released 30 June 2009). The purpose of the pilot project was to determine the requirements for SEB submissions, establish a review framework, and learn about Health Canada’s approach. The pilot project was to be evaluated after three SEBs had been reviewed and stakeholders’ feedbacks were to be sought at the end. The first SEB submitted under this pilot process was Omnitrope, see extended discussion that follows. However, four years later, no other SEBs had been submitted; and because multiple SEBs with indication extrapolation were imminently expected, CADTH terminated the pilot project and began a new consultation with stakeholders to develop a new standardized procedure and process in the fall of 2013 [15]. As a result of theconsultation process, a new Common Drug Review Procedure and Submission Guidelines for Subsequent Entry Biologics was issued in March 2014 [16]. The submission requirements for SEBsrepresent a significant deviation from the existing requirements for non-SEB products.
According to CADTH, CDR recommendations for SEBs will be based on: i) patient and public perspectives on the impact of the drug; ii) safety, efficacy and effectiveness of the drug compared to alternatives; iii) therapeutic advantages relative to current accepted therapy; and iv) cost and cost-effectiveness relative to current accepted therapy [17]. CADTH also noted that while the new procedure is not a pilot project, it would evaluate the process after a few submissions. And as with Health Canada, the CDR does not issue statements regarding interchangeability. Interestingly, however, in recent recommendations for the SEBs Grastofil (filgrastim) and Basaglar (insulin glargine), the CDR’s Canadian Drug Expert Committee (CDEC) noted that patients being treated with the reference biological should be considered for switching to SEB after a discussion between patients and physicians [18, 19].
Currently, pCODR does not have a separate procedure and process in place for the submission and evaluation of SEBs. However, since pCODR is also under the jurisdiction of CADTH, it is expected that the process will be similar to that of the CDR.
In August 2010, the Canadian Premiers announced the pCPA initiative at a meeting of the Council of Federation (COF). The pCPA is a process under the COF’s Health Care Innovation Working Group (HCIWG). The intended goal of the pCPA is to combine the purchasing power of public drug plans across multiple jurisdictions in order to ensure benefits are cost-effective, and to increase access to drug treatment options across jurisdictions. In addition, the pCPA also serves to reduce duplication of negotiations and to address the issue of inconsistent drug listing decisions across the country that were the results of each jurisdiction making individual decisions. All provinces (excluding Quebec) and Yukon (territory) were part of the initial alliance [20]. However, Quebec announced their intention to join the pCPA [21] and have done so.
Formally, the pCPA negotiations are conducted on brand-name drugs and generic drugs. As the pCPA process does not preclude existing evidence-based drug reviews, only (branded) drugs that have been assessed by the CDR and pCODR and received generally positive, i.e. not ‘Do Not List’, recommendations would be considered for negotiations. It should be noted that province-territory specific review may occur following the recommendations of CDR or pCODR in the absence of the pCPA process [22].
Very recently, pCPA released a ‘Subsequent Entry Biologicals (SEBs) First Principles’ document [23]. This document represents a starting point for the pCPA’s engagement with stakeholders to develop a more comprehensive SEB policy framework. Major principles include:
• All SEB and reference biological manufacturer proposals will only be considered through the national pCPA negotiation process rather than individual or selected jurisdictions
• Consistent with its mandate that includes increasing patient access to clinically and cost-effective drug treatment options, the pCPA will encourage a competitive environment that includes SEB market growth and is conducive to long-term cost reductions and sustainability for public drug plans
• The introduction of an SEB must provide a reduction in the drug’s transparent price to benefit all Canadians
• Proposals from reference biological manufacturers will only be considered if they:
• Provide overall national value to public drug plans and do not result in incremental costs to individual jurisdictions
• Provide at least similar overall value compared to the SEB, and must include similar or better transparent price reductions if equivalent listing status is sought
To date, there have been no published submission and evaluation processes specific to SEBs that are available to the public at both the provincial and federal drug plans. However, we believe that many jurisdictions are actively evaluating and adopting their existing submission guidelines to meet the unique aspects posted by SEB submissions.
Omnitrope (Sandoz Canada)
Omnitrope is an SEB of the recombinant growth hormone (GH) somatropin (somatotropin; marketed as Genotropin by Pfizer Canada) produced in Escherichia coli indicated for the treatment of GH deficiency in children and adults [24]. It was the first SEB to receive regulatory approval by Health Canada. Omnitrope was filed during the period in which Health Canada was developing its SEB guideline, as such, it provided the regulatory agency the opportunity to gain the experience needed to validate the concepts and principles behind SEBs [25, 26].
Several issues were encountered during the evaluation of the file and Health Canada had to exercise a certain degree of flexibility in the evaluation of the first SEB. First, Genotropin was used as the reference biological, which at the time, while authorized, was not marketed in Canada. Following a series of discussions, along with the availability of an extensive set of comparability data as well as other reasons, Health Canada eventually accepted the use of Genotropin as a comparator. Second, a reduced preclinical package was accepted in which full pharmacodynamic studies were accompanied by reduced toxicology information but no pharmacokinetic information was provided. Third, while Health Canada preferred equivalence trial, it accepted a complex and non-conventional crossover study as a pivotal trial.
At the time of filing, no trials submitted for Omnitrope were conducted in adults; however, it was considered acceptable to extrapolate the indication of GH deficiency to adults based on the molecule’s mechanism of action, disease pathophysiology, as well as the suitability of the adult study population with GH deficiency since childhood. Health Canada also requested post-marketing commitments that included long-term follow-ups and safety considerations. Overall, the combination of several critical elements led to the regulatory approval of Omnitrope for both the evaluated and extrapolated indications [25, 26].
Consistent with Health Canada policy, and the fact that the reference biological Genotropin was not marketed in Canada at the time, no statement regarding interchangeability and substitution of Omnitrope was made.
Approximately two months following regulatory approval, Omnitrope was submitted to the CDR for HTA. Since the SEB evaluation framework was still in development, a typical recommendation was not given, instead, an ‘Advice’ was issued [27]. In the report, the Canadian Expert Drug Advisory Committee (CEDAC) of the CDR advised the ‘drug plans [to] consider a similar reimbursement policy for Omnitrope as for other growth hormone products’.
Table 1 lists the provincial and federal public reimbursement statuses of Omnitrope. Results indicate that all provinces provide coverage for Omnitrope. Although Omnitrope is not listed on the Non-Insured Health Benefits (for First Nations) Drug Benefit List, it is likely reimbursed on a case-by-case basis. At least two provinces (Prince Edward Island and Saskatchewan) explicitly stated that Omnitrope is not interchangeable.
Inflectra/Remsima (Celltrion Healthcare Co Ltd)
Regulatory background
Inflectra/Remsima, developed as CT-P13 by the Korean company Celltrion, is an SEB of the monoclonal antibody (mAb) against the tumour necrosis factor-alpha (TNF-α), infliximab (marketed as Remicade by Janssen). CT-P13 was the first mAb SEB approved by Health Canada and Celltrion received regulatory approval (notice of compliance; NOC) for Remsima in January 2014 [28]. Hospira Healthcare Corporation, following a subsequent licensing agreement with Celltrion, received an NOC in June 2014 as the manufacturer for CT-P13 under the brand name Inflectra [29]. However, as a result of legal proceeding initiated by Janssen in July 2014, the NOC granted to Hospira was quashed in March 2015 [30]. Although Hospira remains the marketer of Inflectra in Canada, the change in NOC status serves as an inconvenience from a logistical perspective and this highlights one of the ways that innovator companies are dealing with competitors.
At the time of regulatory submission, the finalized version of the Guidance for Sponsors had already been published [7]. Because CT-P13 is a mAb, the processes involved in demonstrating its biosimilarity to Remicade was far more complex compared to Omnitrope [26, 28, 29]. The pivotal trials submitted for regulatoryevaluation included a single phase III therapeutic ‘equivalence’ trial conducted in rheumatoid arthritis (RA) patients and a single phase I pharmacokinetic trial conducted in ankylosing spondylitis (AS) patients, both against Remicade. In Europe, CT-P13 was approved by EMA for the two evaluated indications and was also granted authorization for six other indications (psoriatic arthritis, psoriasis, adult and paediatric Crohn’s disease, as well as adult and paediatric ulcerative colitis) based on extrapolation. In contrast, Health Canada only approved the evaluated indications and two of the extrapolation indications of psoriatic arthritis and psoriasis [28, 29, 31, 32].
Several contentious issues arose surrounding the regulatory approval of CT-P13. Specifically, there were questions regarding whether RA was a sensitive enough disease model for detecting therapeutic differences between products, in part, due to the concomitant use of the immunosuppressive methotrexate [33, 34]. Furthermore, it has been suggested that RA and AS models are not the most sensitive models for measuring immunogenicity [33, 34]. In addition, difference was observed in the most sensitive of the in vitro FcγRIIIa-dependent assay, raising the question of whether CT-P13 can be considered similar to Remicade [33, 34]. Nevertheless, EMA considered the clinical evidence submitted to be acceptable for approval of all requested indications, whereas Health Canada took a slightly more conservative approach in making its decision due to issues surrounding differential FcγRIIIa activity [28, 29, 31, 32].
Hospira, now a Pfizer company, subsequently received regulatory approval for the adult indications of Crohn’s disease (including fistulizing Crohn’s disease) and ulcerative colitis [35]. However, the evidence basis for the anticipated regulatory approval remains to be determined, i.e. whether paediatric data was submitted for regulatory evaluation.
Health technology assessment and pricing negotiation
Both Inflectra and Remsima were submitted to CDR following regulatory approval and represented the first set of SEBs (and first mAb) submitted under the new CDR SEB submission procedure. Remsima was voluntarily withdrawn from the CDR review [36]. In December 2014, the CDEC of the CDR issued the recommendation of ‘List with clinical criteria and/or conditions’ (LWCC) for Inflectra in which the conditions were for the drug to be used ‘in patients for whom infliximab is considered to be the most appropriate treatment option’, and for Inflectra to be listed in the public formularies ‘in a manner similar to Remicade’. The LWCC recommendation applied to all the Health Canada-approved indications for Inflectra (including the two extrapolated indications) [37].
Following the release of the recommendation, Inflectra was submitted for negotiation under the pCPA process and was completed in November 2015 [38]. It is unclear why the process took almost a year but Inflectra represented a first true SEB with multiple indications (approved and subjected to be approved) with substantial budget impact. Therefore, this negotiation may have served as a learning process for the pCPA.
The submitted price of Inflectra represented a ~31% discount [39] relative to Remicade (based on the lowest listed public price in Quebec). However, this level of discount is not seen in other regions. For example, in Norway, the Finnish company Orion Oyi successfully won the tender to supply the country with CT-P13 (also being sold under the name Remsima), with a 69% price discount relative to Remicade, lower than the 51% discount that Hospira was offering [40]. Such discounts are far more aggressive compared to the 15–30% price reduction seen for other biologicals [41]. Although not entirely comparable, the nearly 70% discount is most commonly seen for non-biological generic drugs. This highlights the price pressure that biosimilar companies face. Questions have been raised regarding the sustainability of biosimilar companies under such pricing competition [40].
INESSS’ decision on Inflectra
In February 2015, INESSS made a landmark decision regarding the reimbursement of Inflectra [42]. Recognizing that both Inflectra and Remicade ‘have the same generic name, form and content’, INESSS stated that the lowest price would be the price reimbursed for both infliximab products for the four indications (currently Inflectra has the lowest price on Régie de l’assurance maladie du Québec’s [RAMQ] List of Medications). Patients must pay the difference if they wish to receive Remicade. The most significant observation is that the ‘lowest price method’ is applied to Inflectra, perhaps suggesting a form of interchangeability.
In its deliberation, INESSS recognized that the lack of knowledge (regulatory and scientific) impedes acceptance of SEBs in Canada. However, it hopes that its decision will allow prescribers to appreciate the merit of Inflectra. INESSS has also recognized that factors such as accessibility to certain clinics can limit the uptake of Inflectra. Indeed, in the most recent issue of the formulary (List of Medications) [43], RAMQ stated that the only exception to the lowest price method applicable to infliximab is when prescribers indicate to the pharmacist that Remicade is preferably prescribed to Inflectra because the Remicade perfusion centre is closer to the patient’s home or is more readily accessible, given the patient’s health condition, compared to those for Inflectra. RAMQ stated that this exception has an end date of 3 October 2016.
Other public plans’ decision on Inflectra
After the successful completion of the pCPA negotiation, public plans began to list Inflectra on their formularies. Compared to Quebec, the publicly available list prices of Inflectra across the plans represented between 33–47% discount relative to those of Remicade, which are much higher than Quebec, see Table 2. Perhaps more significantly, many of the public plans preferentially reimburse Inflectra over Remicade for patients new to infliximab, i.e. new infliximab patients will only be covered for Inflectra, and those who failed one infliximab will not be allowed to switch to another infliximab. Some Atlantic provinces also provide reimbursement for Inflectra for indication (psoriatic arthritis) that is not reimbursed for Remicade. This is a clear signal from the drug plans to bolster the success of SEBs.
Despite such generous reimbursement policy, it appears that public plans are still cautious regarding interchangeability. Furthermore, only Ontario has reduced the barrier to access to Inflectra (Limited Use – no prior authorization required) compared to Remicade (Exceptional Access – prior authorization required) while the remaining provinces still require prior authorization to access Inflectra.
Filgrastim (Apotex), Insulin Glargine (Eli Lilly), Etanercept (Merck)
Filgrastim is the recombinant version of the human glycoprotein granulocyte colony-stimulating factor (G-CSF), which is used to treat neutropenia. Amgen currently sells the innovator filgrastim under the trade name Neupogen. Since the patents on Neupogen expired in Europe in 2006, several versions of the filgrastim SEB have become commercially available in the region [44].
Apotex’s filgrastim SEB, Grastofil (co-developed with Intas Pharmaceuticals Ltd), is currently being sold in Europe, and has recently been accepted for filing by the US Food and Drug Administration (FDA) [45]. Grastofil has been approved by Health Canada for all indications approved for Neupogen although the process has not been smooth as indicated by a patent litigation with Amgen [46]. As mentioned above, Grastofil has been assessed by the CDR and has received a LWCC recommendation from CDEC in March 2016 [18].
Recently, Eli Lilly also launched the SEB version of insulin glargine, Basaglar. Similar to Grastofil, CDR has issued a LWCC recommendation and CDEC also included a ‘Of Note’ commentary [19]. Both Grastofil and Basaglar are under pCPA negotiations as of this writing.
Merck Canada has also submitted an SEB version of etanercept (brand name to be determined) and awaiting regulatory approval and CDR recommendation as of this writing [47].
In terms of the spending by private payers, a 2012–2013 Canadian report showed that private insurers paid approximately 35% of all drugs but this was increased to 42% for biologicals for inflammatory conditions, whereas public payers covered 43% for both [5].
The above statistics highlighted both the increasing use of biologicals and the significant financial impact on private payers. Amid these changes, private insurers are seeking ways to offset costs, which include but are not limited to increasing premiums and/or utilizing prior authorizations. Indeed, the introduction of SEBs represents a significant opportunity to reduce cost forprivate insurers. For instance, the Canadian private insurer TelusHealth has specified in its Remicade reimbursement special authorization form that patients must have tried and failed Inflectra prior to being authorized to use Remicade [48]. Janssen, in response to the competition, has entered into a preferred pricing agreement with Express Scripts Canada to provide reduced pricing for Remicade for private drug plans offered by providers such as Manulife and Empire Life [49].
Physicians and medical associations
In a survey of Canadian rheumatologists (81 respondents) [50], 31% were familiar with SEBs. Physicians with greater than 20 years of practice were significantly more likely to be familiar with SEBs compared to respondents with less than 20 years of experience. In addition, approximately two-thirds of the respondents indicated that they were unlikely to offer an SEB as initial therapy if cost was not an issue, mostly due to greater familiarity with brand-name drugs and uncertain long-term safety of SEBs. Even if public and private payers mandate the use of SEBs of an anti-tumour necrosis factor-alpha (anti-TNF-α) biological that physicians would normally not prescribe, e.g. adalimumab is preferred, but an infliximab SEB is mandated, half of the physicians were unlikely to offer them to patients. What is more interesting is that nearly half of the respondents indicated that they would use a non-anti-TNF-α biological to avoid using the SEB. Finally, over half of the respondents disagreed with indication extrapolation to psoriatic arthritis and ankylosing spondylitis, citing clinical and pathophysiological differences versus rheumatoid arthritis.
Based on this survey, although rheumatologists are aware that biologicals are costly for their patients (as shown by three quarters of the respondents indicating that patient assistance programmes have an impact when considering biological therapy), one-third of the respondents demanded at least a 50% price reduction to offset the risk associated with switching to an SEB.
While there are certain limitations to the survey, the results suggest that many physicians still have preference for the originator products, and that significant barriers exist for the wide uptake of SEBs by Canadian rheumatologists [50].
Indeed, considerable discussions have taken place within various medical communities and a number of associations (Canadian Rheumatology Association [CRA]; Ontario Rheumatology Association [ORA]; Canadian Dermatology Association [CDA]; and Canadian Association of Gastroenterology [CAG]) have issued position statements regarding SEBs. The overall consensus of these medical associations appears to be that patients should have access to safe and affordable treatment options. Some do not agree on extrapolation of indication while most also agreed that SEBs should not be considered as interchangeable and no automatic substitution should be allowed. Furthermore, most agreed that distinctive International Nonproprietary Name (INN) should be used for SEBs; and all agreed that post-marketing monitoring/registry should be required [51–54]. The Canadian Association of Medical Oncologists has not yet publicly issued a position statement regarding oncological SEBs.
Patient groups/disease societies
Perhaps those who will be the most directly impacted by SEBs are the patients themselves. Input from patient groups was taken into consideration in the deliberations of both CDR and INESSS [37, 42]. In the Inflectra reports, patients conceded that although therapeutic options are required for those with arthritis, and SEBs offer another biological drug therapy that may be effective for patients who are biological-naïve or who have failed on other biological drugs, many are uncertain whether SEBs actually offer additional therapeutic options. Some patients questioned whether SEBs are tested as rigorously or even perform as well as the reference product, and were even concerned that because not all indications were evaluated, this could be seen as being in a clinical trial in a real situation for verifying the efficacy and safety of SEB for these indications. There was also concern regarding the potential for switching of Remicade/Inflectra due to the same non-proprietary name between these two products.
From a patient access perspective, some expressed concerns about whether SEB manufacturers would offer patient support. Patients expected that Inflectra will be less costly than Remicade and the money saved can potentially be used to increase access to treatments, some patients believe that they will be forced to choose the SEB due to their insurance policy [37, 42].
Several patient groups/disease societies (the Canadian Arthritis Patient Alliance [CAPA], The Arthritis Society, Crohn’s and Colitis Canada, Canadian Skin Patient Alliance [CSPA], as well as various academic institutions) issued their views on the subject of SEBs. Similar to the positions expressed above, all agreed that there should be no automatic substitution for SEBs, that distinctive names should be given, and that post-marketing surveillance should be mandated. However, the view regarding the issue of indication extrapolation is less than unanimous [55–60]. A recent meeting involving a cross-disciplinary group of specialist physicians (dermatology, gastroenterololgy, nephrology, oncology and rheumatology), patients, patient group representatives and pharmacists, reached a consensus that indication extrapolation can be considered but should be based on robust and in-depth clinical data [61].
Despite the perceived value of SEBs, the uptake of SEBs has been relatively slow. According to IMS, the historic (2007) estimation for biosimilar market in the EU was expected to be US$16–20 billion per year by 2010–2011, and it was forecasted that the sales of biosimilars would be US$25 billion by 2020. However, by 2012, the actual sales were only US$0.6 billion [62, 63]. The EU has higher biosimilar uptake compared to Canada, partly due to the early introduction of biosimilars. Within the EU, biosimilars for molecules such as G-CSF and epoetin (EPO) have relatively high uptake percentage, and this has been in part contributed by the role of the payers in these countries.
In the UK, biosimilar G-CSF is hospital prescribed. A single preferred agent is selected via a transparent multifactor tender. Hospital protocols are changed to reflect the winning bid with exception criteria for defined patient groups. Success of uptake in achieving real savings is also published. All these processes led to a ~90% uptake for biosimilar G-CSF in the UK.
In Germany, outpatient physicians treating statutory health plan patients are members of the Kassenärztliche Vereinigung (KV), the professional association of outpatient physicians. There are 17 KVs in total, each covering a geographic region in Germany. There are several strategies that are used to facilitate biosimilar uptake. For instance, biosimilar quotas are utilized for EPO in each of the KV regions to increase uptake. In addition, physicians’ budgets are tracked by prescription utilization management tools and physicians are also provided with education sessions to reinforce the concept of biosimilar medicines. Finally, payers also endorse biosimilars as safe and effective via ‘dear doctor’ letters and address potential concerns. As a result, the uptake for biosimilar EPO is ~60% [64].
In Norway, the combination of the tendering system and a substantial discount has resulted in a more than 90% market share for the infliximab biosimilar as of mid-2016 [65].
Clearly, strong regulatory presence and price incentive are needed to assist the uptake of biosimilars, particularly for countries with well-developed regulations. Overall, the current trend in the US, EU, and emerging markets is that payers and policymakers are expected to increasingly become biosimilar advocates. This is partially reflected in the 2015 European Generic medicines Association–European Biosimilars Group meeting in the UK (April 2015) where both EMA and FDA agreed that biosimilars could be licensed for multiple indicationswithout performing clinical studies for each area. The Vice-Chair of the EMA Working Party on Similar Biological Medicinal Products (BMWP) Dr Martina Weise, FDA’s Center for Drug Evaluation and Research’s Associate Director of Therapeutic Biologics Dr Leah Christl, and EMA Head of Quality Dr Peter Richardson, all stressed that analytical methods and functional assays, e.g. pharmacokinetic and pharmacodynamics, are more sensitive to quality attributes and often much more valuable to regulators, and that clinical studies are the most blunt tool we have to confirm the similarity which are sometimes not needed [66]. FDA’s Dr Leah Christl also went on to state that they have seen sponsors who identified analytical differences between the biosimilar and the reference product but concern was not raised from a regulatory perspective. Finally, Dr Leah Christl also stated that FDA is planning outreach programmes to educate industry and prescribers about the importance of structural and functional analysis over patient studies in proving similarity [66].
Indeed, as stated elsewhere in this paper, policies to promote SEB uptake in the form of preferential reimbursement have been adopted by the Canadian provincial reimbursement bodies. However, without reducing barrier to access, e.g. removal of prior authorization, or allowing interchangeability, the full potential of SEBs in providing true cost savings to public plans may be limited.
The SEB regulatory and reimbursement experience in Canada remain in its infancy. Although substantial economic benefits can be achieved with the use of SEBs, their wider acceptance by payers, prescribers and patients remain to be seen, especially with the first mAb, i.e. Inflectra. More support is needed in order to allow stakeholders to fully comprehend the concept of SEBs so that these therapies can be properly evaluated and utilized.
The authors would like to acknowledge Mr Ferg Mills, MSc, Principal Consultant, Wyatt Health Management, for his valuable input into the preparation of this article. A portion of the information contained in this article has previously been presented at ISPOR North America (2015, 2016) and CADTH Symposium (2016).
Competing interests: Dr Eric Siu is employed by Wyatt Health Management (WHM) and Mr George Wyatt is the owner of WHM. WHM has worked with one or more companies that manufacture the biosimilars listed in this publication. However, WHM did not receive funding or use proprietary information from these sources for the preparation of this manuscript.
Provenance and peer review: Not commissioned; externally peer reviewed.
George Wyatt, BSc, MBA
Wyatt Health Management, Suite 201, 7025 Tomken Road, Mississauga, Ontario L5S 1R6, Canada
References
1. Health Canada. Fact sheet: subsequent entry biologics in Canada [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/activit/fs-fi/fs-fi_seb-pbu_07-2006-eng.php
2. Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol. 2010;10(5):297.
3. Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301-16.
4. Wallia A, Molitch ME. Insulin therapy for type 2 diabetes mellitus. JAMA. 2014;311(22):2315-25.
5. Morgan S, Smolina K, Mooney D, Raymond C, Bowen M, Gorczynski C, et al. The Canadian Rx Atlas. 3rd ed. Vancouver: UBC Centre for Health Services and Policy Research; 2013.
6. Canadian Agency for Drugs and Technologies in Health. Ndegwa S, Quansah K. Subsequent entry biologics – emerging trends in regulatory and health technology assessment frameworks. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/sites/default/files/pdf/ES0284_SEBs_es_e.pdf
7. Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs). 2010 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/seb-pbu/seb-pbu_2010-eng.php
8. Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6(8):469-78.
9. Health Canada. HPaF. Draft – revised guidance document: information and submission requirements for subsequent entry biologics (SEBs). 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/consultation/biolog/submission-seb-exigences-pbu-eng.pdf
10. Health Canada. Consultation on the revised guidance document: information and submission requirements for subsequent entry biologics (SEBs). 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/consultation/biolog/consult-submission-seb-exigences-pbu-eng.php
11. European Medicines Agency. Scientific advice and protocol assistance. 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.ema.europa.eu/ema/index.jsp%3Fcurl%3Dpages/regulation/general/general_content_000049.jsp%26 mid%3DWC0b01ac05800229b9
12. Canadian Agency for Drugs and Technologies in Health. CADTH Common Drug Review (CDR). 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/about-cadth/what-we-do/products-services/cdr
13. Institut National d’Excellence en Santé et en Services Sociaux. About the institute. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.inesss.qc.ca/en/about-us/about-the-institut.html
14. Canadian Agency for Drugs and Technologies in Health. CADTH pan-Canadian oncology drug review. 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/pcodr
15. Canadian Agency for Drugs and Technologies in Health. Common Drug Review: CDR update – Issue 92. 2013 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/cdr-update-issue-92
16. Canadian Agency for Drugs and Technologies in Health. Common Drug Review: CDR update – Issue 100. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/sites/default/files/cdr/filing/templates-new/cdr_Procedure_Submission_Guidelines_Subsequent_Entry_Biologics.pdf
17. Sehgal C. Biologics: what does the future hold? [cited 2016 Sep 1]. Available from: http://www.slideshare.net/CADTH-ACMTS/cadth-biologics-csehgaljune2014
18. Canadian Agency for Drugs and Technologies in Health. Common Drug Review. CADTH Canadian Drug Expert Committee final recommendation. Grastofil. 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/sites/default/files/cdr/complete/SE0446_cdr_complete_Grastofil_March-22-16_e.pdf
19. Canadian Agency for Drugs and Technologies in Health. Common Drug Review. CADTH Canadian Drug Expert Committee final recommendation. Basaglar. 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/sites/default/files/cdr/complete/SE0451_complete_Basaglar-Apr_19-16-e.pdf
20. Council of the Federation. The Council of the Federation: Pan-Canadian Pharmaceutical Alliance: Pan Canadian drug negotiations report. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.pmprovincesterritoires.ca/phocadownload/pcpa/pan_canadian_drugs_negotiations_report_march22_2014.pdf
21. Canadian Intergovernmental Conference Secretariat. Press release – provinces and territories talk health care 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.scics.gc.ca/english/conferences.asp?a=viewdocument&id=2217
22. Council of the Federation Secretariat. The Council of the Federation. Pan-Canadian Pharmaceutical Alliance: Scope of pan-Canadian Pharmaceutical Alliance 2014 [cited 2016 Sep 1]. Available from: http://www.pmprovincesterritoires.ca/phocadownload/pcpa/scope_of_pcpa_process_dec_2014.pdf
23. Pan-Canadian Pharmaceutical Alliance. Subsequent Entry Biologics (SEBs) First principles. Council of the Federation Secretariat; 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.pmprovincesterritoires.ca/phocadownload/pcpa/2016/seb_first_principles_20160401.pdf
24. Sandoz Canada Inc. Product monograph: Omnitrope, somatropin [rDNA origin] for injection. 2013.
25. Klein AV. The first subsequent entry biologic authorized for market in Canada: the story of Omnitrope, a recombinant human growth hormone. Biologicals. 2011;39(5):278-81.
26. Health Canada. Biologics and Genetic Therapies Directorate. Summary basis of decision (SBD). Omnitrope. 2009 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2009_omnitrope_113380-eng.php
27. Canadian Agency for Drugs and Technologies in Health. Common Drug Review. CEDAC final advice-subsequent entry biologic. Omnitrope. 2009 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/media/cdr/advice/cdr_advice_Omnitrope-December-18-2009.pdf
28. Health Canada. Biologics and Genetic Therapies Directorate: Summary basis of decision. Remsima. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_remsima_160195-eng.php
29. Health Canada. Biologics and Genetic Therapies Directorate: Summary basis of decision. Inflectra. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php
30. Norton Rose Fulbright. Wall K. Pharma in brief – Appeals filed for quashed subsequent entry biologic infliximab NOC. 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.nortonrosefulbright.com/knowledge/publications/127641/pharma-in-brief-appeals-filed-for-quashed-subsequent-entry-biologic-infliximab-noc
31. European Medicines Agency. Assessment report. Remsima. EMA/CHMP/589317/2013. 27 June 2013 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002576/WC500151486.pdf
32. European Medicines Agency. Assessment report. Inflectra. EMA/CHMP/589422/2013. 27 June 2013 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002778/WC500151490.pdf
33. Feagan BG, Choquette D, Ghosh S, Gladman DD, Ho V, Meibohm B, et al. The challenge of indication extrapolation for infliximab biosimilars. Biologicals. 2014;42(4):177-83.
34. Lee H. Is extrapolation of the safety and efficacy data in one indication to another appropriate for biosimilars? AAPS J. 2014;16(1):22-6.
35. Health Canada. Notice of compliance, Inflectra. 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://health-products.canada.ca/noc-ac/info.do?no=18169&lang=en
36. Canadian Agency for Drugs and Technologies in Health. Common Drug Review, Submission Status Report, Infliximab (Remsima). 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/infliximab-17
37. Canadian Agency for Drugs and Technologies in Health. Common Drug Review. CDEC final recommendation. Infliximab (Inflectra). 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/media/cdr/complete/cdr_complete_SE0384_Inflectra_Dec-23-14.pdf
38. Council of the Federation. Pan-Canadian Pharmaceutical Alliance: completed negotiations. As of 31 December 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from:. http://www.pmprovincesterritoires.ca/phocadownload/pcpa/pcpa_completed_negotiations_dec31_2015.pdf
39. Canadian Agency for Drugs and Technologies in Health. Common Drug Review. Subsequent Entry Biologic Review Report. Inflectra. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/infliximab-18
40. Kitamura M. Bullied in Norway, Merck sees sales of blockbuster dive. 22 April 2015. Bloomberg. [cited 2016 Sep 1]. Available from: http://www.bloomberg.com/news/articles/2015-04-22/merck-gets-bullied-in-norway-with-remicade-price-war
41. IHS. Benassi F. Norway’s discount on infliximab: a litmus test for biosimilar expansion in Europe? 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://healthcare.blogs.ihs.com/2014/03/20/norways-discount-on-infliximab-a-litmus-test-for-biosimilar-expansion-in-europe/
42. Institut National d’Excellence en Santé et en Services Sociaux du Québec. Avis au Ministre de l’Institut National d’Excellence en Santé et en Services Sociaux – pour la mise á jour des listes de médicaments du 2 février 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Fevrier_2015/_AvisMinistre_Innovateurs_WEB_2015_02.pdf
43. Régie de l’assurance maladie du Québec. List of Medications. 2015.
44. GaBI Online – Generics and Biosimilars Initiative. Biosimilars of filgrastim aplasia [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Sep 1]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-of-filgrastim
45. Apotex. Apotex announces FDA has accepted for filing its biosimilar application for filgrastim (Grastofil™). 2015 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.apotex.com/global/about/press/20150217-2.asp
46. The Food and Drug Law Institute. Norman J, Aumand L. Subsequent entry biologics in Canada. Gowling Lafleur Henderson LLP. 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.fdli.org/docs/canadian/john-norman-subsequent-entry-biologics-in-canada.pdf?sfvrsn=0
47. Canadian Agency for Drugs and Technologies. TBC/etanercept: Common Drug Review Project Status Report. 2016 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: https://www.cadth.ca/etanercept
48. Telus Health. Prior Authorization Program Reimbursement Request Form. For biologic response modifier: Remicade (infliximab). 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.telushealth.com/docs/prior-authorization-forms/remicade_en.pdf
49. Preferred pricing for Remicade® coming in 2015. Manulife Financial.
50. Grabowski D, Henderson B, Lam D, Keystone EC, Thorne C, Jamal S, et al. Attitudes towards subsequent entry biologics/biosimilars: a survey of Canadian rheumatologists. Clin Rheumatol. 2015;34(8):1427-33.
51. Ontario Rheumatology Association. Ontario Rheumatology Association: position paper on subsequent entry biologics. 2012 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://ontariorheum.ca/publications/other-publications.
52. Canadian Rheumatology Association. Canadian Rheumatology Association: position paper on the establishment of a Common Drug Review (CDR) procedure and process for reviewing subsequent entry biologics (SEBs). 2013 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://rheum.ca/images/documents/SEBs_Position_Paper.pdf
53. Canadian Dermatology Association. Canadian Dermatology Association position statement: biosimilars. 2013 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.dermatology.ca/wp-content/uploads/2013/09/Biosimilars-WEB-EN-2013.pdf
54. Devlin SM, Bressler B, Bernstein CN, Fedorak RN, Bitton A, Singh H, et al. Overview of subsequent entry biologics for the management of inflammatory bowel disease and Canadian Association of Gastroenterology position statement on subsequent entry biologics. Can J Gastroenterol. 2013;27(10):567-71.
55. Canadian Arthritis Patient Alliance. Subsequent entry biologics: a position statement (27 August 2014) [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.arthritispatient.ca/files/7514/0918/2653/CAPA_SEB_Eng.pdf
56. The Arthritis Society. The Arthritis Society – position paper: Access to medication:subsequent entry biologics (SEBs). 2014 [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://arthritis.ca/getmedia/0f6db5a6-2a32-4199-b964-acdf0a9f68bd/SEBs-Position-Paper-FINAL-EN.pdf
57. Leveling the field in Canada 2nd edition: moving toward reimbursement equality in biologic therapy for Canadians with rheumatoid arthritis. Vancouver: BC; Arthritis Consumer Experts; 2012.
58. Russell AS, Ahluwalla V, Barnabe C, Jamal S, Offer RC, Olszynski WP, et al. Subsequent entry biologics/biosimilars: a viewpoint from Canada. Clin Rheumatol. 2012;31(9):1289-92.
59. Crohn’s and Colitis Canada. Mistry M. What you need to know about subsequent entry biologics [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.crohnsandcolitis.ca/site/c.dtJRL9NUJmL4H/b.9013439/apps/s/content.asp?ct=13707985
60. Canadian Skin Patient Alliance. The CSPA position on subsequent entry biologics [homepage on the Internet]. [cited 2016 Sep 1]. Available from: http://www.canadianskin.ca/en/education/subsequent-entry-biologics/our-position-on-sebs
61. BIOTECanada. Speaking with one voice: important considerations for SEB policy development. 2014 Jul 29.
62. Sensabaugh SM. Biological generics: a business case. J Generic Med. 2007;4(3):186-99.
63. Morelli G. Biosimilars: evolution and trends. Spanish Generic Medicines Association (AESEG); 2013; Madrid, Spain.
64. Walsh K. Biosimilars’ utilization and the role payers do play in driving uptake in Europe: an industry perspective. 11th EGA International Symposium on Biosimilar Medicines; 25-26 April 2013; London, UK.
65. Spotlight on: biosimilar Enbrel adopted rapidly in Norway; can Europe’s pacesetter share its positive experience with other countries? FirstWord Pharma. [cited 2016 Sep 1]. Available from: https://www.firstwordpharma.com/node/1380212
66. Barry F. Regulators: skip clinical data and extrapolate biosimilar indications. BioPharma. 2015 Apr 29.
|
Author for correspondence: Eric CK Siu, MSc, PhD, Director, Scientifi c Aff airs, Wyatt Health Management, 7025 Tomken Road, Suite 201, Mississauga, Ontario L5S 1R6, Canada |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/current-state-of-subsequent-entry-biologics-biosimilars-in-canada-a-view-from-regulatory-reimbursement-clinician-and-patient-perspectives.html
Author byline as per print journal: Eric CK Siu1,4, MSc, PhD; Anne Tomalin2,4, BA, BSc; Kevin West3,4, BA; Sandra Anderson4, BA, MBA; George Wyatt1,4, BSc, MBA
|
Abstract: |
Submitted: 18 July 2019; Revised: 21 August 2019; Accepted: 21 August 2019; Published online first: 23 August 2019
According to the latest report on Canadian prescription drug spending published by the Canadian Institute for Health Information (CIHI), public drug plan and private-sector spending was forecasted to reach CA$33.7 billion in 2018, an annual increase of ~4.3% [1]. In Canada, drug spending is typically split as public (~40%), private (~40%), and out-of-pocket expenses, e.g. premiums, deductibles: (~20%).
Biologicals are commonly prescribed therapies for the treatment of conditions ranging from diabetes to cancer [2–4], many of which are considered high-cost drugs. In 2017, public drug programmes spent CA$13.5 billion on prescription drugs, and biologicals accounted for 21.6% of that spending [1]. Antitumour necrotic factor (anti-TNF) drugs accounted for the highest proportion of public drug programme spending for the sixth consecutive year at 8.2% (nearly 38% of all biologicals), and were the fifth highest contributor to spending growth; however, anti-TNF drugs were only used by 0.5% of public plan beneficiaries [1]. In the private sector, seven of the top 10 selling high-cost drugs in 2017 were biologicals, with anti-TNF drugs accounting for 11% of the total drug cost [5].
The introduction of biosimilars (previously known as subsequent entry biologics in Canada) provides cost-savings to both public and private drug plans and individuals in an era of financial constraint. According to Canadian Agency for Drugs and Technologies in Health (CADTH), biosimilars of 15 reference biologicals have been launched, or are expected to be launched by 2020 [6]. Furthermore, many are biosimilars of the same reference biological, thus there is a potential for intense competition in the biologicals space. For example, Health Canada has approved/is reviewing seven trastuzumab and two adalimumab biosimilars.
Since our last review, which covered the biosimilars launched in Canada to 2016, Omnitrope® (somatropin), Inflectra® (infliximab), Grastofil® (filgrastim), and Basaglar® (insulin glargine) [7], significant changes have occurred in the Canadian biosimilar environment. Of particular note are the changes in the regulatory and the health technology assessment (HTA) domains, many of which are designed to promote the uptake of biosimilars. This manuscript will highlight these new developments.
The initial Canadian regulatory guideline for biosimilars was published in 2010, shortly after Health Canada completed the review of the first biosimilar, Omnitrope. This review process provided the agency with the opportunity to gain the experience needed to validate the concepts and principles behind biosimilars [8]. Subsequently, in December 2015, Health Canada issued a stakeholder consultation process for a revised biosimilar guideline which was finalized in November 2016 [9]. Some of the key changes between the initial and new guideline were discussed in our previous review [7].
In addition to the new guideline, in September 2015, Health Canada also launched a three-year Scientific Advice Meeting pilot programme to provide drug manufacturers with the ability to discuss their biosimilar with Health Canada early in the development process [10]. However, at the time of this manuscript preparation, Health Canada had not provided any updates regarding this pilot project.
Regulatory Review of Drugs and Devices Initiative
Realizing the rapidly changing healthcare system, Health Canada recognized the need for a regulatory system that is more efficient, that supports the timely access to therapeutic products, and that allows better linkages within the healthcare system as a whole [11, 12]. As a result, Health Canada laid out a Regulatory Review of Drugs and Devices (also known as the lsquo;R2D2’) initiative in 2017 with a series of objectives: i) providing more timely access to drugs and devices, including orphan drugs/drugs for rare diseases; ii) making better use of real-world evidence to support regulatory decisions across a product’s life cycle for both drugs and medical devices; iii) modernizing its operation by increasing transparency and reducing tax-payer burden; and iv) implementing a plan to increase collaboration with both Canadian and international partners [11]. The last objective is particularly relevant for reimbursement: one of the key action items is for Health Canada to work with HTA organizations to reduce the time between regulatory approvals and public plan reimbursement recommendations (HTA is further discussed in the next section). Other action items within this collaboration objective includes Health Canada, and other Canadian health partners, working with international regulators to explore the idea of joint reviews, or the use of foreign reviews, to increase efficiencies and expertise in the review process, and to support access to products otherwise not available in Canada [11].
With the expected increase in new therapies seeking regulatory approval, Health Canada has created several projects that will facilitate the access of these products.
First, one such project was designed specifically to improve access to biologicals (biosimilars and non-biosimilars) by increasing the regulatory review capacity (expected to be fully in place as of the preparation of this manuscript). Ultimately, it is intended that the increased reviewing capacity will lead to a more secure supply of biological drugs with more than one manufacturer, more affordable biological drugs, and earlier access to new treatment options [13].
Second, to decrease the time between regulatory review to reimbursement, Health Canada and HTA organizations (CADTH [including participation by all Canadian federal, provincial, and territorial jurisdictions except the province of Québec; and INESSS in Québec) began to collaborate in 2018 in an aligned review process for all biological and pharmaceutical new drug submissions where the manufacturer intends to seek HTA process on a pre-NOC (Notice of Compliance) basis [14, 15]. This process initially also includes submissions for biosimilars, and new indications for existing drugs made to Health Canada. Prior to this initiative, manufacturers had the option to submit to the CADTH Common Drug Review (CDR) within 90 calendar days [16] prior to the anticipated date of receiving regulatory approval, i.e. pre-NOC submission. However, as part of the alignment process between Health Canada and the HTA organizations, a manufacturer has the opportunity, but not the obligation, to submit to CDR up to six months (or 180 days) before the anticipated date of regulatory approval [16, 17]. The CADTH pan-Canadian Oncology Drug Review (pCODR) has allowed a 6-month pre-NOC submission since its inception, reflecting the seriousness of the therapeutic area and the generally unmet need of improving survival of cancer patients. It is expected that the alignment of Health Canada review and HTA process will reduce duplication and reduce time lags between regulatory approval and reimbursement recommendation [14]. It is important to note that this process, in essence, only lengthens the pre-NOC submission threshold to HTA organizations: the duration of review by Health Canada and HTA organizations do not change and manufacturers must still remain compliant with all submission requirements. The typical Health Canada review timeframe for new drugs or new indications ranges from a target of 180 days for priority review to 300 days for a standard review. Finally, as part of the objective to improve transparency, products that have opted-in to the aligned review process as of 1 October 2018 would be publicly noted as such. Although this process is currently operational, it is expected that all parts of this project will be fully in place by Spring 2020 [18].
Third, Health Canada has also endeavoured to work with HTA organizations to provide concurrent scientific advice to manufacturers early in the drug development process. Currently, Health Canada and HTA organizations work independently to give scientific advice to drug manufacturers. Through this process, it is expected that Health Canada and the HTA organizations will help manufacturers to produce more relevant data from clinical trials to inform better decisions. As a result, this change has the potential to reduce access time to important medications. It is expected that all parts of this project will also be fully in place by Spring 2020 [19].
Initiatives taken to improve and hasten access to new medications are not unique to Canada. For example, European health authorities are becoming far more proactive with funding decisions for new medicines by starting activities well before likely a European Medicines Agency (EMA) approval is anticipated [20–22].
Naming of biologicals (biosimilars and non-biosimilars)
In early 2018, Health Canada and the Institute for Safe Medication Practices (ISMP) Canada conducted an online consultation on the naming of biological drugs. Several options where provided for discussion, including the random four-letter suffix convention that was instituted by the US Food and Drug Administration (FDA). Feedback from 362 respondents (62% from pharmacists and pharmacy-related organizations) showed that one-half do not accept the four-letter suffix option, and more than two-thirds found the current naming convention (brand name plus unique Drug Identification Numbers [DINs]) to be unacceptable for biologicals. Three-quarters of the respondents supported an updated convention of including both brand name and International Nonproprietary Name (INN), citing that this naming option would be compatible with their current practice or environment; would clearly identify products for prescribing, dispensing and tracking of adverse events (AEs); and would reduce the likelihood of confusion and errors for healthcare providers and patients [23]. As a result of the feedback, Health Canada intends to implement an updated naming convention for biologicals that include the product’s brand name, INN, and DINs [24]. It is expected that the updated naming convention will support the clear distinctions between biological drugs for the purpose of AEs tracking.
Public drug plans make extensive use of HTA recommendations as part of the reimbursement decision-making process. Eligible non-cancer prescription drugs approved by Health Canada are reviewed by CDR and INESSS [25, 26], and Health Canada approved cancer drugs are reviewed by pCODR and INESSS [26, 27]. Both the CADTH CDR and pCODR provide reimbursement recommendations that are considered by participating public drug plans outside of Québec, and INESSS makes recommendations to the Québec Government for drug reimbursement in that province. INESSS has extended its collaboration with CADTH through the Health Canada aligned drug review process [15].
In 2018, CADTH initiated new streamlined processes associated with CDR and pCODR that are a part of the CADTH 2018–2021 Strategic Plan [28]. However, in 2019, a significant change took place with regards to the role of CADTH in biosimilar reimbursement [29]. These changes are discussed below.
Common Drug Review (CDR)
CADTH released the first official CDR submission guidelines for biosimilars in March 2014 in anticipation of a new wave of biosimilars entering the Canadian market [7, 30]. This official guideline detailed extensive requirements for a biosimilar submission reflecting the cautious position that it took in the assessment of these products at that time.
CADTH’s approach towards biosimilars changed in 2018 with new submission requirements for biosimilars, as well as a new recommendation framework. In the summer of 2017, CADTH conducted a consultation with stakeholders to update the existing biosimilar evaluative process (CDR Update – Issue 125, released 1 August 2017) [31]. CADTH noted that Health Canada evaluated and approved biosimilars (and their extrapolated indications) based on the totality of evidence demonstrating similarity to the reference biologicals. As such, CADTH felt that a streamlined process would reduce duplication, optimize resources, and support the timely review of biosimilars. The key elements in the proposed revised procedure included a substantially reduced submission package; reduced submission fee; ineligibility for the optional pre-submission meeting; reduced evaluation timeframe (reduced to the targeted 44 to 53 business days from the typical ~180 calendar days); and the lack of deliberation of the dossier by the CDR Canadian Drug Expert Committee (CDEC) and therefore, no CDR recommendation issued, e.g. reimburse, do not reimburse, reimburse with criteria/condition, which is standard for most other submissions [32]. In place of the recommendation, a review record would be posted. Following the consultation process, a new CDR guideline for biosimilars was issued in February of 2018 [33].
Table 1 lists the biosimilar products that have been reviewed by the CDR. Between 2014 and 2017 there were very few submissions (two per year, and one was withdrawn in 2014), and each submission was associated with approximately six months of review time. In 2018, with the introduction of the streamlined process, there were four biosimilar submissions, and the evaluation time was reduced to approximately two months.
In a substantial change in mandate, CADTH announced in May 2019 that it would no longer review biosimilars via its CDR and pCODR (see below) programmes as of 1 June 2019 [29]. This also applied to biosimilar products where the review had been completed after 1 June 2019. CADTH’s reasoning for this decision included the fact that the biosimilar review process may delay access to new biosimilar treatments. In addition, the resources currently devoted to reviewing biosimilars could be diverted to other drug reviews and programme-related issues. CADTH also indicated that the organization’s workload has increased significantly and that biosimilar reviews do not add a lot of value for public drug plans. However, in rare circumstances, e.g. the reference product was not reviewed by CADTH or a drug plan, a CADTH review may still be required for a biosimilar (considered on a case-by-case basis with the drug plans). Although CADTH stated it would no longer review biosimilar submissions, it indicated that manufacturers should still submit their products to the public jurisdictions and the pCPA [29].
Considering public jurisdictions would potentially need to conduct a more thorough review of biosimilars in the absence of a CADTH assessment, it is unclear how much quicker biosimilars would actually reach patients.
pan-Canadian Oncology Drug Review (pCODR)
The CADTH pCODR was much less active compared to the CDR in terms of biosimilar guideline development. This may have been due to the lack of oncology biosimilars entering the Canadian market at the outset. Indeed, the original pCODR guideline for biosimilars was not published until early 2018, at the same time as the revised CDR guideline [34]. Overall, despite the nature of the oncology therapeutic area, the formal submission requirements for oncology biosimilar molecules are highly similar to those for non-oncology biosimilar molecules. At the time of preparation of this manuscript, three biosimilars have been submitted to pCODR, see Table 1: Ogivri®, the BGP Pharma ULC (Mylan) trastuzumab biosimilar to Roche’s Herceptin®, currently under review; Mvasi®, the Amgen bevacizumab biosimilar to Roche’s Avastin®, listed by pCODR; and Pfizer’s bevacizumab.
Mvasi represents an interesting case: it was submitted for two of the Avastin approved indications (non-small cell lung cancer [NSCLC] and metastatic colorectal cancer [mCRC]); however, the Avastin approved indication for NSCLC was not recommended for reimbursement by the interim Joint Oncology Drug Review (iJODR; a precursor to pCODR) by public plans [35]. This was likely due to the lack of perceived therapeutic value as previously reported by INESSS on multiple occasions [36, 37]. In accordance to the updated CADTH guidelines, pCODR did not make a recommendation regarding the reimbursement of Mvasi for either indication. This effectively leaves the funding decision solely to the public payers. Mvasi is currently undergoing pCPA negotiation and it remains to be seen whether Mvasi will be funded for the NSCLC indication. As noted in the previous section, pCODR will no longer review biosimilars as of June 2019.
Institut national d’excellence en santé et en services sociaux (INESSS)
In Québec, INESSS has also instituted a significant change to the HTA process for biosimilars. Unlike CDR, INESSS did not have a biosimilar-specific guideline in place when the first biosimilars came to market. It was not until 2017 that INESSS developed its first set of requirements specific to biosimilars, although they were similar to other new drugs or drugs with new indications. In alignment with CDR, INESSS issued a set of streamlined requirements for biosimilars in 2018, which included the ability for manufacturers to submit to INESSS six months pre-NOC [17].
INESSS’ attitude on biosimilars has been historically more divergent compared to CDR. It was the first provincial jurisdiction to provide what could be considered a landmark positive recommendation on the reimbursement of the infliximab biosimilar, Inflectra [7]. On the other hand, INESSS originally issued a negative recommendation on the reimbursement of the filgrastim biosimilar, Grastofil, due to therapeutic value, despite Health Canada approval and the positive CDR recommendation [38, 39]. The impact of this recommendation on commercialization cannot be underestimated as the province of Québec accounts for nearly one-quarter of the Canadian population. Ultimately, Grastofil was recommended by INESSS for reimbursement in a subsequent evaluation well over one year later [40].
With the streamlined requirements, INESSS also released a new formal evaluation framework. Overall, the framework is in alignment with that of CADTH [41]. As of this writing, all biosimilar products that have received positive CDR recommendations have also received positive recommendations by INESSS. At the time of this writing, INESSS still requires submissions for biosimilars. It is expected that INESSS will continue to review biosimilars, but apply a more streamlined process, which may more closely follow the review it does for generic drugs.
The pCPA was formally established in 2010 with the goal of combining purchasing power for public drug plans to improve access and increase cost-effectiveness of medications. It is interesting to note that several European countries are also attempting to combine multinational purchasing powers to reduce drug cost [42, 43].
In 2016, the pCPA issued the Subsequent Entry Biologics (SEBs) First Principles with the goal of developing a more comprehensive biosimilar policy framework [7, 44].
Since the release of the First Principles, the pCPA completed several negotiations involving biosimilars and received feedback on the First Principles from various stakeholders. As a result, the pCPA developed a series of options that could be considered for inclusion in the policy directions and processes that would govern how negotiations for biologicals would proceed. These options were presented in a consultation meeting that took place in April 2017, with the goal of receiving additional feedback from stakeholders. Taking into consideration the responses received from the consultations, the pCPA developed and released a set of Biologics Policy Directions that applied to pCPA negotiations in late 2018 [45]:
Of note is point 3, negotiation for biosimilar drugs will begin in parallel with the HTA process. INESSS is still reviewing biosimilars, which means that the pCPA will conduct negotiations considering the HTA process. This may change as time goes on, and pCPA has advised that it is currently reviewing their policy. Another notable Policy Direction which has significant commercial impact relates to tiered listings (point 7). As per the pCPA, where a number of biological drug products exist in a therapeutic space, those biological drugs may be tiered, i.e. authorization for some name-brand biological drug products will be based on evidence that other products have used first, at the discretion of the reimbursing drug plan. At least one public jurisdiction has formally laid out a tiered biologicals reimbursement policy, the details of which are discussed in the next section. Whether increasing discounts will be implemented based on the number of existing biosimilars currently reimbursed, a model similar to traditional generics products, remains to be determined as more and more manufacturers enter the market with the same biosimilar. Furthermore, British Columbia has just announced a switching programme (point 8). This is discussed in the Provincial/Federal Formularies section.
Overall, the above set of Policy Directions guides the negotiations between drug manufacturers and the pCPA with the goal lsquo;to develop and pilot a clear and consistent pan-Canadian approach that encourages appropriate use of biologicals in support of a common pCPA mandate to enhance patient access to clinically relevant and cost-effective drug treatment options’ [45]. However, policies beyond those associated with the pCPA, e.g. appropriate use of biologicals and uptake of biosimilars, will need further engagement with stakeholders beyond the pharmaceutical industry, such as patients and clinicians. Furthermore, implementation of the negotiated agreements between manufacturers and jurisdictions still remains subject to jurisdictional legislation and regulation, i.e. separate contracts are applied in each jurisdiction [45].
Partnership with Cancer Care Ontario
The Biologics Policy Directions & pCPA Negotiations paper applies to all biosimilars, non-oncology and oncology. However, the adoption of biosimilars in the oncology setting requires multiple considerations to maintain high quality care and patient outcomes, and provinces differ in their policy, reimbursement and delivery of cancer services in ways that may affect the consistent implementation of biosimilars across the country. Consequently, as part of its biosimilars strategy, the pCPA partnered with Cancer Care Ontario to develop the pan-Canadian Oncology Biosimilars Initiative in order to promote the uptake of biosimilars for cancer treatment and to ensure that the implementation and use of therapeutic oncology biosimilars are appropriate and cost-effective across Canada. To kick-start this initiative, a pan-Canadian Oncology Biosimilars Summit was held in November of 2018 [46]. Learnings from the implementation of biosimilars in other jurisdictions and therapeutic areas were shared with participants (patients, patient advocacy organizations, clinicians, healthcare administrators, and government officials) from across Canada. Based on the feedback received from the summit, the vision, goals, and revised strategic objectives of the Initiative have been clearly delineated, see Figure 1 [46]. The strategic objectives informed the development of an Action Plan, which provides an overview of the anticipated pre-implementation steps to ensure the successful adoption of oncology biosimilars [46, 47]. Some of the pre-launch activities are already underway [47].
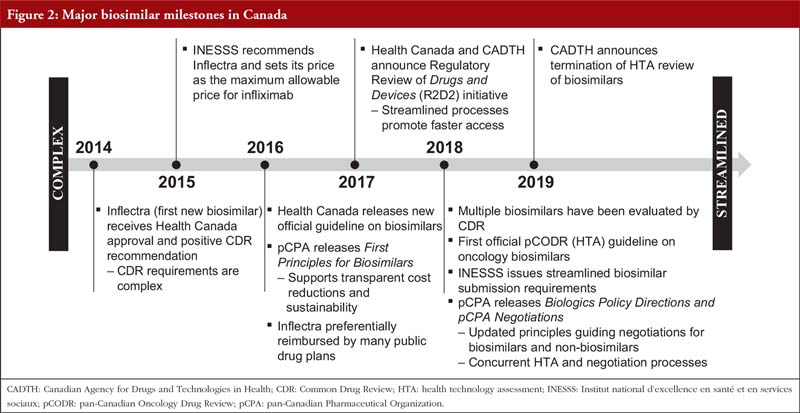
The Canadian milestones for biosimilars in the context of Health Canada, CADTH, INESSS and pCPA are summarized in Figure 2. Overall, it can be seen that the journey of biosimilar products has evolved from a complex and time-consuming process, to a far more streamlined and efficient process. It is also evident that along with these changes, the time from regulatory filing to completion of pCPA negotiations could be substantially shortened for biosimilars (by approximately one year, see Figure 3). However, in order to realize the significant advantage offered by the new streamlined processes, submitters of biosimilars need to engage in this highly efficient process, which requires a keen understanding of all facets of the components involved from regulatory filing to pricing negotiations. The selection of a knowledgeable, integrated consultancy can provide significant benefits to the marketers of biosimilars that could result in accelerated speeds to market for their products.
CDR/pCODR-participating jurisdictions typically rely on CDR/pCODR to provide HTA processes and recommendations of new pharmaceutical products (or new indications) including biosimilars, for their reimbursement decisions. In light of the change made of CADTH, manufacturers have to submit to these jurisdictions directly. This, although considered by CADTH to promote faster access to biosimilars, may require additional resources by both the public drug plans and the biosimilar manufacturer since the drug plans have additional requirements (and sometimes inter-jurisdictional differences) for products that are not eligible for CDR/pCODR assessments. It is unclear if public drug plans will institute (or revise, in British Columbia’s case) a separate set of processes for biosimilars. It is likely that we will see more developments in this space over the next year.
In an unprecedented move, British Columbia (BC), Canada’s most western province, launched its Biosimilars Initiative on 27 May 2019 that promotes switching among a number of biologicals and biosimilars [48–50]. Specifically, in phase 1 of this initiative, patients that are on a select reference product/indication should be switched to the biosimilar during the transition period. At the end of the transition period, originator products would only be covered on a case-by-case basis. The Initiative is designed to increase uptake of biosimilars, which has lagged behind that of other OECD (Organization for Economic Co-operation and Development) nations. Additional comments on uptake are found in the next section. The savings associated with the use of biosimilars have been, and will be, used to lower premiums and co-pays for some BC residents and to provide budget room for new drugs. Although BC is currently the only province to require switching, the impact of this decision will be significant as the remaining provinces will likely follow suit. From the patients’ perspective, a careful coordination between prescribing physicians, pharmacists, and patient support programmes is necessary to ensure patients receive timely, as well as equal, services and access to the prescribed molecules in the form of biosimilars as with the originators.
Manitoba was the first Canadian province to publish a Tiered Biologics Reimbursement Policy [51]. The policy classifies molecules into Tier 1 (biosimilars or biologicals for which no biosimilars exist), and Tier 2 (biologicals for which no biosimilars exist), for any given indication, and also provides prescription restrictions. As an example, according to this policy, all biological-naïve patients must first be prescribed a Tier 1 product, and if the therapy fails, must be prescribed a second Tier 1 product. Only after the second failure can a Tier 2 product be prescribed. Beyond this tier classification, there is no preferential reimbursement of one biosimilar over another where multiple products of the same molecule exist. An important distinction here is that these products are all antibody-based molecules, and no switching is allowed between the reference biological and the biosimilar (or between different biosimilars) of the same molecule at this time. Other jurisdictions also have similar reimbursement schemes where biosimilars are preferentially reimbursed over the reference biologicals.
It is important to note that the Biosimilar Initiative for BC is not the same as automatic substitution or interchangeability. Currently, no jurisdictions have implemented policies to support interchangeability/automatic substitution. FDA has recently issued the finalized guidance for demonstrating the interchangeability of biosimilars with their reference products [52]. However, policy efforts for biosimilar and interchangeable products in the US are still ongoing [53]. It is likely that interchangeability will not play a role in Canada until the US finalizes its policy.
Estimates provided by Canada’s Patented Medicine Prices Review Board (PMPRB) suggested that in the third year following biosimilar entry, potential savings could range between 13%–43% for acute-use products, e.g. filgrastim, epoetin alfa, and 8%–43% for chronic-use products, e.g. infliximab, adalimumab [6]. The low estimates represent an average-uptake, low-discount scenario, and the high estimates represent a high-uptake, high-discount scenario [6]. Based on the latest publicly available list prices, the discounts offered by biosimilars launched post-2010 relative to reference products range from 17% (Grastofil) to 50% (Renflexis), see Table 2, although it is unclear what the confidential prices are for the reference biologicals. Interestingly, the first biosimilar, Omnitrope (launched several years before Inflectra), had prices that ranged from more costly to less costly relative to the reference product, Genotropin, see Table 2. This could be due to a drop in the price of the reference product. Despite the potential to generate significant savings, the uptake of biosimilars (with the exception of Grastofil) in Canada have been modest [6, 54–57]. Indeed, Canada is significantly behind in terms of adopting biosimilars compared to many OECD countries, e.g. the adoption rate for biosimilar infliximab was 1% in Canada in 2016 versus 82% in Norway, and 90% in Denmark in 2015 [46].
CADTH published a report in October 2018 regarding the mechanisms that various international jurisdictions utilize to promote biosimilar uptake, which can be broadly categorized into supply-side policies and prescribing incentives. Educational and awareness initiatives were also identified at the national or organization level to facilitate biosimilar utilization. Overall, no specific mechanisms that would support the uptake of biosimilars in Canada were proposed by CADTH due to a myriad of factors [58]. Additional information regarding policies for promoting biosimilar uptake can be found in a separate publication [59].
In a biosimilars forum hosted by the Institute of Health Economics (IHE) in March 2017, key stakeholders from the public and private sectors, as well as clinicians, academics, and patient and provider associations gathered to lsquo;understand options to categorize and consider biosimilars, as well as a process to engage stakeholders to identify place in therapy and further evidence development that may be required’, and to lsquo;identify an approach to knowledge exchange that will enable a common view and shared agreement amongst patients and clinicians regarding appropriate and intended use of biosimilars’, based on the biosimilar experience from NHS Scotland [55]. One of the take-aways from this forum that is relevant to increasing biosimilar uptake, revolved around the need to achieve a healthy co-existence and competition among all biological agents, which would help to facilitate cost-savings and achieve continued product innovation. Specifically, this could include the reduction in time to market for biosimilars and better mechanisms for fostering competition and achieve sustainable cost-savings. As discussed previously, processes continue to be streamlined, which could significantly decrease the time for biosimilars to reach patients. However, more appropriate mechanisms that manage the adoption/utilization of biosimilars (beyond preferential reimbursement) may be needed to achieve such an objective.
Another potential mechanism to promote uptake involves switching, and this relates to the issue of therapeutic equivalence, which was discussed during the pCPA consultations [45]. Overall, the industry is supportive of Health Canada’s position on switching, that is, the decision to switch a patient being treated with a reference biological to a biosimilar should be made by the treating physician in consultation with the patient; and should take into account available clinical evidence and any policies of the relevant jurisdiction. However, considering that current jurisdictions, i.e. provincial, territorial, and federal plans, do not have policies that support interchangeability for biosimilars (unlike oral generic drug products), and also have reimbursement criteria that do not clearly promote switching, the onus for this decision rests on physicians and patients. An example relating to the latter point can be seen from the Manitoba Tiered Biologics Reimbursement Policy [51]. Specifically, it is stated that patients cannot be switched from branded infliximab or etanercept products to other infliximab or etanercept products, i.e. biosimilars, if they have lsquo;previously trialed and deemed unresponsive to therapy’. What this means is that there is a potential opportunity to switch from a reference product to the biosimilar product, if patients are responsive to the reference product. A key challenge associated with the decision to switch from the reference product to a biosimilar is associated with the confidence of both physicians and patients. In the IHE forum, it was suggested that through NHS Scotland’s experience, therapeutic monitoring of outcomes, e.g. collecting real-world evidence, achieved with biologicals was key to physician and patient confidence. This is an important driver for acceptance and utilization of biosimilars, and for better and more appropriate utilization of biologicals in general, that should be considered and implemented as part of routine care rather than in the context of research [55]. Clearly, availability of real-world evidence not only increases physician/patient confidence; but could also be supportive of the pCPA Policy Directions and the pan-Canadian Oncology Biosimilars Initiative on switching [45–47]. It will also be supportive of the Health Canada Regulatory Review of Drugs and Devices (R2D2) initiative for real-world evidence to identify opportunities for enhanced use of real-world evidence throughout the drug life cycle [11, 60].
The availability of resources to support education and to provide services that support patient care, e.g. treatment optimization and adherence, are the typical initiatives of well-resourced reference biological manufacturers. Manufacturers, and even physicians, believe that molecules should be considered in combination with the added services that are offered when assessing value [45, 55], and that competition should not be solely based on the price of the molecule alone. Therefore, the availability of similar services by biosimilar manufactures should enable increased uptake of biosimilars [55, 56]. While the provision of these services may be feasible for reference biological manufacturers, this may not be the case for biosimilar manufactures for a variety of reasons. In response to this challenge, Biosimilars Canada, an industry association, has recently developed a centralized patient support service platform that allows biosimilar manufacturers to leverage common resources to provide the support services needed for their patients [61]. This could facilitate the market entry of additional biosimilar manufacturers and is a model other jurisdictions may consider to manage the challenges of bringing a biological to market in a competitive marketplace with barriers of entry set high by innovator offerings.
Substantial efforts are taking place to promote the uptake of biosimilars. The prime examples from international jurisdiction highlight the fierce competition for market share. In 2015, Orion offered a 69% discount for biosimilar infliximab compared to the originator Remicade’s tender price in Norway [62], which is far more substantial compared to the typical discount of 15%–30% for biosimilars [63]. Other manufacturers have provided significant discounts in other jurisdictions [64].
Private insurers have raised concerns about the ever-increasing number of high-cost drugs entering the market. The ability of plan sponsors to continue to provide coverage for high-cost drugs through private insurers is being challenged. For instance, Canada is second to the US in terms of the percentage of biologicals as a portion of all prescriptions, which increased from 8.3% in 2013 to 21.2% in 2017 [65]. With the launch of Inflectra in 2016, some private insurers provided preferential coverage of the biosimilar over the reference product for naïve patients [7]. Since that time, in what could be considered as a pivotal step for the Canadian private insurance industry, the private payer/adjudicator, Green Shield Canada, completed the pilot phase of a Biosimilar Transition Program, an initiative designed to allow for the voluntary switching of patients currently being treated with the reference products Remicade or Enbrel, to the corresponding biosimilar products. Development of this programme was based on emerging clinical evidence (including the landmark NOR-SWITCH study [66], and was done in consultation with a Canadian patient organization. Overall, 41% of participants voluntarily switched from the reference product to the biosimilar, and 26% stayed on the reference biological but were reimbursed at the biosimilar level. Nine per cent stayed on the reference biological due to valid clinical reasons, and only 3% switched to completely different molecules. Twenty-one per cent had not yet submitted claims. Through this programme, it was shown that an average savings of CA$8,500 per member per year was realized for the population participating in the pilot. As a result of this outcome, the Biosimilar Transition Program has been opened to all plan sponsors [57]. Importantly, this programme could also potentially provide real-world evidence on the safety and efficacy of switching from a Canadian perspective that could further promote biosimilar uptake by both private and public payers.
As with any new therapies, concerns are always raised regarding their safety and efficacy, and there was no difference when the first infliximab biosimilar was launched [7]. Since that time, the attitudes of some physicians and patients towards biosimilars have become substantially more receptive. For example, in 2012, the Ontario Rheumatology Association (ORA) was cautious regarding its recommendation of the use/implementation of biosimilars [67]. However, in the most recent position statement issued in 2018, the ORA openly supported non-medical switching from reference products to biosimilars [68].
Furthermore, a national patient-led organization, Arthritis Consumer Experts, is supportive of switching [69]. Payers and biosimilar manufacturers continue to emphasize education for physicians and patients to help them better understand biosimilars.
Significant changes have taken place in the Canadian regulatory and reimbursement environments for biosimilars since the launch of Inflectra, with many of these efforts and policies developed, e.g. the requirement of patients to switch to biosimilars in the BC, to further promote the uptake of biosimilars. As Canadian prescribers, patients and payers gain more confidence with biosimilars through accumulation of real-word safety and efficacy evidence, the promise of savings offered by these products may be fully realized. These savings are important to help sustain the healthcare system, and to provide resources for new medications in Canada. And, changes are likely to continue.
The authors would like to acknowledge France Lelievre, PharmAcce FDL, for her valuable input and Roberta Wood, Senior Medical Writer, Innomar Strategies, for her professional review of this manuscript. No funding was received for the preparation of this manuscript. The material contained in this manuscript has not been presented at conferences.
Competing interests: Eric CK Siu, Anne Tomalin, Kevin West, Sandra Anderson, and George Wyatt are all employees of Innomar Strategies. Innomar Strategies is a consulting firm that has business dealings with both originator and biosimilar manufacturers. No consulting fees were accepted relating to the current work. The authors have no other conflicts of interest to declare.
Provenance and peer review: Not commissioned; externally peer reviewed.
1InnomarConsulting
2TPIreg
3HealthForward
4Innomar Strategies, Oakville, ON, L6L 0C4, Canada
References
1. Canadian Institute for Health Information. Prescribed drug spending in Canada, 2018: a focus on public drug programs [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cihi.ca/sites/default/files/document/pdex-report-2018-en-web.pdf
2. Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301-16.
3. Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol. 2010;10(5):297.
4. Wallia A, Molitch ME. Insulin therapy for type 2 diabetes mellitus. Jama. 2014;311(22):2315-25.
5. Patented Medicine Prices Review Board. Private drug plans in Canada: high-cost drugs and beneficiaries, 2005 to 2017 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: http://www.pmprb-cepmb.gc.ca/view.asp?ccid=1366&lang=en
6. Patented Medicine Prices Review Board. Potential savings from biosimilars in Canada [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.pmprb-cepmb.gc.ca/view.asp?ccid=1304
7. Siu ECK, Wyatt G. Current state of subsequent entry biologics (biosimilars) in Canada: a view from regulatory, reimbursement, clinician, and patient perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):105-13. doi:10.5639/gabij.2016.0503.028
8. Klein AV. The first subsequent entry biologic authorized for market in Canada: the story of Omnitrope, a recombinant human growth hormone. Biologicals. 2011;39(5):278-81.
9. Health Canada. Guidance document – information and submission requirements for biosimilar biologic drugs [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs-1.html
10. Health Canada. Notice to stakeholders – subsequent entry biologics scientific advice meeting pilot [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/notice-subsequent-entry-biologics-scientific-advice-meeting-pilot.html
11. Health Canada. Improving the regulatory review of drugs and devices [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices.html
12. Health Canada. Consultations on improving the regulatory review of drugs and devices [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/programs/consultation-regulatory-review-drugs-devices.html
13. Health Canada. Improving access to biosimilars and biologics [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices/improving-access-biosimilars-biologics.html
14. Health Canada. Notice to industry: aligned reviews between Health Canada and health technology assessment organizations [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices/notice-aligned-reviews-health-canada-health-technology-assessment-organizations.html
15. Canadian Agency for Drugs and Technologies in Health. Health Canada, CADTH, and INESSS collaborate to align drug review processes Ottawa [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/news/health-canada-cadth-and-inesss-collaborate-align-drug-review-processes
16. Canadian Agency for Drugs and Technologies in Health. CADTH common drug review will accept submissions up to six months Pre-Notice of Compliance (NOC) [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://cadth.ca/news/cadth-common-drug-review-will-accept-submissions-six-months-pre-notice-compliance-noc
17. Institut national d’excellence en santé et en services sociaux. AVIS AUX FABRICANTS DE MEDICAMENTS. Nouvelles informations concernant revaluation des medicaments aux fins d’inscription Quebec [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_fabricants/20180404_Avis_fabricants_pre-AC_et_biosimilaires.pdf
18. Health Canada. Aligned reviews for certain drugs [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices/aligned-review-of-certain-drugs.html
19. Health Canada. Early scientific advice to manufacturers [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices/early-scientific-advice-manufacturers.html
20. Malmström RE, Godman BB, Diogene E, Baumgartel C, Bennie M, Bishop I, et al. Dabigatran – a case history demonstrating the need for comprehensive approaches to optimize the use of new drugs. Front Pharmacol. 2013;4:39.
21. Godman B, Bucsics A, Vella Bonanno P, Oortwijn W, Rothe CC, Ferrario A, et al. Barriers for access to new medicines: searching for the balance between rising costs and limited budgets. Front Public Health. 2018;6:328.
22. Eriksson I, Wettermark B, Persson M, Edstrom M, Godman B, Lindhe A, et al. The early awareness and alert system in sweden: history and current status. Front Pharmacol. 2017;8:674.
23. Health Canada. What we heard report – stakeholder consultation on the naming of biologic drugs [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/naming-of-biologics-what-we-heard.html
24. Health Canada. Notice to stakeholders – policy statement on the naming of biologic drugs [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/biosimilar-biologic-notice-to-stakeholders-drugs-naming-of-biologics.html
25. Canadian Agency for Drugs and Technologies in Health. CADTH Common Drug Review (CDR) [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/about-cadth/what-we-do/products-services/cdr
26. Institut national d’excellence en santé et en services sociaux. About the institut [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.inesss.qc.ca/en/about-us/about-the-institut.html
27. Canadian Agency for Drugs and Technologies in Health. About the pan-Canadian Oncology Drug Review (pCODR) [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/pcodr/about-pcodr
28. Canadian Agency for Drugs and Technologies in Health. 2018–2021 Strategic plan. Transforming how we manage health technologies in support of better health, better patient experience, and better value [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/sites/default/files/corporate/planning_documents/CADTH_2018_2021_Strategic_Plan_Overview.pdf
29. Canadian Agency for Drugs and Technologies in Health. Pharmaceutical Reviews Update – Issue 8 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://cadth.ca/cadth-pharmaceutical-reviews-update-issue-8
30. Canadian Agency for Drugs and Technologies in Health. Common Drug Review: CDR update – Issue 100 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/pharmaceutical-review-update
31. Canadian Agency for Drugs and Technologies in Health. Common Drug Review: CDR update – Issue 125 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/pharmaceutical-review-update
32. Canadian Agency for Drugs and Technologies in Health. Proposed revisions to the biosimilar review process for its common drug review and pan-Canadian oncology drug review programs [cited 2019 Aug 21]. Available from: https://www.cadth.ca/media/cdr/templates/consultations/CADTH_Biosimilar_consultation.pdf
33. Canadian Agency for Drugs and Technologies in Health. Procedure and submission guidelines for biosimilars. Common drug review [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/sites/default/files/cdr/process/Procedure_and_Guidelines_for_CADTH_CDR.pdf
34. Canadian Agency for Drugs and Technologies in Health. Revisions to CADTH’s biosimilar and resubmission processes, other administrative changes and new fee guidance for applications to CADTH’s pharmaceutical review programs [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://cadth.ca/news/revisions-cadths-biosimilar-and-resubmission-processes-other-administrative-changes-and-new-fee
35. Canadian Agency for Drugs and Technologies in Health. pCODR. Drugs reviewed under the joint oncology drug review process from 2007 to 2011 provincial funding summary [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/sites/default/files/pcodr/pcodr-ijodr-drugs-provfund.pdf
36. Institut national d’excellence en santé et en services sociaux. Conseil du medicament. Fevrier 2010 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.inesss.qc.ca/en/themes/medicaments/drug-products/manufacturer-information-centre/notices-to-manufacturers.html
37. Institut national d’excellence en santé et en services sociaux. Conseil du medicament. Juin 2010 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.inesss.qc.ca/en/themes/medicaments/drug-products/manufacturer-information-centre/notices-to-manufacturers.html
38. Canadian Agency for Drugs and Technologies in Health. Canadian Drug Expert Committee final recommendation. Grastofil [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/sites/default/files/cdr/complete/SE0446_cdr_complete_Grastofil_March-22-16_e.pdf
39. Institut national d’excellence en santé et en services sociaux. Avis au ministre de l’institut national d’excellence en santé et en services sociaux. Pour la mise à jour des listes des médicaments du 16 décembre 2016 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: http://collections.banq.qc.ca/ark:/52327/bs2741926
40. Institut national d’excellence en santé et en services sociaux. Avis au ministre de l’institut national d’excellence en santé et en services sociaux. Avis transmis au ministre le 15 mai 2018. [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Juin_2018/20180515_AvisMinistre_WEB.pdf?sword_list%5B0%5D=neupogen&no_cache=1
41. Institut national d’excellence en santé et en services sociaux. Evaluation of Biosimilars Quebec [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.inesss.qc.ca/en/themes/medicaments/drug-products/evaluation-process-and-criteria/evaluation-of-biosimilars.html
42. O’Mahony JF. Beneluxa: What are the prospects for collective bargaining on pharmaceutical prices given diverse health technology assessment processes? Pharmacoeconomics. 2019;37(5):627-30.
43. KCE Report 283. Horizon scanning for pharmaceuticals: proposal for the BeNeLuxA collaboration: Belgian Health Care Knowledge Centre; 2017 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: http://www.beneluxa.org/sites/beneluxa.org/files/2017-07/Horizon%20scanning_ScientificReport_full.pdf
44. The pan-Canadian Pharmaceutical Alliance: Subsequent Entry Biologics (SEBs) First Principles Ottawa, ON: Council of the Federation Secretariat; 2016.
45. The pan-Canadian Pharmaceutical Alliance. Biologics Policy Direction & pCPA Negotiations. Ottawa, ON: Council of the Federation Secretariat; 2018.
46. Cancer Care Ontario. Pan-Canadian Oncology Biosimilars Summit. Proceedings Report [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cancercareontario.ca/sites/ccocancercare/files/assets/PanCanadianBiosimilarsSummitProceedingsReport_0.pdf
47. Cancer Care Ontario. Pan-Canadian Oncology Biosimilars Initiative Action Plan [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cancercareontario.ca/sites/ccocancercare/files/assets/PanCanadianOncologyBiosimilarsActionPlan_0.pdf
48. British Columbia. Biosimilars Initiative for patients. 2019 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www2.gov.bc.ca/gov/content/health/health-drug-coverage/pharmacare-for-bc-residents/what-we-cover/drug-coverage/biosimilars-initiative-patients
49. British Columbia. B.C. expands use of biosimilars to offer coverage for more treatment options. 2019 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://news.gov.bc.ca/releases/2019HLTH0080-001072
50. British Columbia. Biosimilars Initiative Pharmacist Guide [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www2.gov.bc.ca/assets/gov/health/health-drug-coverage/pharmacare/biosimpharmguide.pdf
51. Manitoba Health, Seniors and Active Living. Notice – Tiered Biologics Reimbursement Policy Winnipeg, MB: Council of the Federation Secretariat; 2018 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.gov.mb.ca/health/mdbif/docs/bulletins/bulletin100.pdf
52. U.S. Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product. Guidance for industry [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.fda.gov/media/124907/download
53. U.S. Food and Drug Administration. Statement from Acting FDA Commissioner Ned Sharpless, M.D., on policy advancements to help bring interchangeable biosimilars to market [homepage on the Internet]. [cited 2019 Aug 21]. Available from: [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.fda.gov/news-events/press-announcements/statement-acting-fda-commissioner-ned-sharpless-md-policy-advancements-help-bring-interchangeable
54. Innomar Insight Series: Biosimilars in Canada Toronto, ON: Innomar Strategies; 2018 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.innomar-strategies.com/insights/insight-series-biosimilars-in-canada-a-market-access-update
55. Institute of Health Economics. Towards a framework for biosimilar evidence and knowledge exchange. Summary report of the IHE Biosimilars Forum [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.ihe.ca/publications/towards-a-framework-for-biosimilar-evidence-and-knowledge-exchange-summary-report-of-the-ihe-biosimilars-forum
56. Kauth G. Rising biosimilar uptake touted amid lsquo;continuously evolving’ evidence for safety, efficacy. Benefits Canada. 2018 Jun 18.
57. Green Shield Canada. GSC’s biosimilar transition program completes a successful pilot. Winter 2018. 2018 Dec 1. Follow the Script [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://assets.greenshield.ca/greenshield/GSC%20Stories%20(BLOG)/Follow%20the%20Script/2018/english/Follow%20the%20Script_Winter%202018.pdf
58. Canadian Agency for Drugs and Technologies in Health. International Policies on the appropriate use of biosimilar drugs. 2018 Oct 25 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.cadth.ca/international-policies-appropriate-use-biosimilar-drugs-0
59. Moorkens E, Vulto AG, Huys I, Dylst P, Godman B, Keuerleber S, et al. Policies for biosimilar uptake in Europe: an overview. PLoS One. 2017;12(12):e0190147.
60. Health Canada. Strengthening the use of real world evidence for drugs Ottawa, ON: Health Canada; 2018 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices/strengthening-real-world-evidence-medical-devices.html
61. Biosimilars Canada. Biosimilars Canada Announces Patient Support Program Platform. 2018 Nov 22 [homepage on the Internet]. [cited 2019 Aug 21]. Available from: http://biosimilarscanada.ca/news/nov222018.html
62. Matusewicz W, Godman B, Pedersen HB, Fürst J, Gulbinovič J, Mack A, et al. Improving the managed introduction of new medicines: sharing experiences to aid authorities across Europe. Expert Rev Pharmacoecon Outcomes Res. 2015;15(5):755-8.
63. The cost of slow biosimilar uptake in the US: an Infliximab case study. Pharmaceutical Technology. 2018 Aug 22.
64. Sagonowsky E. AbbVie’s massive Humira discounts are stifling Netherlands biosimilars: report. FiercePharma. 2019 Apr 2.
65. Moulton D. Biosimilars have potential to offer relief for rising drug plan costs. Benefits Canada. 2018 Oct 5.
66. Jø¸rgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304-16.
67. Ontario Rheumatology Association. Position paper on subsequent entry biologics in Canada [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://ontariorheum.ca/images/uploads/content_documents/ORA_SEB_Position_Statement_final_Dec_2012.docx
68. Ontario Rheumatology Association. Updated: ORA position – biosimilar non-medical switching [homepage on the Internet]. [cited 2019 Aug 21]. Available from: https://ontariorheum.ca/updated-ora-position-biosimilar-non-medical-switching
69. Arthritis Consumer Experts. Joint Health Biosim Exchange – Biosimilars Policy. 2019.
1For the purpose of this policy, ‘Biologicals’ include both biosimilar and non-biosimilar products.
|
Author for correspondence: Eric CK Siu, Senior Manager, InnomarConsulting, Innomar Strategies, 3470 Superior Court, Oakville, ON L6L 0C4, Canada |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2019 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/an-ever-evolving-landscape-an-update-on-the-rapidly-changing-regulation-and-reimbursement-of-biosimilars-in-canada.html
Copyright ©2025 GaBI Journal unless otherwise noted.