|
Introduction: Biosimilars were first introduced in Europe in 2006 and then in the US in 2015. An online webinar on the successful uptake in Europe and the US was held to discuss measures taken for improving biosimilar uptake, and trends in uptake. |
Submitted: 9 August 2022; Revised: 16 September 2022; Accepted: 22 September 2022; Published online first: 26 September 2022
In collaboration with the Alliance for Safe Biologic Medicines (ASBM), the Generics and Biosimilars Initiative (GaBI) organized and hosted the first in a series of webinars on biosimilars entitled ‘Key factors for successful uptake of biosimilars: Europe and the US’. This webinar set out to discuss measures taken for improving biosimilar uptake, trends in uptake, and the potential role to be played by healthcare providers and patients in Europe and the US.
Biosimilars have been in the market for 16 years since their first launch in 2006 in Europe. At the time of the webinar, there were 88 approvals (including 16 biosimilars withdrawn after approval) in the EU [1] and 36 in the US [2] where the first biosimilar was approved in 2015 [2], representing almost 90% of the world market. Today, more and more biosimilars are entering the healthcare system and there remains the need for broader sharing of information and regular discussions to clarify and alleviate the concerns over biosimilars use for physicians and patients, specifically on the issues of prescribing practice and biosimilars switching.
In this online event, held on 29 June 2022, the contributors discussed key elements contributing to the wide uptake of biosimilars in Europe and the US and physicians’ trust in prescribing and switching of biosimilars. Presentations of concrete examples and experiences of challenges and uptake policies of biosimilars in Europe, and the current status of market access and successful uptake measures implemented for biosimilars in the US, were given. These allow evaluation of the role of healthcare providers (physicians, pharmacists) and health policymakers in biosimilars use.
The webinar was moderated by Steven Stranne, JD, MD, and partner at Foley Hoag LLP, who has a unique perspective as a physician and lawyer. The Keynote Presentation, ‘Impact of biosimilars in the US healthcare system and the path forward’, was given by the Honourable Eric David Hargan, JD, former United States Deputy Secretary of Health and Human Services (HHS). Additional presentations were given by Mr Michael S Reilly, Esq, Executive Director of ASBM; Dr Ralph McKibbin, past President of the Pennsylvania Society of Gastroenterology and the Society of Gastroenterology and Digestive Disease; Professor Philip J Schneider, Professor at Ohio State University College of Pharmacy; Mr Chad Pettit, Executive Director of Marketing, Biosimilars Business Unit at Amgen; and Mr Andrew Spiegel, Esq, Executive Director of the Global Colon Cancer Association.
Overall, the panel was made up of an academic clinician with specialty in gastroenterology, a pharmacist, a patient advocate, and a market access expert. They shared their experiences with biosimilars, highlighting successes and challenges, their perspectives on prescribing and switching of biosimilars and measures to increase biosimilar adoption, including the role of healthcare providers.
Learning objectives
The overall learning objectives of the webinar were outlined as follows:
Registrant statistics
There were a number of expert speaker presentations followed by a Q&A and an in-depth panel discussion. The presentations are available online [3].
The webinar was opened by Mr Reilly with input from the moderator, Dr Steven Stranne.
Keynote presentation: Impact of biosimilars in the US healthcare system and the path forward
Mr Eric Hargan started by asking the question, ‘Why is increasing biosimilar uptake important?’. He then described how a large proportion of US drug spending is due to biological medicines and this has increased over time as a percentage. As such, the US HHS, keeps a close eye on spending growth due to biologicals and biosimilars. The savings that biosimilars could offer to healthcare systems are of particular relevance. It is also thought that overall, more biosimilars will lead to more savings.
The savings due to biosimilars are estimated to be US$38.4 billion or 5.9% of projected total US spending on biologicals from 2021 to 2025, according to a study published in the American Journal of Managed Care [4]. In addition, more aggressive biosimilar uptake and competition could trigger larger cuts, with savings estimated to be as large as US$124.5 billion from 2021 to 2025 under the most-optimistic scenario. The study also estimates that most of the expected savings from biosimilars would be caused by downward pressure on the brand-name biologicals they compete with, rather than lower biosimilar prices.
Learning from Europe
It was also noted that Europe is widely considered the world leader in the approval and commercialization of biosimilars; and is ahead of the US regarding their regulation, introduction and adoption.
In the European Union (EU), a legal framework for approving biosimilars was established in 2003 [1]. This framework means that biosimilars can only be approved centrally via the European Medicines Agency (EMA) and not nationally [5]. EMA first developed guidelines for the approval of biosimilars via an abbreviated registration process during 2005 to 2006 [6]. Omnitrope (somatropin) was the first product approved in the EU as a biosimilar in 2006 [7] and the first biosimilar monoclonal antibodies (Remsima, Inflectra; both for infliximab) were approved in 2013 [8].
To date, EMA has recommended the approval of 88 biosimilars (including several different marketing authorizations of the same molecule) within the product classes of:
However, 16 of these have been withdrawn leaving 72 biosimilars approved in Europe at the time of the webinar.
Although the US has lagged behind Europe and did not have the regulatory pathway established until much later, it is now catching up and approved 28 biosimilars in the first five years (vs 11 approved by EMA in the first five years) [1, 2, 9].
US biosimilar approvals
FDA approved its first biosimilar in March 2015 [2]. As of approximately 12 years after implementation of the biosimilar approval pathway, 36 biosimilars have been approved by FDA, with most of those approvals granted in the last three years. Of these, more than 20 are currently on the market and available to patients. Mr Hargan noted that many of the products approved as biosimilars in Europe are approved in the US as ‘follow-on biologicals’ via the earlier, 505(b)(2) pathway, e.g. somatropin, insulin, teriparatide. These medicines are available to US patients but are not considered biosimilars.
In the US, biosimilars have gained a significant share in the majority of therapeutic areas in which they have been introduced (see Figure 1): 80% for filgrastim biosimilars, 70% for trastuzumab and bevacizumab biosimilars, and 55% for rituximab biosimilars. However, infliximab biosimilars have had the most limited adoption, with approximately 20% market share.
 In addition, US biosimilar uptake rates are now very similar to European rates (although these vary from country to country within Europe). For example, in Europe, the countries with the highest total biosimilar volume were Denmark: 63%; UK: 45%; Germany 40%; France 34%, Belgium and Switzerland tied at 14%. Regarding filgrastim/pegfilgrastim uptake, 16 European countries had > 90% biosimilar utilization in 2018, (however, in Ireland this was just 27%). Anti-TNF biosimilars (adalimumab, etanercept and infliximab) biosimilar uptake was high in Norway and Denmark (81% and 96%, respectively), while every other country’s utilization was less than 50%.
In addition, US biosimilar uptake rates are now very similar to European rates (although these vary from country to country within Europe). For example, in Europe, the countries with the highest total biosimilar volume were Denmark: 63%; UK: 45%; Germany 40%; France 34%, Belgium and Switzerland tied at 14%. Regarding filgrastim/pegfilgrastim uptake, 16 European countries had > 90% biosimilar utilization in 2018, (however, in Ireland this was just 27%). Anti-TNF biosimilars (adalimumab, etanercept and infliximab) biosimilar uptake was high in Norway and Denmark (81% and 96%, respectively), while every other country’s utilization was less than 50%.
Furthermore, variations in uptake rates between the US and Europe are influenced by government involvement, reimbursement structures and tender procurement policies.
Savings in the US
Biosimilars are now generating substantial savings in the US. Mr Hargan highlighted that generally, biosimilars launch at wholesale acquisition cost (WAC) 15% to 37% lower than their reference products and up to 40% below the reference product’s average sales price (ASP). As more become available, the increased competition has been shown to drive down prices of both biosimilars and innovator biologicals.
In 2019, biosimilars saved the US healthcare system US$2.5 billion and in 2020, that figure more than tripled to US$7.9 Billion.
It was highlighted that price was key when it came to boosting biosimilars uptake in the US. As is the case in Europe, as more and more biosimilars launch in a given product class, competition drives prices downward, discounts increase, and biosimilar market share in the US goes up. For example:
As it becomes routine to have 3, 4, or 5 biosimilars approved for a reference product, it is expected that this trend, and savings, will continue.
The future for US biosimilars – interchangeable biosimilars
Mr Hargan concluded by noting that six adalimumab biosimilars have been approved, including one ‘interchangeable’ biosimilar (meaning it can be substituted at the pharmacy level without physician involvement) [10].
An interchangeable biosimilar is a US-specific higher regulatory standard that requires more data and usually includes switching studies.
An interchangeable biosimilar:
1) Must be a biosimilar (‘highly similar’ to reference product).
2) Must have same clinical result expected as with reference product.
3) Must create no additional risk to patient when switching back and forth between itself and reference product.
4) May be substituted for the reference product in a pharmacy without the intervention of the prescriber.
The interchangeable biosimilar, Cyltezo (adalimumab-adbm), will become available in 2023 after the originator product’s patent protection expires. Together with (insulin glargine-yfgn) that was approved as an interchangeable biosimilar in July 2022, these are the first two interchangeable, patient-administered biosimilars in the pharmacy side and not infused in a clinical setting. It is anticipated that this increased competition will lead to continued savings. Mr Hargan highlighted that this will also be a test of to what degree an ‘interchangeable’ designation affects physician prescribing and formulary design of payers. He noted that, in terms of this there are potential for reimbursement and regulatory changes, and asked, ‘Will all products compete on a level playing field?’
European prescribers trust in prescribing and switching biosimilars
Mr Michael Reilly outlined how the ASBM has carried out a number of prescriber surveys across the globe between 2012?2022 [11-16].
As was highlighted by Mr Hargan, Mr Reilly also noted that Europe is widely acknowledged as a global leader in biosimilars for successfully developing a robust and sustainable biosimilars programme. To understand Europe’s success, it is critical to understand that European physicians are generally in power to prescribe in the best interest for patients.
Mr Reilly outlined the findings of the 2019 European prescribers survey [11] which was a refresher of that carried out in 2013 [14].
The 2019 survey revealed that European physicians increased their familiarity between 2013 and 2019 and, after 13 years of experience with biosimilars in Europe, physicians:
Overall, as familiarity and comfort with biosimilars increased, so did the importance to physicians of maintaining control of treatment decision.
Online questions
Q1: It is good to see how penetration rates are consistently high in various geographies. What about variances in time it takes to reach such penetration levels?
A1 (Reilly): The results of the surveys would suggest that involving physicians and patents when developing policies that have an impact on treatment decisions is critical. It is also important for these policies to provide the flexibility to meet individual patient needs that can sometimes be different from patient to patient. The extent to which these issues are considered play an important role in penetration levels for biosimilars.
Q2: There are some widely anticipated biosimilars expected to hit the market in 2023, what do you think will be the impact of those, if any?
A2 (Reilly): More biosimilars in the marketplace will create more price competition and lower costs for treatment with biological medicines. This has been shown in market research where the more competitors there are, the more prices come down.
US physicians’ perspective in biologicals/biosimilars prescribing and substitution
Dr Ralph McKibbin presented the results of a 2021 ASBM survey of US physicians [17]. The survey examined:
The study revealed that US physicians are:
Overall, the interchangeable designation, makes the majority of physicians more comfortable with prescribing and substitution.
Dr McKibbin also highlighted that, when comparing different surveys from across the globe, there is strong agreement that it is very important or critical that the physician, with the patient, decides which treatment option to use, rather than a third party.
European and US physicians agree that it is very important or critical that payers (public and private) reimburse/cover multiple products, including the originator biological as well as the different biosimilars. They also agree that it is very important or critical that payers (public and private) reimburse/cover multiple products, including the originator biological as well as the different biosimilars.
Regarding the access scenarios, US physicians prefer the approach of Western Europe (where multiple products including innovator and biosimilars are reimbursed, biosimilars may be encouraged for new patients, no automatic substitution permitted) over that of Canadian provinces that have implemented forced substitution (only the government-chosen biosimilar or biosimilars are reimbursed, new patients must be prescribed this product, and current patients must switch).
A comprehensive report of the survey is published in GaBI Journal [17] or the poster with video walkthrough is viewable at www.safebiologics.org.
Online questions
Q1: Considering the 2021 US physicians survey results showing the physicians’ concerns about switching to biosimilars, would they be voluntarily switched to a biosimilar if a patient is being treated with an off-patent originator?
A1 (McKibbin): While we did not specifically ask about off-patent originator medications, in clinical practice they would be viewed as identical to the drug while it was still under patent protection with no change in production mechanisms or testing. The concerns expressed were related to the safety and efficacy of the biosimilar medications.
Q2: What is the clinical importance of some patients’ unawareness/mistrust on biosimilars (in particular in non-medical switching) on adherence/nocebo/other outcomes?
A2 (McKibbin): On the surface this is a simple question but there are so many issues wrapped up in this. While the placebo effect is commonly thought to be related to patients ‘believing they are on treatment’ so there is an improvement in their condition, but there can also be Pavlovian type mechanisms developed from previous experiences. Similarly, the nocebo effect is a recently used ‘buzz word’ which some use to suggest that prescribers are using loaded language to suggest that biosimilars might be less effective thereby resulting in side effects or lack of efficacy. Branding by marketers with direct-to-consumer advertising of originator medicines greatly influences the patients as well. More than six billion dollars are spent yearly on this type of advertising. Industry data shows that as many as 42% of patients will ask for a medication by name. I believe that the advertisers feel this investment will ‘pay off’. If a physician informs a patient that there is a ‘highly similar’ medication which can or will be substituted for condition. Will the patient have the same level of confidence? What if this is a life and death situation such as cancer treatment agents? Marketers have a long history of building brands to inspire confidence. I would suggest, by example, to think of brand name versus generic or store brand frozen vegetables. Do we in our hearts think that they are identical or just similar? Getting patients to be fully confident in biosimilars likely requires looking at all of the forces influencing them.
Sustainable biosimilars market in Europe – policy considerations
In his presentation, Professor Philip Schneider summarized how Europe has the largest biosimilar market in the world (approximately 60% of the global biosimilar market). As of June 2022, 70 biosimilars of 15 originator biological medicines had marketing authorization in Europe. (~50 unique products, under different brands). Here, biosimilar market share is as high as 91% for older products (before the approval of the first monoclonal antibody biosimilar in 2013) and as high as 43% for newer products (approved post-2013). In light of this, European countries, with their large biosimilar markets and diverse healthcare systems, serve as valuable examples of different approaches to biosimilar policy.
He highlighted the findings of the 2020 white paper, ‘Policy recommendations for a sustainable biosimilars market: lessons from Europe’ [18], that had the overall objective to identify principles which can be applied to develop an efficient and sustainable biosimilar market. It examined findings and recommendations of five previous studies and reports on biosimilar sustainability in Europe.
The 2020 white paper revealed that there are differences in biosimilar policy within the 28 EU Member States and the three European Economic Area (EEA) countries, these are:
Concerning national tender markets, Norway and Denmark are the only European countries to pursue a national tender policy for biosimilar products, such as adalimumab, etanercept, or infliximab. Denmark will only reimburse the manufacturer with the lowest bidding price for a particular molecule, for a 12-month period. This potentially requires physicians to switch patients every 12 months. In Norway’s tender process the choice is ultimately left to the physician and all lower ranked products are still reimbursed if prescribed, i.e. switching is not explicitly mandated as it is in Denmark. Tenders in Norway tend to be in effect for one year.
It is clear that across all European markets, biosimilars have:
While the policies by which this has been achieved vary between countries, all major European markets share the following principles:
The six principles of the ‘Gold Standard’
Professor Schneider noted that there are six principles that support the ‘Gold Standard’ for biosimilars as outlined in the white paper, these are:
1. Policies should be designed to incentivize and reward innovation in all types of biologicals.
2. Healthcare financing must take into account societal benefits derived from biological medicines, as well as the unique characteristics of biologicals.
3. Procurement practices must provide for multiple suppliers and a minimum term of 12 months.
4. Physicians must have autonomy to choose the most appropriate medicine for their patient, including making decisions on switching, which must also be consented to by the patient; no automatic substitution.
5. Mandatory brand-name prescribing to avoid unintended switches and a robust pharmacovigilance system to report adverse drug reactions (ADRs).
6. Policies with potential to undermine sustainability, such as measures which induce biosimilar uptake or promote preferential treatment, thereby limiting physician choice, should be avoided.
The white paper identified three must-have principles:
1. Physicians should have the freedom to choose between off-patent originator biologicals and available biosimilars and to act in the best interest of their patients based on scientific evidence and clinical experience.
2. Tenders should be designed to include multiple value-based criteria beyond price, e.g. education, services, available dose strengths, and provide a sufficient broad choice (multi-winner tenders versus single-winner tenders) to ensure continuity of supply and healthy competition.
3. A level playing field between all participating manufacturers is the best way to foster competition; mandatory discounts which place artificial downward pressure on manufacturers do not engender a sustainable market environment.
Professor Schneider concluded that while countries seek to replicate Europe’s success with biosimilars, some are ignoring the principles which made it possible. For example, there are forced-switching policies enacted in both Canada and Australia.
Forced switching in Canada
Some Canadian provinces (Alberta, British Columbia, Quebec and New Brunswick, have begun to forcibly switch patients to the government-chosen biosimilar.
Forced switching in Australia
The Australian government has also force-switched patients, including stage IV cancer patients. Reimbursement practices have caused manufacturers to cancel planned launches and even remove their products from the country.
In addition to raising concerns among patients and physicians, these forced-switching policies may jeopardize the long-term sustainability of these countries’ biosimilar markets.
Online questions
Q1: Do you consider approval with only Quality/CMC [chemistry, manufacturing, and controls] and PK [pharmacokinetics] comparability as ‘lower standard’?
A1 (Schneider): The scientists tell us analytics are more sensitive than clinical studies in detecting differences in similar biological molecules, so they might answer this question. Having the real-world experience demonstrated by clinical trials would make most healthcare professionals and patients more comfortable and confident in biologicals may be affected by reducing or even eliminating these studies. This issue places increased importance on post-marketing surveillance programmes and pharmacovigilance, which is why ASBM has been such a strong supporter of distinguishable names for biological medicines.
Q2: It is very informative to see physician’s reactions to various dynamics including specific points of discomfort. How does this discomfort affect actual uptake of the biosimilar if it’s driven for example by a payer’s formulary design?
A2 (Schneider): The experience in Australia where the Pharmaceutical Benefits Advisory Committee mandated switching to a biosimilar resulted in significant pushback from physician and patient groups and significantly slowed the adoption of the biosimilar. Physicians and patients need to be part of the policymaking process when it comes to those that affect individual patient care decisions.
Measures leading to successful uptake and the current state of market access of biosimilars in the US
Mr Chad Pettit described how Amgen is developing its biosimilars portfolio in the therapeutic areas of oncology, haematology and chronic inflammatory diseases. The company currently (at time of writing) has 11 biosimilar candidates in its portfolio, including some approved and marketed biosimilars.
He highlighted that there are four elements that are crucial for fostering a robust and sustainable marketplace for biosimilars:
The US biosimilar marketplace is now well established and accelerating across many therapeutic areas. At the time of the webinar there were 36 approved and 21 launched biosimilars on the US market.
Figure 2 shows the number of biosimilars approved and launched each year from 2015 to 2022. There was a dramatic increase in biosimilar launches from 2018 to 2020 compared to prior years.
The slowdown of biosimilar approvals in 2020 and 2021 observed in Figure 2 was likely due to several factors, some of which were pandemic related. Over the next few years, the US marketplace with biosimilars should recover from this decline in activity, with new approvals and launches expected to increase at pre-2020 rates. Despite the decline in the number of approvals during the 2020 to 2021 timeframe, the number of development programmes that are participating in the FDA Biosimilar Development Program has continued to rise, with 77 in March 2019, 79 in March 2020, 90 in March 2021 and 97 in December 2021.
Mr Pettit also pointed out that biosimilars in the US market is advancing at a faster rate than the EU market during a comparable time period. In the eight years after the EU approved the first biosimilar (2006), there were 15 approved biosimilars. By contrast, in the eight years after the US approved the first biosimilar (2015), there were 36 approved biosimilars, see Figure 3.
Figure 3 shows the cumulative number of biosimilars approved in the EU versus the US, beginning with the year the first biosimilar was approved. The slowdown in EU approvals between years 4 and 7 was likely due to several factors that may include intellectual property timelines and duration of development programmes.
In addition, biosimilars are helping reduce healthcare costs in the US by providing significant wholesale acquisition cost (WAC) and average sales price (ASP) savings at launch and through price competition, resulting in the opportunity for additional savings over time.
As shown in Figure 4, manufacturers are launching biosimilars at a WAC that is generally lower than that of the reference product (biosimilars’ ASP becomes available two full quarters after launch):
As expected, competition usually results in lower ASP for both reference products and biosimilars, leading to additional savings. Figure 5 shows that, in most cases, the prices of biosimilars declined once ASP was established and continued a steady downward trend. The ASPs for reference products have been declining over time, leading to further opportunity for healthcare savings. It also shows that the prices of biosimilars are decreasing at a compound annual growth rate (CAGR) of 9% to 22% and the prices of most reference products are decreasing at a CAGR of 4% to 20%.
The rate of biosimilar uptake is generally increasing over time, as depicted in Figure 6. Biosimilars have gained significant share in the majority of therapeutic areas where they have been introduced. Additionally, first-to-launch biosimilars have tended to capture a greater portion of the segment compared to later entrants. For therapeutic areas with biosimilars launched in the last three years, the average share was 74%. For therapeutic areas with biosimilars launched prior to 2019, the average share after three years was 38%.
Biosimilar competition is contributing to decreased spending
Mr Pettit also shared Figure 7 which shows the estimated decrease in total drug spend after biosimilar competition was introduced. The change in drug spend shown is the difference between the projected reference product spend (based on historical trend) versus the actual spend following biosimilar launch. Beginning in Q1 2019, drug spending for most classes continues to decrease. The cumulative savings in drug spend for classes with biosimilar competition is estimated to have been US$18 billion over the past six years. Trends show an increase in savings per quarter, and in Q1 2022 alone, savings in drug spend are estimated to be US$3 billion.
The future for biosimilars
Over the next few years, the growing number of biosimilars will likely lead to a rapid evolution in the US biosimilars marketplace, including:
These changes are likely to cement the role of biosimilars as viable and integral US treatment options. Biosimilars will find new audiences in different prescriber specialties, pharmacists, payers, and patients, and may change the patient support programme landscape and interactions at the pharmacy counter.
Patients’ perspective on biosimilars use – Europe and the US
Mr Andrew Spiegel gave an overview of patients and biological medicines, describing how over one billion patients around the world have benefited from a biological medicine.
Overall, this has led to improved outcomes for colorectal cancer (CRC) patients. Biosimilars offer patients new treatment choice and reduced costs.
The patient community is excited about biosimilars, but it is key to ensure that biosimilar policies work for patients, which means, it is key to maintain:
There are concerns over loss of treatment stability due to unnecessary switching. Specifically, patients on biological therapies generally have, for example, chronic diseases; and thus, changes in treatment can have a significant impact. Many patients take years to find a medicine that works for them to help control disease, and in many cases, biological medicines may be the most or even the only effective treatment. As such, for patients that are on a biological that is working for them, decisions related to switching therapy should be carefully considered. Changes in therapy could lead to an immune response and/or a loss of response to the new and old therapy, exposing patients to a scenario with no, or fewer, or more serious treatment options.
Biosimilars training programmes
In light of this, Mr Spiegel highlighted that, in order to facilitate uptake of biosimilars, and switching where this is an option, education is key. He noted that biosimilars training programmes for patient advocacy groups can play an important role here.
He gave an overview of the Global Colon Cancer Association Biosimilars Training Program [25], which was a 6-hour interactive training programme for patient advocacy organizations and advocates representing all diseases to fill a gap in education and awareness. It included:
During the biosimilar training programme, several representatives from patient advocacy organizations in the US, Europe, Canada and Australia shared the experiences of patients in their countries. It was clear that these experiences varied significantly:
For Europe, patient advocacy representatives, Charis Girvalaki, European Cancer Patient Coalition and Zorana Maravic, Digestive Cancers Europe, described how European governments generally permit the physician to choose the most appropriate biological medicine, between the originator and several biosimilars and that all products will be reimbursed. Generally, lower cost medicines, usually biosimilars, are encouraged for new patients, and some quotas are in place to encourage this. Discussions about switching are made collaboratively and education of patients to build trust in biosimilars has been a priority. In addition, savings attributed to biosimilars are being visibly reinvested into the system, e.g. more healthcare workers.
For the US, the patient advocacy representatives were Mr Spiegel himself and Ms Anna Hyde, Arthritis Foundation. They explained that biosimilars are attaining significant market share, and competition is creating savings. Overall, physician and patient confidence in biosimilars is high, although there are concerns about non-medical switching by third parties (such as private health insurers or pharmacy benefit managers). Substitution laws at the state level, supported by patients, have attempted to address these concerns. Only ‘interchangeable’ biosimilars can be automatically substituted at the pharmacy level. In addition, State law also ensures that the prescribing physician is aware of any substitution that occurs. Many states are working on legislation to restrict how and how often patients may be switched by private insurers, e.g. step therapy.
For Canada, the patient advocacy representative was Ms Gail Attara, Gastrointestinal Society. She advised that four provinces have adopted forced-switching policies: British Columbia (BC), Alberta, Quebec, and New Brunswick. Within this, BC’s policy was the most strict–no exceptions and Alberta offered minimal exceptions, for children and pregnant women. The patient communities, particularly the gastrointestinal (GI) community, joined by their physicians, pushed back heavily on these policies to no avail. Surveys by patient organizations showed many problems including side effects, and many patients discontinuing treatment altogether. The patient community was able to gain some concessions in negotiations with the Quebec government, including many exceptions for age, pregnancy, high-risk patients, mental health, geographical and logistical concerns, e.g. change in infusion clinic location.
For Australia, the patient advocacy representative was Ms Julien Wiggins, Bowel Cancer Australia. In 2015, Australia broke with other advanced nations and allowed automatic substitution of biosimilars, over the objection of patients and physicians. Physicians often blocked these forced substitutions, leading to very low uptake/market share. Several manufacturers have pulled their products – one for liability reasons, after the government began automatic substitution, another after an unexpected, deeper-than normal price cut. Forced switching is now occurring with stage IV cancer patients and there is no grandfathering of current patients. Overall, patients are disappointed and bitter as biosimilars were sold to patients to expand choice, with many products listed alongside each other to choose from – this has not happened. Patients ask the question, ‘They have replaced one monopoly with another … was this by design?’
Mr Spiegel concluded with the following summary:
Online questions
Q1: Please clarify the patients’ nocebo effect and how can the biosimilars marketplace achieve the best outcomes for patients and cost savings for the healthcare system?
A1 (Spiegel): These two questions are interrelated. It is an issue of building confidence in order to drive uptake, price competition, and savings. The nocebo effect is when a patient experiences a negative response to a medication due to the expectation of a negative outcome, for example, they might believe the medicine is of lower quality, and thus might not work well for them. It becomes something of a self-fufilling prophecy. This is similar to the better-known placebo effect – wherein a patient is likely to report a positive experience after being conditioned to expect a positive outcome – even if the so-called ‘medicine’ is simply a sugar pill. So how do we build confidence in biosimilars? First, approval standards regarding safety, efficacy, purity, must not be lower for biosimilars than for originator products – or this may create a negative impression of biosimilars as a whole – that they are medicines which could not be approved but for lowered standards. Second, transparency – particularly in substitution – builds patient (and physician) confidence in biosimilars. We want to know which medicine we are being prescribed, and which we receive at the pharmacy or infusion centre. For example, patients have long been supportive of biosimilars having distinct non-proprietary names to clearly distinguish them from the reference biologicals upon which they are based, as well as other biosimilars to that product. Finally, good communication between patients and their healthcare team is essential. Patients want to know (and want their physicians to know) if they receive a biosimilar substitution so that their response to treatment is accurately captured in the patient record and the effects, good or not-so-good, are attributed to the correct product. The patient-physician relationship must remain central in treatment decisions, including if and when to switch between biologicals – as treatment plans are not one-size fits all. Patients trust their physicians and we have seen that while confidence in biosimilars is very high among physicians, there is less acceptance of forced-switching, which should be avoided if patients are to feel they are getting the treatment that is best for them, rather than what is best for the payer. Together, these principles will lead to high confidence and positive expectations for patients about biosimilars, and thus better outcomes.
Following the presentations, the panel members discussed a number of questions. These discussions were moderated by Dr Stranne and are summarized here.
Question 1: Is there a point where the standards imposed on biosimilars should be revisited and loosened to promote more competition?
Mr Hargan was first to address this question and noted that lowering standards to promote competition is not a route currently being considered for biosimilars. He highlighted that biosimilars are a relatively young class of drugs and as such, patient confidence is very important. So, particularly at present, lowering standards is not a good option. He noted, ‘We are starting to see savings due to biosimilars introduction without lowered standards, so lowering them does not seem necessary. Overall, we want more competition, and we are getting it without lowering standards’.
Mr Pettit added that lowering standards will not achieve anything except ‘hurting the system’. The confidence of patients and physicians is needed, and lowering standards will damage this. In addition, lowering standards is unlikely to achieve any additional savings. He noted that, looking at all the different classes of medicines, cost savings in all are fairly consistent once a few biosimilars are introduced. In the future, further savings may be made by investing more in biosimilars in more therapeutic areas. He added, ‘It is not about lowering the bar to get lots of biosimilars in a class, but it is about creating opportunities for investors in many classes of biosimilars’. He also noted that, in the past, regulators had been put under pressure to lower the standards of drugs, e.g. during the AIDs epidemic, but this is not something that was considered then; and should not be now.
Mr Reilly highlighted that there have been many conversations about reducing standards. EMA has argued that this is a moral question as some countries with no access to drugs need access, and perhaps this does require a lowering of standards in some circumstances. However, the ASBM position is: ‘no easy point of access’. This means that, countries with fewer resources should not receive medicines that are substandard compared to those received by wealthier nations. This is in opposition to EMA and WHO’s position. In relation to this, he asked the question, ‘How far do you lower standards before you make a mistake that will undermine confidence?’
Dr McKibbin noted that, from a physician’s perspective, ‘people want confidence in healthcare system and changing access methods is one thing but lowering standards is another which cannot be done.’
Professor Schneider added that currently there is debate on reducing emphasis on clinical trials in favour of analytics. The argument is that analytics are more effective in proving the position of some biosimilars (in proving biosimilarity), than lengthy and expensive clinical trials. He suggested that there is an ongoing debate on whether the elimination of clinical trials lower the regulatory standards for approval of biosimilars.
Mr Spiegel highlighted that the biosimilars bubble is growing and, ‘the last thing we want is to burst that bubble due to a bad experience of a product that has lower standards’. He noted that, in South America, some countries were calling for lower standards and he asked the question, ‘why should you have lower standards because you do not have as much money as other countries such as Canada, Europe and the US?’ Countries with fewer means should not be in receipt of lower standard drugs, this is not just unfair but dangerous. He added that a bad experience anywhere will affect the whole global biosimilars market.
Dr McKibbin added that we need trust in the system and should make any changes in a stepwise fashion.
Mr Reilly concluded that it is surprising that EMA and WHO regulators argue for lower standards in some regions but never within the EU itself. This could be considered a case of ‘not in my back yard’, or nimbyism.
Question 2: What sort of medical education topics, initiatives, are key to help drive physician acceptance of biosimilars use?
Dr McKibbin advised that the cornerstone of acceptance is transparency. This should be present in every step of the process. Physicians have a responsibility to have an informed conversation with patients, and to understand risks and benefits, he added ‘I read the menu and the patient orders’.
It is very important to have all information on a drug, particularly safety data. In terms of generics, in some cases, the medication can be the same, but the binders may be different, and this can cause issues, e.g. with coeliacs. So, physicians need additional information on manufacturing, processing, and pharmacovigilance. We also need to track all aspects, such as where agents are added and look for side effects; and ensure that reporting bad outcomes is reliable.
Overall, transparency is needed at each step, e.g. between the doctor, pharmacists, pharmacy benefit managers.
Mr Reilly highlighted that, at the conception of the ASBM, key to this was data sharing. In the case of Australia where policymakers created a force-switch policy with an opt out option. They did not believe that physicians would take the extra step to mark ‘DISPENSE AS WRITTEN’ (DAW), to prevent switching. However, it became apparent that 98% of physicians were marking DAW as there was an absence of data that would support switching to biosimilars, and they did not have confidence in the switches. He noted that, when described the US’s interchangeable biosimilars concept, many Australian physicians expressed that they would have a different perspective on switching as the interchangeable products have additional data supporting the biosimilars. Overall, where biosimilars only programmes have been pushed, the absence of negative data has often been highlighted, stating, ‘having data out there is key’.
Question 3: Please comment on when is the appropriate time for an emerging market to start working on collecting data on biosimilars and, with your knowledge, how would you roll that out in a region/country that is starting to grapple with this?
Data demonstrating the safe use, including the safe switching, of biosimilars is absolutely paramount to building physician and patient confidence, and thus, to building a successful biosimilar market from the very beginning stages. We have seen this time and again with physicians we have surveyed across the globe. For example, when asked if switching studies should be conducted before biosimilar substitution should be permitted, overwhelming majorities of physicians in Canada (82%) and Australia (81%) agreed. Physician societies in these countries – including the Canadian Gastroenterological Association (CGA) and Australian Rheumatology Association (ARA) – have repeatedly echoed these views in their own policy statements.
Recent US survey data show that the benefits of data are not merely theoretical. The FDA’s additional data requirements for a biosimilar to be classified ‘interchangeable’, i.e. automatically substitutable at a US pharmacy, make the majority of physicians (57%) more likely to prescribe the biosimilar. Further, it makes the majority (59%) more comfortable with a third-party biosimilar substitution in place of the prescribed reference product.
Too often, however, policymakers confuse or conflate the mere absence of negative data with what physicians really want – the presence of positive data. It is also important that the right data be gathered. Generally speaking, physicians have expressed a desire for robust clinical studies and greater post-marketing surveillance to demonstrate there has been no loss of efficacy or additional adverse events due to switching. Policymakers, however, especially in countries which artificially try to boost biosimilar market share through forced substitution, have either ignored these requests or offered unusual or inappropriate metrics – such as tracking increased hospitalizations. This mismatch has led to biosimilar markets struggling to gain physician support, as we have seen in Canada and Australia, jeopardizing both confidence and long-term sustainability of their markets. Emerging markets should learn from these missteps and support robust data collection about patient outcomes as a key feature of their biosimilar programmes.
Question 4: How does regulatory policy involving biologicals and biosimilars play into the rising concern over potential shortages?
Professor Schneider had just attended an FDA webinar on shortages where a report by an FDA taskforce on drug shortages [26] was shared. This included root causes of drug shortages based on analysis and also made recommendations.
The root causes for drug shortages were:
Recommendations included:
Mr Hargan noted that on the logistical side, in the US, onshoring of medical supplies is likely to increase following the shortcomings highlighted in the pandemic. These were highlighted pre-pandemic but were not acted upon in time which led to shortages. We also now see a paradox where we are more likely to see shortages of low-cost drugs.
Mr Pettit added that from a manufacturer’s perspective, they compete on price, plus things such as manufacturing and reliability of supply. In reality, not all manufactures play ‘all keys on the keyboard’. Some policies that tilt the playing field or restrict competition, do not allow for a robust competitive marketplace and these can trigger situations where shortages are more likely, e.g. all or nothing tenders in Europe that make only a single product available. He stressed that manufacturers are all competing and all have different levels of investment in how they manufacture and the way they ensure reliable supply.
Concurrent online Q&A
Due to the nature of the webinar, the audience had the opportunity to ask questions throughout the day, by submitting them online during the presentations and Q&A session.
Question 1: Directed at Mr Reilly, ‘I do not agree that uptake in Canada for filgrastim 93% market share (MS), pegfilgrastim 95% MS, bevacizumab 85% MS, trastuzumab 70% MS, adalimumab just been launch before force switching infliximab was 5% MS only from 2015 to 2020 now more than 50%’.
Mr Reilly responded that the IQVIA presenter ended the presentation by showing a slide of US and EU and referenced Canada. The issue was whether or not forced switch is sustainable and also the degree to which Canada is diametrically opposite from lessons learned from EU and the US. I would welcome any feedback and data that you would like to share, particularly at the upcoming webinar that will focus on non-medical switching.
Question 2: Following Professor Schneider’s presentation clarification was requested about the Canada policy and forced switching
Professor Schneider responded that focus on Canada’s policy has been on the forced switch aspect of it which takes the physician out of the equation. IQVIA recently presented data in another webinar showing that Canada is doing poorly on uptake relative to the US and EU even with a forced switch approach. This was also a lesson learned from Australia when they began with a forced switch approach but allowed the physician to write DAW [Dispense as Written], wherein they defaulted to DAW. Canada is open to multi-biosimilar switching not to just one biosimilar.
Question 3: Following Dr McKibbin’s presentation it was noted that the surveys are not based on the same period of time (2016 and 2017 Canada, to 2021 comparing different countries). In the short lifetime of a biosimilar this is a huge timeframe, how is this reflected in the results?
Dr McKibbin acknowledged that the surveys were conducted over a range of time and work has constantly been done to update these. The US survey was redone in 2021, the EU in 2019 and we are just updating Canada now. We only compare them to demonstrate clear trends.
Question 4: Directed at Mr Reilly, do you have some surveys in Norway and Denmark where the system is different versus major other markets?
Mr Reilly responded that the EU surveys were over the five western EU countries (Spain, Italy, UK, France and Germany) with the most recent survey including Switzerland. We are certainly open to surveying those countries but there is a significant cost for these surveys. Do you believe that it would provide useful information and worth doing?
Concerning the question of ‘If we are looking to Europe as a model of biosimilar adoption and use, what are the opinions in regard to animal toxicology studies, extensive phase III studies, interchangeability studies which are either not performed or performed on a more efficient and expedited fashion in Europe?’ This topic will be dealt with in an upcoming webinar.
The webinar provided the opportunity to gain insight on key elements contributing to the wide uptake of biosimilars in Europe and the US. Surveys were presented that described the physicians’ trust in prescribing and switching of biosimilars. The challenges and uptake policies of biosimilars in Europe were outlined, and the status of market access of and successful uptake measures implemented for biosimilars in the US was explored. In addition, the webinar highlighted issues in regions such as Canada and Australia, where forced-switching policies have been introduced. The webinar also provided the opportunity to evaluate the role of healthcare providers (physicians, pharmacists) and health policymakers in biosimilars use, and identify educational needs to enhance knowledge on biosimilars use globally.
The Generics and Biosimilars Initiative (GaBI) wishes to thank all speakers and moderator in delivering the presentations, implementing the panel discussion and clarifying information when finalizing the meeting report, as well as Mr Michael S Reilly for his strong support through the offering of advice and information during the preparation of the webinar.
The authors would like to acknowledge the help of the webinar speaker faculty and all participants, each of whom contributed to the success of the webinar and the content of this report, as well as the support of the moderator in facilitating meaningful discussion during the panel discussions, and contributing to the finalization of this meeting report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI JournalEditor, in preparing and finalizing this meeting report manuscript.
Speakers
The Honourable Eric D Hargan, JD
Ralph McKibbin, MD, FACP, FACG, AGAF
Chad Pettit, MBA
Michael S Reilly, Esq
Professor Philip J Schneider, MS, FASHP, FASPEN, FFIP
Andrew Spiegel, Esq
Moderator
Steven Stranne, MD, JD
Speakers and moderator had provided feedback on the article content and panel discussion, read and commented the revised content of the manuscript, and approved the final report for publication.
Competing interests: The webinar was funded by ASBM.
Provenance and peer review: Not commissioned; externally peer reviewed.
Michael S Reilly, Esq
Professor Philip J Schneider, MS, FASHP, FASPEN, FFIP
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Sep 16]. Available from: www.gabionline.net/biosimilars/general/biosimilars-approved-in-europe
2. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in the US [www.gabionline.net. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Sep 16]. Available from: www.gabionline.net/biosimilars/general/biosimilars-approved-in-the-us
3. Key factors for successful uptake of biosimilars: Europe and the US [webinar]. Alliance For Safe Biologic Medicines (ASBM) and Generics and Biosimilars Initiative (GaBI). 29 June 2022. Available from: https://gabiworkshop.wixsite.com/asbm1-1
4. Mulcahy A, Buttorff C, Finegold K, El-Kilani Z, Oliver JF, Murphy S, et al. Projected US savings from biosimilars, 2021-2025. Am J Manag Care. 2022;28(7):329-35.
5. European Medicines Agency. Marketing authorisation [homepage on the Internet]. [cited 2022 Sep 16]. Available from: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation
6. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Sep 16]. Available from: www.gabionline.net/guidelines/EU-guidelines-for-biosimilars
7. GaBI Online – Generics and Biosimilars Initiative. Biosimilars use in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Sep 16]. Available from: www.gabionline.net/Reports/Biosimilars-use-in-Europe
8. GaBI Online – Generics and Biosimilars Initiative. EMA approves first monoclonal antibody biosimilar [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Sep 16]. Available from: www.gabionline.net/biosimilars/news/EMA-approves-first-monoclonal-antibody-biosimilar
9. Generics and Biosimilars Initiative Journal (GaBI Journal). A white paper: US biosimilars market on pace with Europe. 2020;9(4):150-4. doi:10.5639/gabij.2020.0904.025
10. U.S. Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product. Guidance for industry. 2019 [homepage on the Internet]. [cited 2022 Sep 16]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-demonstrating-interchangeability-reference-productguidance-industry
11. Feldman MA, Reilly MS. European prescribers’ attitudes and beliefs on biologicals prescribing and automatic substitution. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(3):116-24. doi:10.5639/gabij.2020.0903.020
12. Jensen AR. Biosimilar product labels in Europe: what information should they contain? Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):38-40. doi:10.5639/gabij.2017.0601.008
13. Murby SP, Reilly MS. A survey of Australian prescribers’ views on the naming and substitution of biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):107-13. doi:10.5639/gabij.2017.0603.022
14. Dolinar RO, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018.
15. Reilly MS, Gewanter HL. Prescribing practices for biosimilars: questionnaire survey findings from physicians in Argentina, Brazil, Colombia and Mexico. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):161-6. doi:10.5639/gabij.2015.0404.036
16. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
17. McKibbin RD, Reilly MS. US prescribers’ attitudes and perceptions about biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2022;11(3):96-103. doi:10.5639/gabij.2022.1103.016
18. Reilly MS, Schneider PJ. Policy recommendations for a sustainable biosimilars market: lessons from Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):76-83. doi:10.5639/gabij.2020.0902.013
19. Xcenda. Biosimilar approval and launch status in US. April 2022 [homepage on the Internet]. [cited 2022 Sep 16]. Available from: https://www.xcenda.com/biosimilars-trends-report
20. U.S. Food and Drug Administration. Biosimilar product information. 2022 [homepage on the Internet]. [cited 2022 Sep 16]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
21. European Medicines Agency. Authorised medicines: biosimilar [homepage on the Internet]. [cited 2022 Sep 16]. Available from: https://www.ema.europa.eu/en/medicines/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar
22. Data on file, Amgen; Reference Product and Biosimilars – WAC and ASP Price; May 2022
23. Data on file, Amgen; Reference Product and Biosimilar Share Trends; May 2022.
24. Data on file, Amgen; Biosimilars Spend Analysis; May 2022.
25. Global Colon Cancer Association and World Patients Alliance. Biosimilars Training for Patient Advocacy Organizations. Available from: www.learnbiosimilars.org/
26. U.S. Food and Drug Administration. Drug shortages. Root causes and potential solutions [homepage on the Internet]. [cited 2022 Sep 16]. Available from: https://www.fda.gov/media/131130/download
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2022 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/key-factors-for-successful-uptake-of-biosimilars-europe-and-the-us.html
Author byline as per print journal: Professor Philip J Schneider1, MS, FASHP, FASPEN, FFIP; Michael S Reilly2, Esq
|
Canada has approved a total of 36 biosimilars. While the approval of biosimilars is regulated at the national level, decisions about biosimilar substitution are made at the provincial level. Four Canadian provinces, representing around 50% of the population in Canada, have now implemented policies requiring non-medical switching of biosimilars – switching from a patient from an originator biological to a biosimilar primarily for economic reasons. In this article, we compare biosimilar substitution policies in Canada to policies in Europe and the US, finding an enhanced focus on clinical and marketplace factors in these regions. We also find evidence that in some cases non-medical switching may pose a risk to patients and suggest that Canada could learn from more mature markets, such as those in Europe, where switching policies better consider patient needs, preserve physician choice and promote market competition. |
Submitted: 29 May 2021; Revised: 22 July 2021; Accepted: 26 July 2021; Published online first: 30 July 2021
The rising cost of health care is a matter of global concern. Drug pricing has been a growing part of the discussion about this issue and the high cost of new biologicals medicines has contributed to the concern. While these therapies have been medical breakthroughs, access to them because of cost has been a concern, as has the potential for bankrupting the funding for health care. Health authorities and payers are scrambling to find solutions to this dilemma.
Biosimilars – copies of biological drugs that are similar but not identical to the product on which they are based – have proliferated significantly over the past decade as the patents for several biologicals have expired. Biosimilars have been developed across a variety of therapeutic areas globally, including oncology and inflammatory diseases. The rationale for creating biosimilars is to promote competition among manufacturers to lower prices, thereby increasing access to expensive biological medicines. A competitive marketplace typically produces price competition in general, and for biological medicine this is no exception. The unexpected challenge has been realizing the potential savings.
There has been the temptation to apply the lessons learned from the creation of generic versions of simple molecules to biological medicines and biosimilars. This is understandable because of the substantial saving resulting from price competition, price reductions, and the ability to freely substitute a much less expensive generic version is dispensed when a more expensive brand name product is prescribed. This model is not possible for biologicals because while highly similar, biosimilars are not identical to the reference product with which they compete. This has prompted many studies of the comparability of biosimilars to their reference product, including switching studies where patients are switched from the reference product to a biosimilar. These studies have confirmed the similarity of biosimilars to reference products but may have been inconclusive about important clinical differences that may affect patients being treated. As a result, there have been a variety of different regulatory and policy approaches to the use of biosimilars around the world.
Regulatory and policy approaches to the use of biosimilars are usually developed with the goal of reducing drug costs by stimulating their use. These approaches should, but do not always have a goal in assuring the effectiveness and safety when biosimilars are used in the clinical setting. Other considerations include maintaining an environment for future innovation, sustaining a competitive market, and assuring a reliable supply chain. Good regulations and policies consider all these factors.
As of 30 April 2021, Canada has approved 36 biosimilars, and another 13 are under review [1]. While this is on par with the Food and Drug Administration’s (FDA) approval of 30 biosimilars [2] and the European Medicines Agency’s (EMA) approval of 73 biosimilars (13 of which are not considered biosimilars in the US) [3], Canada’s recent implementation of policies requiring a non-medical switch to biosimilar medicines in British Columbia, Alberta, New Brunswick, and Quebec stands in contrast to biosimilar substitution practices in the US and Europe and does not consider some of the factors used in other countries to foster the effective and safe use of biosimilars as ways to control rising healthcare costs are considered.
Health Canada originally issued their regulatory guidelines for biosimilars, Information and Submission Requirements for Biosimilar Biologic Drugs, in 2010 [4]. Since then, 36 biosimilars based on 13 reference products have been approved, see Table 1. To improve the efficiency of the regulatory review of drugs and devices and to support timely access to biological products, Health Canada issued a Regulatory Review of Drugs and Devices initiative (also known as ‘R2D2’) in 2017 [5]. A key objective of R2D2 was to work with local health partners, including health technology assessment organizations, to reduce the time between Health Canada approvals and reimbursement recommendations.
While the approval of biosimilars is regulated at the national level by Health Canada, decisions about biosimilar substitution are made at the provincial level. Policies requiring the switching of a patient’s medicine from an originator biological to a biosimilar primarily for economic reasons is referred to as ‘non-medical switching’ and have been avoided by Canadian payers until recently. Newer initiatives to realize potential savings opportunities offered by biosimilars have reflected a shift in the perspective of provincial payers, and four of Canada’s provinces, that are inhabited by approximately 50% of the country’s population, have begun implementation of biosimilar switching programmes, see Table 2. Other provinces are expected to consider similar biosimilar initiatives soon.
In May 2019, British Columbia announced that it would forcibly switch more than 20,000 of its arthritis, psoriasis and diabetes patients from their originator biological medicines to the government’s choice of preferred biosimilar products. In September of the same year, it was announced that an additional 1,700 patients with inflammatory bowel disease would be switched [6,7]. The province of Alberta announced plans to switch at least 26,000 patients, including those being treated with infliximab for ulcerative colitis and Crohn’s disease, from biological therapies (etanercept, infliximab, insulin glargine, filgrastim, pegfilgrastim) to biosimilars by the summer of 2020 [8]. The COVID-19 pandemic has delayed the implementation of these programmes. The recent implementation of forced non-medical switching policies by Canadian payers represents a departure are different from biosimilar substitution practices in the US and Europe in that they focus on economics, not clinical or marketplace factors.
In the US, pharmacy-level substitution of a biosimilar for an originator biological without physician consent is only permitted for biosimilars that have been designated ‘interchangeable’ by FDA. To receive an interchangeable designation, the product must meet additional requirements beyond being biosimilar, which translates to more clinical development – including switching studies. The rationale for this policy is based on the acknowledgement that biosimilars are similar but not identical to the reference product and to protect patients, additional evaluation of comparability is needed. Prior to 26 July 2021, no biosimilars in the US had been approved as interchangeable, therefore, non-medical switching has not existed in the US§.
In contrast, EMA does not have recommendations on interchangeability but the decision-making authority on substitution policies rests with the individual EU Member States. While automatic substitution is prohibited in most European countries, a few financially constrained countries in Central and Eastern Europe, e.g. Estonia, Latvia, Poland and Serbia, allow pharmacy-level substitution [9]. Particularly in Bulgaria, Poland and Serbia, tendering procedures are applied for purchasing biologicals and dictate which product a patient will receive. However, the prescription of switching to a biosimilar medicine in Europe most commonly occurs under the supervision of a physician in consultation with the patient. These policies are based on the responsibility and accountability of the physician for the care of their patient, while acknowledging the need to consider cost when making treatment decisions.
Successful biosimilar markets in the EU and US have demonstrated that forced medical switching is unnecessary to achieve high uptake of biosimilars and the associated savings. While the global biologicals market is dominated by the US, which has a share above 50%, Europe is the leader in biosimilar approval and commercialization [10]. The EU was the first to establish a regulatory framework in 2004 and has the largest biosimilar market in the world, representing ~60% of the global biosimilar market [10]. Biosimilars have attained market shares as high as 91% for older products (before the approval of the first monoclonal antibody biosimilar in 2013) and as high as 43% for newer products (approved post-2013) in some European markets. The success of the European biosimilar markets reveals three common principles: physician choice, not mandating automatic substitution, and promotion of competition [11-13]. This fosters a robust and sustainable biosimilar market with multiple suppliers in any given product class. Tenders are designed to include value-based criteria in addition to price and award multiple contracts not single-winner tenders to ensure continuity of supply and healthy competition [12]. Experience in the EU suggest that competition not regulation results in cost savings without compromising patient care [13].
The introduction of major reimbursement policy changes by Canadian payers that is intended to increase the use of biosimilars, has been criticized by various groups in Canada. Physicians, medical societies and patients themselves have argued that non-medical switching will adversely affect patient care. Furthermore, these policies conflict with principles believed to be the foundation for a sustainable biosimilars market.
At the behest of Quebec’s Ministry of Health and Social Services, the Institut National d’Excellence en Santé et Services Sociaux (INESSS) conducted a state-of-knowledge report about the risks associated with non-medical switching and the interchangeability of biologicals [14]. The systematic review on which this report is based indicated that the available scientific data are insufficient to support the safety of switching between originator biological and biosimilar, particularly in inflammatory bowel diseases and oncology, and that larger studies are needed to address the uncertainty associated with switching between biologicals in these indications. Furthermore, the report indicated that non-medical switching is generally not supported by clinicians due to the potential destabilization of complex patients, along with many other patient-related concerns. While clinicians in Quebec support the use of biosimilars in patients who have not used the corresponding reference product, they believe that switching to a biosimilar should be carried out under medical supervision. A survey of Canadian prescribers of biologicals conducted by the Alliance for Safe Biologic Medicines indicated that 83% of physicians across 13 therapeutic specialties considered it ‘very important’ or ‘critical’ that the prescribing physician decide the most suitable biological for their patients [15]. Most prescribers were not comfortable with a third-party switching a patient’s medicine for non-medical reasons.
In a joint statement from the Canadian Association of Gastroenterology and Crohn’s and Colitis Canada, non-medical switching was not recommended for patients who are stable on biological treatment [16]. Further, gastroenterologists across Canada do not support automatic substitution of any kind but supported starting treatment-naïve patients on biosimilar products if they had active disease and the price differential between the originator biological and the biosimilar is significant. The Gastrointestinal Society stated that reimbursement policies must recognize and respect the physician’s right to prescribe based on clinical evidence and a patient’s right to choose the therapy that is best for them [17].
The Biosimilars Working Group of Canada, a key collaboration of diverse non-profit organizations, registered health charities, and healthcare advocacy coalitions dedicated to ensuring good outcomes for patients, stated that the cost-driven objective of the forced-switch policy is worrisome as it fails to put physician wisdom, patient choice, appropriateness of care, accessibility, and affordability at the forefront of health policy [18]. When Canadian researchers surveyed patients with gastrointestinal diseases and their caregivers to determine their views on the use of biological and biosimilar drugs, 95% of those surveyed believed it was important that decisions regarding choice of medication be determined solely by the treating physician in collaboration with the patient [19].
The burden on healthcare systems is clear according to the Gastrointestinal Society’s report on the impact of British Columbia and Alberta’s non-medical switching policies. Patients reported experiencing delays with biosimilar doses, with some reporting shortages in availability, inadequate education on biosimilars, as well as the physical and mental toll they experienced in navigating their new treatment pathways [20].
It can be concluded that many Canadian physicians feel that non-medical switching of biological products poses a risk to patients [14-17]. As prescribers of biosimilar medicines, these physicians feel that it is critical for them to retain the right to make treatment decisions that best benefit their patients, see Table 3. While the opinions of these Canadian physicians are at odds with the forced non-medical switching policies of Canadian drug plans in British Columbia, Alberta, New Brunswick and Quebec, they are aligned with the views of experienced prescribers from the US and Europe [11,14].
Health policy decisions in Canada affecting the clinical use of biosimilars focuses primarily on economic factors and a strategy to reduce rising healthcare costs. While this is a laudable aim, it does not consider other factors that are important when making regulatory decisions. Factors that must be considered include the effectiveness and safety of therapy, the supply chain, and sustaining a healthy market for innovation and price competition. Mandatory switching for non-medical reasons does not consider that not all patients respond the same way to medications and that this can increase the financial burden on healthcare systems. In a recent systematic literature review conducted to evaluate the economic impact of non-medical switching, Liu et al. [20] identified 17 studies that reported an overall increase in real-world costs associated with non-medical switching. Higher rates of surgery (11%) increased steroid use (13%) and biosimilar dose escalations (6% to 35.4%) were cited as some of the reasons for the cost increases. Most studies evaluating the economic impact of non-medical switching consider only drug costs; however, a comprehensive evaluation should incorporate all elements of healthcare service needs [21].
For example, Alberta’s switching policy resulted in many unintended consequences, the health and financial impacts of which the province has said it needs to investigate [22]:
In Alberta, patients were switched from infliximab, etanercept, pegfilgrastim, and filgrastim originator biologics. They were also switched from insulin glargine originators, although in the United States there are no official biosimilars for these products yet.
Alberta ran into some problems. For example, some pharmacies began stocking only preferred biosimilars, which meant that patients and their providers could not choose. Alberta has attempted to remedy this problem.
The government has succeeded in switching over 60% of patients from reference infliximab to biosimilar infliximab, but there has been opposition. In addition, more than 15% of patients moved to a different biologic and roughly 15% dropped or terminated their government coverage, [Alberta Health Assistant Deputy Minister, Chad Mitchell] said. The government is attempting to find out the reasons for these coverage departures and treatment changes.
For the originator insulin glargine product Lantus, more than 40% of patients moved to a different biologic after switching was initiated, and 15% of beneficiaries dropped or terminated their coverage. Mitchell said 10% to 20% of beneficiaries in other biosimilar categories terminated coverage. More investigation is needed to understand the significance of these trends, he said.
Non-medical switching to biosimilar products may also have a negative impact on patient safety in cases where administration devices for self-administered biosimilars differ from the reference product [22,23]. Without proper guidance from a healthcare provider, biosimilar products available in different presentations compared to their reference products could lead to inappropriate use by patients or caregivers – again highlighting the need for physician responsibility in treatment decisions.
While the European biosimilars market has been credited with higher uptake compared to the US market, rates of uptake differ from country to country in Europe and can vary significantly by product class. A report by KPMG commissioned by Medicines for Europe to analyse the procurement of medicines in hospitals in eight European countries highlighted the variability in biosimilar sales against originator in these different Member States [24]. An average of hospital biosimilar volume in March 2019 showed that Denmark achieved 63% overall biosimilar volume, with the UK coming in second at 45%. Germany had 40% biosimilar volume, France had 34%, and Belgium tied with Switzerland for last place among the countries studied at 14%. In a recent assessment of the impact of biosimilar competition in Europe, 16 European countries were reported to have achieved > 90% biosimilar utilization for filgrastim and pegfilgrastim in 2018, while utilization in Ireland was just 27%. Among anti-tumour necrosis factor biosimilars (adalimumab, etanercept and infliximab), Norway and Denmark had 81% and 96% biosimilar uptake, respectively, while every other country’s utilization was less than 50% [25]. Variations in adoption rates among individual European countries as well as across therapeutic areas are influenced by government involvement, reimbursement structures and tender procurement policies.
In the US, biosimilars have gained significant share in the majority of therapeutic areas in which they have been introduced, ranging on average from 20% to 25% within the first year of launch, with some projected to reach greater than 50% within the first two years [26,27]. As expected, first-to-market biosimilars tend to capture a greater portion of the segment compared to later entrants. Filgrastim biosimilars have been on the market the longest at five years and have achieved a 72% share, while bevacizumab and trastuzumab biosimilars have approximately 40% share. Rituximab and infliximab have had the most limited adoption, with approximately 20% market share [25].
Canada desires a robust biosimilar market share like that observed in Europe. While there may be short-term savings from non-medical switching, a long-term consequence of this policy may be decreased competition resulting from fewer products launches and a negative impact on patient safety. Potential drug shortage issues may develop in the absence of multiple suppliers of biologicals in each product class. There would also likely be undermining the confidence of both physicians and patients in biosimilars that creates an additional barrier to biosimilar uptake.
The successful uptake of biosimilars in Europe was not accomplished by limiting the choice of biological or forced non-medical switching, but through preserving choice for physicians and patients and by promoting competition among all products approved by regulatory authorities. To foster its success in creating a sustainable biosimilars market, Canadian payers can learn from the lessons learned in more mature markets and implement evidence-based transition policies that consider patients’ needs primary.
This paper is funded by the Alliance for Safe Biologic Medicines (ASBM).
The ASBM is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion.
The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Professor Philip J Schneider is a member of the International Advisory Board of Alliance for Safe Biologic Medicines (ASBM) since 2012 without compensation. From September 2014, Emeritus Professor Schneider has been the Chair of the International Advisory Board of ASBM and is paid a small stipend for that role. Mr Michael S Reilly, Esq is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002 to 2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
§On 26 July 2021, the US FDA approved its first interchangeable biosimilar, the insulin glargine product Semglee. To be designated interchangeable, a biosimilar must provide additional data to FDA demonstrating that a patient switched repeatedly between the biosimilar and the originator product can expect the same clinical result without additional risks, compared to a patient who remained on the originator product. As of April 2021, all US states permit interchangeable biosimilars to be substituted at the pharmacy level without prior physician authorization.
Professor Philip J Schneider1, MS, FASHP, FASPEN, FFIP Michael S Reilly2, Esq, Executive Director
1College of Pharmacy, The Ohio State University, 500 West 12th Avenue, Columbus, OH 43210, USA
2Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Wojtyra U. Update on biosimilars in Canada – April 2021. JD Supra. 2021 May 3. [homepage on the Internet]. [cited 2021 Jul 22].Available from: https://www.jdsupra.com/legalnews/update-on-biosimilars-in-canada-april-7703939/
2. U.S. Food and Drug Administration. Biosimilar product information. FDA-approved biosimilar products [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
3. European Medicines Agency. Medicines [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar
4. Health Canada. Guidance document: information and submission requirements for biosimilar biologic drugs [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs-1.html
5. Health Canada. Improving the regulatory review of drugs and devices [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.canada.ca/en/health-canada/corporate/transparency/regulatory-transparency-and-openness/improving-review-drugs-devices.html
6. AJMC. British Columbia becomes the first Canadian province to mandate a switch to biosimilars [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.centerforbiosimilars.com/view/british-columbia-becomes- the-first-canadian-province-to-mandate-a-switch-to-biosimilars
7. AJMC. British Columbia starts second phase of switching patients to biosimilars, this time for IBD [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.centerforbiosimilars.com/view/british-columbia- starts-second-phase-of-switching-patients-to-biosimilars-this-time-for-ibd
8. Mushtaq A, Kazi F. Switching to biosimilars: boon or bust? Lancet Gastroenterol Hepatol. 2020;5(4):341-2.
9. La Noce A, Ernst M. Switching from reference to biosimilar products: an overview of the European approach and real-world experience so far. EMJ. 2018;3(3):74-81.
10. Schneider PJ, Reilly MS. Policy recommendations for a sustainable biosimilars market: lessons from Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):76-83. doi:10.5639/gabij.2020.0902.013
11. Reilly MS, Spiegel A. 1633PD – Biosimilar substitution: European prescriber perspectives. Poster presentation at: Annual Congress of the European Society of Medical Oncology; 27 September–1 October 2019; Barcelona, Spain.
12. Feldman MA, Reilly MS. European prescribers’ attitudes and beliefs on biologicals prescribing and automatic substitution. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(3):116-24. doi:10.5639/gabij.2020.0903.020
13. Vulto AG, Vanderpuye-Orgle J, van der Graaff M, Simoens SRA, Dagna L, Macaulay R, et al. Sustainability of biosimilars in Europe: a Delphi panel consensus with systematic literature review. Pharmaceuticals (Basel). 2020; 13(11):400.
14. INESSS. Report: Innocuité de la substitution et de l’interchangeabilité des médicaments biologiques (Safety of substitution and interchangeability of biologic drugs). Mai 2020 [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Rapports/Medicaments/ INESSS_Biosimilaires_EC.pdf
15. Alliance for Safe Biologic Medicines. Survey: Canada prescribers and biosimilars. October 2017 [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://safebiologics.org/wp-content/uploads/2017/09/2017-Canada-Prescriber- Survey-Final-Report.pdf
16. Moayyedi P, Benchimol EI, Armstrong D, Yuan C, Fernandes A, Leontiadis GI. Joint Canadian Association of Gastroenterology and Crohn’s Colitis Canada position statement on biosimilars for the treatment of inflammatory bowel disease. J Can Assoc Gastroenterol. 2020;3(1):e1-e9.
17. Gastrointestinal Society. Canadian Society of Intestinal Research. Balancing act: cost containment vs patient health outcomes [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://badgut.org/balancing-act/
18. Biosimilars Working Group. Biosimilars Working Group responds to phase II of the BC PharmaCare Biosimilars Initiative [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://biosimilaroptions.ca/2019/09/18/reaction-to-biosimilars-initiative/
19. Attara G, Bailey R, Bressler B, Marshall J, Panaccione R, Aumais G. P433. Canadian patient and caregiver perspectives on subsequent entry biologics/ biosimilars for inflammatory bowel disease. J Crohns Colitis. 2016;10:S319.
20. Gastrointestinal Society. Forced to switch: Canadian biosimilar experience survey report. December 2020 [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://badgut.org/biosimilars-survey-report-2020/
21. Liu Y, Yang M, Garg V, Wu EQ, Wang J, Skup M. Economic impact of non-medical switching from originator biologics to biosimilars: a systematic literature review. Adv Ther. 2019;36(8):1851-77.
22. AJMC. Mandatory biosimilar switching pays off for Canada [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.ajmc.com/view/mandatory-biosimilar-switching-pays-off-for-canada
23. Ebbers HC, Schellekens H. Are we ready to close the discussion on the interchangeability of biosimilars? Drug Discov Today. 2019;24(10):1963-7.
24. KPMG. Improving healthcare delivery in hospitals by optimized utilization of medicines: a study into 8 European countries. Commissioned by Medicines for Europe. 2019 [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2019/10/ 20190903_Hospital-Reform-Study_final.pdf
25. IQVIA. White paper. The impact of biosimilar competition in Europe. Oct 2020 [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://ec.europa.eu/docsroom/documents/31642/attachments/1/translations/en/renditions/native
26. Amgen. 2020 Biosimilar trends report [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.amgenbiosimilars.com/-/media/Themes/Amgen/amgenbiosimilars-com/Amgenbiosimilars-com/spandf/USA-CBU-80723-2020-Amgen-Biosimilar-Trends-Report.pdf
27. IQVIA. Biosimilars in the United States 2020-2024. Competition, savings and sustainability. October 2020 [homepage on the Internet]. [cited 2021 Jul 22]. Available from: https://www.iqvia.com/-/media/iqvia/spandfs/institute-reports/iqvia-institute-biosimilars-in-the-united-states.pdf
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2021 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-critical-review-of-substitution-policy-for-biosimilars-in-canada.html
Submitted: 9 July 2020; Revised: 9 August 2020; Accepted: 19 August 2020; Published online first: 26 August 2020
Healthcare systems across the globe face resource and budget constraints. Biosimilar drug products offer less expensive alternatives to brand-name originator drug products and can thus offer some relief to healthcare costs. Biosimilars are highly similar and have no clinical meaningful differences; but are not identical to originator biologicals. As countries seek to control health costs and expand access to biological therapies, building physician confidence in biosimilars is critical to promoting their use and reaping the cost benefits.
The European Union (EU) and the European Medicines Agency (EMA) have led the development of a regulatory framework for biosimilars. In 2005, EMA established the first biosimilars approval pathway that was distinct from generics approval [1]. Since then, EMA has developed and refined a comprehensive set of regulatory guidelines on which biosimilar applications are reviewed and approved or rejected. By the end of 2019, 58 biosimilars of 15 originator biological medicines have a marketing authorization in Europe [2]. The European biosimilars market is currently the largest in the world, representing approximately 60% of the global biosimilar market and growing consistently year on year [3].
At present, once authorized, EMA applies a ‘same-label’ (generic) approach to biosimilar product labels [4]. However, there are concerns over whether this is sufficient to ensure appropriate drug switching and product traceability. There is ongoing debate about what information is appropriate in the naming and labelling of biosimilars. In the US, FDA released its requirements for the non-proprietary naming of biological products in January 2017 [4]. Prior to this, the Alliance for Safe Biologic Medicines (ASBM) carried out surveys of Australia and EU prescribers and US pharmacist perspectives on the naming of these products. Overall, both groups believed that naming should make biosimilars distinguishable from originator products [5–7]. The ASBM is an organization composed of diverse healthcare groups including patients, physicians and medical innovators. It is funded by its many member partners that are made up of international organizations and companies [8].
The interchangeability of biosimilars is viewed differently in countries across the world. This is particularly marked by the approaches to interchangeability and substitution in the US and Europe [9]. ‘In the US, insurance mandates can result in formulary changes requiring patients to be switched from a reference product to a biosimilar strictly for cost reasons’ . In Europe, automatic substitution of originator biologicals with biosimilars is rare as this practice excludes physicians from decisions regarding the treatment of patients. There have been a number of surveys and workshops carried out across the world (Australia [5], Europe [4, 6], South America [10] and the US [11]) that have asked for prescriber opinions on prescribing practices, naming and labelling of biologicals. In terms of naming, prescribers in Australia, Europe and the US, overall, agreed that there is a need for distinguishable non-proprietary names to be given to all medications. In South America, knowledge about biosimilars varied in different countries surveyed (Argentina, Brazil, Colombia and Mexico) and revealed gaps in understanding and in the use of distinguishable names for biologicals.
In 2019, the ASBM commissioned 15-minute web-based surveys to be carried out by biological prescribers in six Western European countries (France, Germany, Italy, Spain, Switzerland and the UK) to document their perspectives on biological substitution. This survey mirrors their previous European prescriber survey conducted in 2013 [6] (both survey reports can be found at www.safebiologics.org/surveys).
Overall, the 2019 survey showed that awareness of biosimilars in the countries had increased since the 2013 survey. Specifically, more physicians (90%) rated themselves as being ‘Familiar’ or ‘Very familiar’ with biosimilars than did in 2013 (76%). A strong majority of respondents (82%) felt that it is either ‘Very important’ or ‘Critical’ for them to decide which biological medicine is dispensed to their patients, representing a 10% increase over the results of the 2013 survey. Again, a strong majority of respondents (84%) considered authority to prevent a substitution either ‘Very important’ or ‘Critical’ , another 10% increase over the 2013 findings. In 2019, physicians remained uncomfortable with switching a stable patient to a biosimilar for non-medical reasons. Since the 2013 survey, there has also been a sharp increase in physicians who are highly uncomfortable with a non-medical substitution performed by a third party.
It is hoped that the findings of this study may serve as a resource for other countries in developing biosimilar policies that can build physician confidence in biosimilars. Confidence that will increase biosimilar uptake and reduce government expenditures on biological products.
In March 2019, 579 prescribers practising in six specified European countries (France, Germany, Italy, Spain, Switzerland and the UK) completed the 15-minute web-based survey that was administered in the respondents’ native language – French, German, Italian, Spanish, or English. The survey was commissioned by the ASBM and was a refreshed version of that carried out in 2013 [6]. The questionnaires were developed as a collaboration among ASBM management, ASBM membership and Industry Standard Research (ISR) management. No ‘validation’ was conducted as the instruments did not measure higher level ‘constructs’ . They are purely direct measures of opinion and attitude.
Potential respondents were identified in – and recruited from – a large, global, commercial database/panel of healthcare professionals. The response rate was high because people in this database/panel have already indicated a willingness to participate in market research. In addition, their specialties were known prior to recruitment, which decreased the rate of disqualification, as if someone was identified as representing a specialty that did not qualify for the study, they were not invited.
Respondents were paid a stipend for their participation. Stipends ranged from US$37.00 to US$48.00, depending on the specialty.
Prescriber eligibility criteria
Online survey
The surveys were administered by ISR. In summary, prescribers were asked to rate:
ISR provided statistical significance tests by country and practice area for most questions. The information from these tests made it possible to determine which answers were most significant amongst prescribers from different countries and working in different practice areas.
Information on survey participants
Participants were sourced from six countries and across 10 therapeutic areas. The detailed breakdown of this information is as follows. Table 1 provides details of the survey sample disposition.
A total of 579 responses were received:
The breakdown of the practitioner’ s primary therapeutic areas is as follows:
Neurology (14%), Rheumatology (14%), Gastroenterology (13%), Ophthalmology (12%), Nephrology (12%), Endocrinology (11%), Dermatology (10%), Oncology (6%), Immunology (5%) and Haematology-Oncology (3%).
The largest group of prescribers (47%) practice in a hospital setting, with the remainder in academic medical centres (23%), private/family practice (18%), multi-specialty clinics (8%), community settings (3%), and other settings (1%).
Respondents’ mean experience level was 15.5 years in practice. Forty per cent of responders had been practising medicine for 11–20 years, 24% for more than 21 years, 23% for 6–10 years, and 13% for 1–5 years.
Seventy-nine per cent of responders said they commonly treat patients who are using biological medicines prescribed by another healthcare provider.
Respondents use different sources to learn about the details of a medicine for prescribing and monitoring, see figure 1.
All data refer only to those who completed the survey. All data were analysed in MS Excel and checked manually.
Familiarity with biosimilars
Familiarity with biologicals versus biosimilars
When asked about familiarity with biological medicines, 58% of prescribers said they are ‘Very familiar’ , and have a complete understanding of them, compared to 41% who said the same about biosimilars. Thirty-seven per cent of prescribers said they are “Familiar”, with a basic understanding of biologicals, compared to 49% who said the same about biosimilars. And 4% had heard of biologicals but could not define them, compared to 8% who said the same about biosimilars. All prescribers had heard of biologicals whereas 2% of prescribers have not heard of biosimilars.
Since the 2013 European prescribers study, familiarity with biosimilar medicines increased from 76% to 90%; and a prescriber’ s awareness that a biosimilar may be approved for several or all indications of the reference product on the basis of clinical trials in only one of those indications increased from 63% (2013) to 83% (2019.) Strongest familiarity with biosimilars was among prescribers in Italy, Spain and Germany (48%, 47% and 44% are very familiar/have complete understanding). Prescribers in Switzerland had the lowest familiarity with only 31% stating they are very familiar/have complete understanding of biosimilars; 19% of Swiss prescribers either could not define biosimilars or have never heard of biosimilars.
Strongest familiarity with biological medicines was among Rheumatology and Gastrointestinal prescribers (96% and 88% are very familiar/have complete understanding) when compared to the other practice areas. Strongest familiarity of biosimilars was also among Rheumatology, Gastrointestinal and Endocrinology prescribers (70%, 61% and 60% are very familiar/have complete understanding).
Preferred route to familiarity
Of the respondents (n = 517) that said they were very familiar/familiar with biosimilar medicines. The top five sources of information were: 1) scientific publications (70%); 2) national medical conferences/symposia (70%); 3) international medical conferences/symposia (61%); 4) self-study (42%); and 5) CME/IME (40%).
The sources varied among the countries. For example, prescribers in the UK became more familiar with biosimilars through self-study (66%) and scientific publications (56%), while in Spain scientific publications (73%) and CME/IME (66%) were the most utilized.
The top five sources to learn about biosimilars among the respondents (n = 62) who had never heard of, nor could define biosimilar medicines were: 1) scientific publications (68%); 2) international medical conferences/symposia (61%); 3) national medical conferences/symposia (55%); 4) CME/IME (37%); and 5) reference product company sponsored education (35%).
There were no significant differences in the preferred method for becoming familiar with biosimilars among practitioners in different countries.
Biosimilar approval awareness
Prescribers in Italy (94%) had significantly higher biosimilar approval awareness compared to the rest of the countries. The specialties with the highest biosimilar approval awareness were Rheumatology (96%), Endocrinology (95%), Oncology (94%) and Gastrointestinal (92%) prescribers. All had significantly higher awareness than other specialties.
Adverse drug reaction reporting: mechanism, recording, information required, barriers
The survey showed that four out of five prescribers are legally required to report adverse drug reactions (ADRs) that are brought to their attention.
Italian prescribers rated the highest percentage for being required to report ADRs (96%), and French prescribers the lowest (69%). The practice areas in which the highest number of prescribers are required to report ADRs were Oncology (91%) and Immunology (90%).
More than half (54%) of prescribers said they are most likely to report an ADR to the National Competent Authority (NCA). The UK is significantly more likely to report to a combination of the NCA, the Marketing Authorization Holder (MAH), i.e. the manufacturer, and EMA (54%) as opposed to the NCA alone (29%).
ADR report mechanisms, time spent and follow-up
Email was utilized by almost half (49%) of prescribers (n = 550) to report ADRs to the NCA or MAH. However, when looking at specific countries, prescribers in Germany (58%) and the UK (52%) had a majority preference for paper. Prescribers in France (57%) and Italy (60%) had a preference for email, while Spain (53%) preferred a web-based tool/app.
Two-thirds (65%) of prescriber respondents said that the amount of time spent on filing a report ranged from 10 to 20 minutes, with 25% requiring less than 10 minutes and 10% requiring more than 20 minutes (average 36 minutes) While prescribers do file detailed reports, the time varies among the specialties. Dermatology (38%) prescribers need less time to file compared to other practice areas, whilst Neurology (19%), Immunology (17%) and Nephrology (14%) prescribers need more than 20 minutes to file the ADR report.
In terms of follow up from the NCA or MAH, 24% of prescribers responded they always receive follow-up, 21% very often, 30% sometimes, 19% rarely and 6% never. Prescribers in Switzerland have one of the highest rates of follow up from reporting entities (Always, 35%) compared to several other countries.
Information included in the ADR reports
When ADR reports are filed for a biological medication, 92% of practitioners responded that information about the ADR experienced by the patient are included, 84% include brand name of the biological suspected to have caused the incident, 80% include date and time of report, 72% include the non-proprietary name of the biological suspected to have caused the incident, 69% include batch number of the biological suspected to have caused the incident, including the manufacturer of the product suspected to have been associated with the reaction.
Prescribers in Italy (79%) are better about including batch number in the ADR report; Germany (74%) prescribers are better about including the manufacturer of the product; prescribers in the UK (90%) are better about including date and time.
When asked about how frequently the NCA or MAH follow-up to request the brand name or manufacturer of the product, 55% of prescribers responded either always or very often, 28% said sometimes, while 18% said rarely or never.
Fifty-five per cent of practitioners said that the level of detail required in ADR reports deters them from reporting minor events. When looking at the country specific data, prescribers in France are significantly more deterred from reporting minor events, while those in Italy are significantly less deterred.
Barriers to reporting ADRs
Fifty-five per cent of the prescribers responded that the amount of information necessary to report an adverse drug reaction deters them from reporting minor events. France (74%) is significantly more deterred from reporting minor events, while Italy (38%) is significantly less deterred.
More than half (56%) of prescribers responded that reporting infrastructure, e.g. the mechanism of reporting ADRs, was the biggest barrier to accurate reporting; another 20% responded no barriers exist. When looking at the country specific data, prescribers in Spain identified reporting infrastructure (70%) and lack of integration of electronic health records (55%) as barriers to accurate reporting more so than most countries.
Nearly all prescribers responded that they were somewhat confident (62%) or highly confident (36%) in the European pharmacovigilance system’ s ability to accurately identify the specific product at the brand-name level that might be responsible for the ADR. However, prescribers in the UK were less confident in the European pharmacovigilance system than the other countries surveyed, with only 24% reporting they were ‘highly confident’ the system would be able to accurately identify the product responsible.
Frequency of including batch number when reporting adverse events was mixed; 37% always, 27% very often, 20% sometimes, 17 % rarely/never. The survey showed that prescribers in Italy (55%) were best about including batch number (always) when compared to most of other countries. Of the prescribers who said they only included batch number sometimes, rarely, or never, more than half (53%) of prescribers responded that the reason for this was due to not having it available at time of reporting.
Automatic substitution, switching and physician choice
A high majority of prescribers (82%) feel very strongly about having control over what is prescribed and dispensed to their patients.
Opinion on sole authority for prescribers
Most prescribers agreed that it is either critical or very important (82%) that they had the sole authority, together with their patients, to decide on the most suitable biological medicine for their disease. When looking at each country, it is significantly more critical to have sole authority in deciding medicine for prescribers in Italy (94%), Switzerland (91%) and Germany (84%). When looking at specific fields, it was most important/critical to have sole authority in deciding biological medicine for Immunology (86%), Dermatology (86%) and Ophthalmology (86%) prescribers. It was least important/critical for Haematology-Oncology prescribers, 20% of whom considered it slightly/not important, compared to an average of 2% across all specialties which thought this, see figure 2.
It is significantly more critical to have sole authority in deciding medicine for Immunology, Rheumatology, Dermatology and Endocrinology.
Government tenders
Most prescribers stated that they believe it is very important or critical (63%) that government tenders for biosimilars are awarded to multiple suppliers. Prescribers in Spain and the UK, while considering this very important, do not think it is as critical for government tenders to be awarded when compared to the other countries surveyed. Only 7% and 9% considered this ‘critical’ compared to an 18% average across all other respondents.
Most prescribers agreed that it is either critical or very important (83%) that factors besides price to be taken into account in national tender offers, e.g. reliability of supply, patient support services, manufacturer reputation.
Prescriber authority to deny substitution
Most prescribers agreed that it is either critical or very important (84%) that, in a situation where substitution by a pharmacist was an option in their country, they have the authority to designate a biological medicine as ‘DISPENSE AS WRITTEN’ or ‘DO NOT SUBSTITUTE’ . It was significantly more critical for those in Switzerland (94%) to have authority to deny substitution for a biological medicine, and least so for those in the UK (73%), compared to those in the other countries. It was significantly less important for Haematology-Oncology prescribers to be able to deny substitution when compared to almost all other practice areas, see figure 3.
Identifying medicines
Eighty-five per cent of prescribers said that, when prescribing medicine including biologicals, they identify the medicine in the patient record by brand name. When looking at country and practice area responses, UK (68%) and Oncology (56%) identify medicine in a patient’ s record by brand name significantly less than those in other countries and practice areas, see figure 4.
Forty-three per cent of prescribers responded they rarely or never prescribe biological products by non-proprietary name only. When compared to the other countries, prescribers in Switzerland (40%) are most likely never to use the non-proprietary name of a product. When compared to those in other practice areas, Dermatology (32%) and Rheumatology (28%) prescribers are more likely never to use the non-proprietary name of a product, see figure 5.
When asked about how confident a prescriber can be in their ability to know exactly what product is dispensed to a patient when using a non-proprietary name, 63% were very or somewhat confident, while 38% were slightly confident or not confident at all. Prescribers in Switzerland (26% are not confident at all) noted that they are significantly less confident in knowing what is dispensed when a non-proprietary name is used than those in Italy, Spain and the UK. Prescribers in the fields of Dermatology (55%) and Rheumatology (42%) are significantly less confident in knowing what is dispensed when a non-proprietary name is used compared to those in several other practice areas, see figure 6.
Dispensing in pharmacies
When asked about biological products dispensed directly to patients in a pharmacy, 61% of prescribers said that they were either very confident or somewhat confident that, if the pharmacy dispenses a drug that is different from the one that is prescribed (whether it is biosimilar 1, 2, or 3 or even the reference product), they have the ability to identify exactly what drug was dispensed to the patient. Thirty-nine per cent were either slightly confident or not confident at all. Prescribers in the UK (73%) said they are significantly more confident in knowing what is dispensed by pharmacy than those in Germany (49%) and Spain (60%); and those in Switzerland (24% not confident at all) are significantly less confident than several countries. It was shown that Oncology (82%) prescribers are significantly more confident in knowing what is dispensed than those in almost all of the other practice areas, see figure 7.
Eighty-three per cent of prescribers said it was critical or very important to be notified by the pharmacist if a patient has received a biological other than the one prescribed, if the patient was receiving chronic (repeated) treatment. It was shown to be significantly more critical for prescribers in Switzerland (80%) to be notified that a different biological was prescribed than for those in all other surveyed countries. It was also shown that it is significantly more critical for Rheumatology (84%) prescribers to be notified that a different biological was prescribed than those in several other practice areas; and it is significantly less important for Haematology-Oncology prescribers to be notified.
Only 5% of prescribers thought it was totally acceptable for a pharmacist to determine which biological (reference product or biosimilar) to dispense to a patient at the initiation of treatment. Fifty-eight per cent thought this was acceptable if the pharmacist’ s ability to determine the product was agreed to by clinicians in advance, and 37% thought it not acceptable. It was shown to be significantly not acceptable for a pharmacist to make the decision for prescribers in Spain (52%) and Switzerland (51%) when compared to the other countries surveyed. It was shown to be significantly not acceptable for a pharmacist to make decision more so for Rheumatology (60%) and Dermatology (52%) prescribers compared to those in other practice areas, see figure 8.
Prescribing biosimilars and switching
Seventy-four per cent of prescribers agreed that the correct definition for a ‘naïve’ patient is: a patient that has never received any biological treatment from this class of medicines. Eighty-four per cent of prescribers said they were very comfortable or somewhat comfortable in prescribing biosimilars to treat naïve patients. Prescribers in France, Germany, Italy, Switzerland and the UK are significantly more comfortable (very) than those in Spain (18%) in prescribing a biosimilar to a naïve patient. Rheumatology (60%) prescribers are more comfortable (very) than those of many other practice areas in prescribing a biosimilar to a naïve patient; Ophthalmology (10%) prescribers are the least comfortable.
Comfort level decreases when asked about switching a stable patient to a biosimilar versus to a naïve patient. While 17% are uncomfortable in prescribing a biosimilar to a naïve patient, see figure 9; twice as many (40%) are uncomfortable with switching a stable patient from an originator to a biosimilar. Spain (54%) prescribers are the least comfortable with switching a stable patient to a biosimilar. Haematology-Oncology prescribers are more comfortable switching a stable patient from an originator to a biosimilar than those in several other practice areas; Ophthalmology and Rheumatology prescribers are less comfortable, see figure 10.
Comfort level decreases further when asked about switching a patient to a biosimilar for non-medical reasons. More than half of prescribers (58%) said they are uncomfortable with switching their patients to a biosimilar for non-medical reasons. Prescribers in France are significantly more comfortable (very) switching a patient to a biosimilar for non-medical reasons than several other countries; prescribers in Italy and Spain are the least comfortable. Haematology-Oncology prescribers are significantly more comfortable (very) switching a patient to a biosimilar for non-medical reasons than those in most other practice areas, see figure 11.
Even more prescribers are uncomfortable (73%) when asked about a third party initiating such a switch. In the UK and France, prescribers were shown to be most comfortable with switching their patients (40% and 35% comfortable, respectively), while in Spain, prescribers are the least comfortable with having a third party make the switch (14%). Haematology-Oncology prescribers were shown to be significantly more comfortable with a third party switching a patient to a biosimilar for non-medical reasons (60% versus an average of 27%) than those in several other practice areas, see figure 12.
In summary, the survey reveals that European physicians have increased their familiarity with biosimilars since last surveyed in 2013. After 13 years of experience with biosimilars in Europe, physicians:
The survey study was sponsored by Alliance for Safe Biologic Medicines (ASBM) and administered by Industry Standard Research, LLC.
This paper is funded by the ASBM.
The ASBM is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’ s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Dr Madelaine Feldman is the Chairperson of the Alliance for Safe Biologic Medicines. She has participated in advisory boards for Gilead, Lilly, Pfizer and Samsung. Mr Michael S Reilly, Esq, is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Madelaine Feldman, MD, FACR
Michael S Reilly, Esq
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Aug 9]. Available from: www.gabionline.net/Guidelines/EU-guidelines-for-biosimilars
2. Bird E. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(1):37-44. doi:10.5639/gabij.2020.0901.007
3. Schneider PJ, Reilly MS. Policy recommendations for a sustainable biosimilars market: lessons from Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):76-83. doi:10.5639/gabij.2020.0902.013
4. Jensen AR. Biosimilar product labels in Europe: what information should they contain? Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):38-40. doi:10.5639/gabij.2017.0601.008
5. Murby SP, Reilly MS. A survey of Australian prescribers’ views on the naming and substitution of biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):107-13. doi:10.5639/gabij.2017.0603.022
6. Dolinar RO, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018
7. Schneider PJ, Reilly MS. Naming and labelling of biologicals – the perspective of hospital and retail pharmacists. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(4):151-5. doi:10.5639/gabij.2016.0504.040
8. Safe Biologics. Members partners [homepage on the Internet]. [cited 2020 Aug 9]. Available from: www.safebiologics.org/member-partners/
9. Derbyshire M. USA and Europe differ in interchangeability of biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):183-4. doi:10.5639/gabij.2017.0604.039
10. Reilly MS, Gewanter HL. Prescribing practices for biosimilars: questionnaire survey findings from physicians in Argentina, Brazil, Colombia and Mexico. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):161-6. doi:10.5639/gabij.2015.0404.036
11. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/european-prescribers-attitudes-and-beliefs-on-biologicals-prescribing-and-automatic-substitution.html
Author byline as per print journal: Professor Philip J Schneider1, MS, FASHP, FASPEN, FFIP; Michael S Reilly2, Esq
|
Abstract: |
Submitted: 24 October 2019; Revised: 6 February 2020; Accepted: 17 February 2020; Published online first: 24 February 2020
Approximately 25% of all new medicines approved in recent years and in development today are biologicals [1]. The biosimilar market in Europe is the largest in the world, representing approximately 60% of the global biosimilar market and growing consistently year on year [2].
Biosimilars are large and structurally complex molecules obtained from a biological source. Due to inherent biological variability, they can be reproduced to a high degree of – but not complete – similarity [3]. Compared to generics, their development requires additional quality and comparability studies as well as clinical studies on immunogenicity, safety and efficacy [4]. As a consequence, the cost to develop and gain approval for a biosimilar medicine ranges between US$100 million to US$200 million [5].
The European Union (EU) and the European Medicines Agency (EMA) developed a legal framework for the review and development of biosimilars in 2004 [6]. Based on this framework, EMA has a comprehensive set of regulatory guidelines for biosimilar review. One of the most challenging areas in the regulation of biosimilars is switching. So-called ‘switching studies’ where the originator medicine is substituted with the biosimilar candidate and vice versa are not part of the existing European regulatory approval requirements. Neither are studies where biosimilar medicines referencing the same originator medicine are substituted with each other. As a consequence, limited sufficiently powered randomized clinical trial data exist to inform on the risks of switching such as immunogenicity, treatment-emergent adverse events, or lack of efficacy. Because of these factors, the authors of the NOR-SWITCH study recommend caution in generalizing their findings to other biological agents. They also stated that more studies are needed ‘to examine multiple-sequenced as well as back-and-forth switches’ [7].
In summary, a biological medicine should generally not be substituted with another biological or biosimilar medicine that is made using a different bio-manufacturing process. Switching does occur in practice, but the decision to change a patient’s therapy (switch [4]) generally resides with the treating physician in consultation with the patient.
Because of the differences between generic and biosimilar medicines, different rules apply to their clinical use, e.g. procurement practices, prescriber authority and pharmacist involvement. These differences, along with the relative novelty of biosimilars, have resulted in a limited number of biosimilars being made available to date compared with small molecule generics. The limited amount of data available on switching between originator medicines and biosimilars and in-between biosimilars in particular results in a major challenge for all stakeholders – and physicians, patients and payers in particular – to establish policies and frameworks for efficient and patient-friendly long-term sustainable biosimilar markets. These policies must take into consideration: the uniqueness of biosimilar medicines, the educational needs of both physicians and their patients, the maintenance of physician choice and clinical decision-making, the guarantee of drug supply to ensure continuous patient care, pricing rules that do not discriminate against either originator or biosimilar medicines, purchasing mechanisms (procurement frameworks) which encourage companies to compete, and maintaining the correct balance of incentives for providers and prescribers while delivering savings.
Europe’s legislative and regulatory leadership in biosimilars is evident in European biosimilar sales. While the global biologicals market is dominated by the US, which has a share above 50%, the global biosimilar market has been even more so dominated by the European market, which accounted for close to 90% of global biosimilar sales between 2012 and 2016, see Figure 1.
With the launch of 22 biosimilars on the US market since 2017 [8], Europe’s share has been dropping towards 60%, see Figure 1. Although in absolute terms the share of the global biosimilar market as a percentage of the global biologicals market was below 1% in 2016 (compared with 35% of global generics sales as a percentage of global non-biologicals sales), this figure has grown closer to 2.5% and is expected to grow further in the future.
While European biosimilar markets are more mature than those in the US, opportunities for biosimilar companies are smaller in Europe given the overall lower spending on biologicals. In the longer term, the US market represents the greater economic opportunity for these companies [9].
Several research papers and position statements defining the factors and conditions required for sustainable biosimilar markets have been published in recent years, with the intent to inform policymakers and payers and to support them to develop, assess and refine biosimilar policies in the European Economic Area (EEA, i.e. EU Member States plus Iceland, Liechtenstein and Norway). These publications [10, 14] originate from diverse stakeholders, offering a comprehensive perspective on the necessary conditions for long-term sustainable biosimilar markets, see Appendix 1 on research papers and position statements defining the factors and conditions required for sustainable biosimilar markets in Europe, 2014−2019.
Based on these publications, we believe a ‘gold standard’ for a sustainable European market for off-patent biologicals can be derived, containing six policy requirements:
(1) Policies should be designed to incentivize and reward innovation in all types of biologicals.
(2) Healthcare financing must take into account societal benefits derived from biological medicines, as well as the unique characteristics of biologicals.
(3) Procurement practices must provide for multiple suppliers and a minimum term of 12 months.
(4) Physicians must have autonomy to choose the most appropriate medicine for their patient, including making decisions on switching, which must also be consented to by the patient; no automatic substitution.
(5) There should be mandatory brand- name prescribing to avoid unintended switches as well as a robust pharmacovigilance system to report adverse drug reactions (ADRs).
(6) Policies with potential to undermine sustainability, such as measures which induce biosimilar uptake or promote preferential treatment, thereby limiting physician choice, should be avoided.
When comparing the findings and recommendations from these papers, several key conditions to achieve sustainable biosimilar markets can be identified and may be considered as ‘must haves’ for the long-term success of these markets. These are:
(1) Physicians should have the freedom to choose between off-patent originator biologicals and available biosimilars and to act in the best interest of their patients based on scientific evidence and clinical experience.
(2) Tenders should be designed to include multiple value-based criteria beyond price, e.g. education, services, available dose strengths, and provide a sufficient broad choice (multi-winner tenders versus single-winner tenders) to ensure continuity of supply and healthy competition.
(3) A level playing field between all participating manufacturers is the best way to foster competition; mandatory discounts which place artificial downward pressure on manufacturers do not engender a sustainable market environment.
Elements or conditions recommended by some but not all of the above studies are:
(1) The provision of early access and swift market entry of approved biosimilars upon the innovator’s loss of exclusivity to expand access to treatment for patients while lowering costs.
(2) Incentives, e.g. gainsharing, where a portion of generated savings are returned to prescribers and/or an institution and prescribing targets should be carefully designed so as not to restrict physician choice nor limit competition between originator biologicals and their biosimilars.
(3) The pursuit of a multi-disciplinary team approach when designing payer purchasing policies to ensure the perspectives and needs of different stakeholders, i.e. payers, manufacturers, physicians and patients, are taken into consideration.
(4) Safeguarding the interest of patients and serving their needs as best as possible remains a critical consideration for health authorities, in particular considering the increasing numbers of self-administered biosimilars.
Biosimilar policy varies within the 28 EU Member States and the three EEA countries.
Policies have evolved over time and are likely to continue to evolve based on experience, clinical data and the growing number of authorized biosimilars. Current policies are characterized by different supply and/or demand-side incentives as well as different degrees of competition and are determined either at the national, regional or hospital level, or a mixture of these.
Independent of the kind of policy which is pursued, data suggest that biosimilar price and use also depends on therapeutic area and the time the biosimilar has been on the market. This diversity is reflected in Figure 2, that shows differences in price reductions as well as volume increases for both originator and biosimilar relative to the period prior to when the first biosimilar within a product group was launched [15].
National tender markets
Norway and Denmark are the only European countries to pursue a national tender policy for biosimilar products such as adalimumab, etanercept, or infliximab. Denmark’s policy has been ambitious and restrictive, in that only the manufacturer with the lowest bidding price for a particular molecule will be reimbursed for a 12-month period, potentially requiring physicians to switch patients every 12 months (unless they provide clinical arguments not to do so). In comparison, Norway, which has a history of anti-tumour necrosis factor (anti-TNF) tendering, tenders all molecules in a single tender split by indication as well as application, i.e. infusion or intravenous (IV) (self-administered) based on bi-annual cost according to each product’s prescribing information. Norway then ranks products based on those bi-annual costs in each indication and urges physicians to prescribe the most economical product for new patients. However, the choice is ultimately left to the physician and all lower ranked products are still reimbursed if prescribed, i.e. switching is not explicitly mandated as it is in Denmark. Tenders in Norway tend to be in effect for one year.
Regional and hospital tender markets
In Sweden, hospital administered products are funded by county councils in each of the 21 Swedish counties. Each county runs their own tenders by molecule thus making the market more sustainable for all competitors, as they can compete in more than one tender. Resulting tender prices are communicated among hospital doctors with the intent to promote economic prescribing. As in the case of Norway, all available products continue to be reimbursed if prescribed and, while switching is encouraged, the decision remains with the treating physician and her patient. Tenders tend to run for 24 months and may be re-opened as new competitors enter the market. A recent study of infliximab prescribing in Sweden [16] showed that biosimilar infliximab prescribing varied between regions depending on the absolute difference in price between the originator and the biosimilar products, the opinion of key opinion leaders and clinic heads, local guidelines and hospital initiatives.
With the intent to increase competition, maintain choice for physicians and their patients and avoid shortages of supply from a single manufacturer, Italy passed a national law in December 2016 (2017 Budget Law) which forbids automatic substitution in the case of biologicals and gives priority to the physician’s freedom of prescription. Based on this law, if there are no more than three suppliers of a specific molecule on the market, each is available to physicians to be prescribed. In cases where there are more than three suppliers, the three lowest priced products may be prescribed. In both cases, prices are determined in hospital tenders for both IV and subcutaneous (SC) products in each of the Italian regions. For stable patients, physicians may choose to continue the current treatment under the condition that the respective medicine participated in the tender process.
As in Sweden, hospital tenders in France only apply to IV products. Tenders tend to promote a single winner based on price with a duration of 12 months. Physicians are encouraged to prescribe the most economical product for new patients as well as for stable patients, even though the decision to switch a patient is left at the physician’s discretion. However, while a single switch from the originator to a considerably lower priced biosimilar is seen as less controversial, multiple switches are looked upon unfavourably, unless a very substantial discount to the previously lowest priced biosimilar applies. Given that hospitals are paid a lump sum per patient per case, lower priced products offer a profit incentive to hospitals.
In Spain, similar to Italy, hospitals tender both IV and SC products by molecule, all products remain available for physicians to choose from, and all products are reimbursed. In addition to hospital tenders, some regions have pursued biosimilar prescribing targets, however, physicians’ responsibility to provide the best treatment for their patients remains paramount. Furthermore, physicians are liable for their treatment choices as regards patient outcomes.
In Poland, medicines are prescribed in the hospital. Every hospital is obliged to tender each molecule annually and only the winning bidder can generally expect to be used and reimbursed during each 12-month tender period. As a result, competition has been particularly fierce, and the longer-term sustainability of the biosimilar market may be affected. In addition, product choice for physicians and their patients is limited, both for new and stable patients.
In the UK, National Health Service (NHS) England in their September 2017 publication ‘Commissioning framework for biological medicines (including biosimilar medicines)’ stated that ‘… at least 90% of new patients will be prescribed the best value biological medicine within three months of launch of a biosimilar medicine, and at least 80% of existing patients within 12 months, or sooner if possible’. In England, which comprises 80% of UK citizens, NHS England has provided guidance in their Commissioning Framework for Biological Medicines. Based on tenders in each of the four regions within England, prices for originators and their biosimilars are determined for both IV and SC products. Commissioners are encouraged to promote switching where appropriate, incentivize prescribing and monitor biosimilar uptake. While all approved biologicals remain available for prescribing by physicians, various mechanisms have been put in place to promote the use of the best value biological medicine [17].
Retail and hospital contract markets
Germany, where both SC and IV biologicals are primarily prescribed by private doctors, is the largest retail market in Europe. Pricing is free allowing for price competition both at the list price level as well as towards individual sick funds based on discount agreements. German Statuary Health Insurance (GKV), which represents dozens of health insurers and the German National Association of Statutory Health Insurance Physicians (KBV), agrees national prescription volume targets for biosimilars. However, such quotas vary widely by region and molecule, as regional associations are not obliged to abide by national targets and are free to set higher or lower regional targets. To achieve set quotas, private doctors’ prescribing practices are monitored to encourage compliance. Despite these incentives, physicians remain free in their choice among all available products and are expected to base their decision on clinical data, experience and patient needs. All available products are reimbursed. For biologicals prescribed in the hospital, prices are typically determined by contracts agreed between hospitals and manufacturers.
In France, SC biologicals are prescribed by private doctors and reimbursed nationally. While certain quotas do apply, similar to Germany, French physicians are free to choose the best product for their patients and all available products are reimbursed.
In Sweden, hospital tenders apply only to IV products, but SC biologicals are prescribed in the hospital and reimbursed centrally. Prices for SC biologicals are determined in so-called managed entry agreements by way of three-party negotiations between The Dental and Pharmaceutical Benefits Agency, county councils and manufacturers. After an agreement is reached, prices apply nationwide. Managed entry agreements have durations of up to two years and may be re-visited when new competitors arrive. Savings resulting from these agreements are shared between the government (40%) and the counties (60%). Physicians are free to prescribe the most suitable product for their patients while taking economic arguments into consideration. Based on two recent studies of infliximab and etanercept in Sweden [16], see Table 1, biosimilar prescribing varied between regions to a larger degree in the case of infliximab probably due to a limited price difference between originator and biosimilar etanercept in comparison with infliximab. Overall, as in the case of infliximab, etanercept volume has increased, likely driven by a lower threshold to prescribe and by higher and more appropriate dosing.
Understanding, acceptance, familiarity and use of biosimilars in Europe have evolved in recent years. Based on 13 years’ experience with biosimilars in off-patent biologicals markets in Europe, it can be stated that biosimilars have: (1) increased competition; (2) reduced unit cost of both originator and biosimilars compared to price levels prior to the arrival of biosimilars; (3) increased volume consumption of molecules with biosimilar competition thus expanding market access and optimizing patient dosing; and (4) alleviated budget pressures by providing headroom to fund novel treatment solutions.
While the policies by which this has been achieved vary between countries, all major European markets share the following principles:
(1) Automatic substitution for biologicals is forbidden.
(2) All approved biologicals, i.e. originators and their biosimilars, are available on the market and are reimbursed when prescribed.
(3) Reimbursement decisions on novel treatment solutions are independent from biosimilar use and uptake.
(4) The time from market approval to first product sales for biosimilars is shorter than the time to first sales of novel medicines [18].
Biosimilars have attained market shares in some European markets as high as 91% for older products (before the approval of the first monoclonal antibody biosimilar in 2013) and as high as 43% for newer products (approved post-2013) [15]. However, this does not necessarily mean that countries with a more moderate biosimilar market share are less competitive. On the contrary, these markets tend to benefit not only from the use of lower cost biosimilars but equally from lower cost originators. Furthermore, markets with multiple manufacturers tend to be better positioned for the long term since market participation and price levels allow for a return on investment which will accommodate the development and launch of future biosimilars, the generation of real-world data and more patient-friendly devices and services. Last but not least, such markets allow physicians to choose among available products and act in the best interest of their patients based on clinical evidence, experience and patient preferences.
As countries decide on their biosimilar policies, they must strike the right balance between a desire for substantial immediate savings and more sustainable savings in the long term, as well as future innovations. The number of future off-patent biologicals offers an attractive savings outlook which should not be jeopardized by a short-term race to the bottom of the prices of today’s biosimilars. Policymakers should carefully design procurement measures such as tenders and contracting to ensure: (1) a wide and non-discriminatory choice of products; (2) selection criteria beyond price such as services and supply; (3) the avoidance of (forced) multiple switches [19]; and (4) the involvement of physicians, pharmacists and other stakeholders in the process. Any demand-side incentives to promote biosimilar prescribing, such as biosimilar market share targets, financial prescriber incentives or sanctions, should also be designed with care regarding their size and level of enforcement in order to ensure appropriate product use. This ultimately needs to be a clinical decision made by the treating physician for an individual patient on the basis of shared decision-making.
Creating a new market is a slow process. It took the generics sector over a decade to establish itself as the major volume driver in developed markets. After more than a decade of biosimilar experience in Europe, biosimilars have gained wider acceptance and the use of newly approved products has increased. While many blockbuster products with European sales above US$1 billion have lost market exclusivity during this decade, there will only be three such products going off patent in the decade to come. The majority of products will have sales between US$100 million to US$1 billion [20]. For future off-patent originator biologicals to offer a compelling investment for multinational biosimilar companies, regulatory and payer policies which help to foster a fair and sustainable environment will be crucial. Advocacy from patient and industry groups as well as healthcare providers will be important to convince market gatekeepers to develop a market with these characteristics.
The European experience is testament to the value of multi-stakeholder participation and engagement at every stage of the process, from the regulatory framework through education all the way to procurement policy decisions and treatment guidelines. Trust is at the core of matters such as the appropriate use of complex medicines for the treatment of chronic and life-threatening diseases; only an approach which is transparent and includes all stakeholders will facilitate the long-term acceptance of biosimilar products.
In their pursuit of savings and increased patient access in off-patent biologicals markets, stakeholders will need to strike the right balance between both short- and long-term savings, as well as the maintenance of patient care and physician and patient trust. Based on the European experience to date, the most suitable policies appear to be those which provide continuous, unbiased information on biosimilars, stimulate manufacturer competition, guarantee a sufficiently broad choice of products, are non-discriminatory towards either originator or biosimilar medicines and allow the treating physician to choose the most appropriate product in consultation with their patient. Given the required investment to develop a biosimilar medicine, their market access and participation should be supported by medical guidelines and incentives such as prescribing targets for biosimilars. While the use of biosimilars for bio-naïve patients is not controversial, some uncertainty remains about switching patients currently being treated with an originator product to a biosimilar (or vice versa), from one biosimilar to another, and about switching on multiple occasions [21]. Any incentives must take these uncertainties into account in order for prescriber and patient confidence in biosimilar medicines to evolve.
This paper is funded by the Alliance for Safe Biologic Medicines (ASBM).
The ASBM is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Professor Philip J Schneider is a member of the International Advisory Board of Alliance for Safe Biologic Medicines (ASBM) since 2012 without compensation. From September 2014, Emeritus Professor Schneider has been the Chair of the International Advisory Board of ASBM and is paid a small stipend for that role. Mr Michael S Reilly, Esq, is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor Philip J Schneider1, MS, FASHP, FASPEN, FFIP
Michael S Reilly2, Esq, Executive Director
1College of Pharmacy, The Ohio State University, 500 West 12th Avenue, Columbus, OH 43210, USA
2Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Will biologics surpass small molecules in the pharma race? BiopharmaTrend.com. 2018 July 11.
2. IQVIA. MIDAS MAT Q4 2018. Middle East & Africa pharmaceutical market insights [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/nemea/mea/edition-13-mea-pharmaceutical-market-quarterly-report.pdf
3. European Commission. Biosimilars in the EU: a paradigm in regulatory science [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://ec.europa.eu/docsroom/documents/38050
4. European Medicines Agency. Biosimilars in the EU, Information guide for healthcare professionals, 2017. [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
5. Belloni A, Morgan D, Paris V. Pharmaceutical expenditure and policies: past trends and future challenges. 2016. OECD Health Working Papers, No. 87, OECD Publishing, Paris.
6. European Commission. Directive 2001/83/EC, as amended by Directive 2003/63/EC and Directive 2004/27/EC
7. Jørgensen KK, Olsen IC, Guro L, Goll GL, Lorentzen M, Bolstad N, et al. Kvien TK, on behalf of the NOR-SWITCH study group. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. The Lancet. 2017;389(10086):2304-16.
8. U.S. Food and Drug Administration. FDA-approved biosimilar products 10 [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
9. IQVIA, The global use of medicine in 2019 and outlook to 2023, January 2019 [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.iqvia.com/insights/the-iqvia-institute/reports/the-global-use-of-medicine-in-2019-and-outlook-to-2023
10. Medicines for Europe. Factors supporting a sustainable European biosimilar medicines market [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2016/03/GfK_Final_Report-_Factors_Supporting_a_Sustainable_European_Biosimilar_Medicines_Market.pdf
11. European Federation of Pharmaceutical Industries and Associations. EFPIA policy principles for off-patent biologic medicines in Europe [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://efpia.eu/media/15409/efpia-policy-principles-for-off-patent-biologic-medicines-in-europe-september-2015.pdf
12. Medicines for Europe. Simon Kucher & Partners. Payers’ price & market access policies supporting a sustainable biosimilar medicines market [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2016/09/Simon-Kucher-2016-Policy-requirements-for-a-sustainable-biosimilar-market-FINAL-report_for-publication.pdf
13. IQVIA. Advancing biosimilar sustainability in Europe [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.iqvia.com/insights/the-iqvia-institute/reports/advancing-biosimilar-sustainability-in-europe
14. European Federation of Pharmaceutical Industries and Associations. Towards a sustainable European market for off-patent biologics [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://efpia.eu/media/412909/towards-a-sustainable-european-market-for-off-patent-biologics-pugatch-consilium.pdf
15. European Commission. IQVIA. The impact of biosimilar competition in Europe. September 2018 [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://ec.europa.eu/docsroom/documents/31642
16. Moorkens E, Simoens S, Troein P, Declerck P, Vulto AG, Huys I. Different policy measures and practices between Swedish counties influence market dynamics: Part 1—Biosimilar and originator infliximab in the hospital setting. BioDrugs. 2019;33:285-97.
17. National Health Service England. Commissioning framework for biological medicines. September 2017 [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.england.nhs.uk/wp-content/uploads/2017/09/biosimilar-medicines-commissioning-framework.pdf
18. IQVIA. Advancing biosimilar sustainability in Europe [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.iqvia.com/institute/reports/advancing-biosimilar-sustainability-in-europe
19. Scherlinger M, Schaeverbeke T. ‘To switch or not to switch’: the missing piece in the puzzle of biosimilar literature? Ann Rheum Dis. 2019;pii: annrheumdis-2018-214908.
20. Arias A. Future biosimilars opportunities. 2019 [homepage on the Internet]. [cited 2020 Feb 6]. Available from: https://www.iqvia.com/blogs/2019/12/future-biosimilar-opportunities
21. Simoens S, Le Pen C, Boone N, Breedveld F, Celano A, Llombart-Cussac A, et al. How to realize the potential of off-patent biologicals and biosimilars in Europe? Guidance to policymakers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):70-4. doi: 10.5639/gabij.2018.0702.014
In 2014, consultancy GfK Market Access published a study undertaken on behalf of the European Generics Association (EGA) now Medicines for Europe on Factors supporting a sustainable European biosimilar medicines market [10]. In this study, four elements for a Sustainability Policy Framework were identified, namely: (1) Education and Understanding; (2) Experience and Use; (3) Sustainable Pricing; and (4) Rational Decision-making. The core considerations under each of these elements were:
(1) Education and Understanding
(2) Experience and Use
(3) Sustainable Pricing
(4) Rational Decision-making
In 2015, the European Federation of Pharmaceutical Industries and Associations (EFPIA) published their Policy principles for off-patent biologic medicines in Europe [11] ‘to help healthcare systems to design and implement policies that can successfully create competitive off-patent biological markets’. The four principles identified by EFPIA and the core aspects in each of them were:
(1) Overarching principles for European off-patent biological markets
(2) Principles for creating sustainable competition
(3) Principles for prescribing frameworks
(4) Principles for specific market mechanisms
In 2016, consultancy Simon & Kucher published a report undertaken on behalf of Medicines for Europe titled Payers’ price & market access policies supporting a sustainable biosimilar medicines market [12]. In their comprehensive study based on data and interviews with payers and manufacturers, Simon & Kucher identified the following principles for a sustainable biosimilar medicines market:
(1) Biosimilars increase competition thus increasing access for more patients and funding for new innovative medicines
(2) Pricing and procurement policies should ensure continuous market participation of several manufacturers to maintain healthy competition
(3) Biosimilar policies should allow physicians to choose from different treatment alternatives
(4) Tender decisions should not be based only on price. They should take into consideration multiple value-based influencing factors, e.g. supply, education, services
(5) Procurement policies should allow parallel sourcing to sustain competition and supply
(6) Pricing and procurement policies for biosimilar markets must be commercially attractive to sustain competition and biosimilar investment long term
(7) Pricing and market access policies enforcing lower prices (compared to their originators) have to be accompanied by specific guidance on biosimilar use and prescribing incentives
(8) Voluntary and mandatory price discounts without volume compensation do not offer biosimilar manufacturers a sustainable market environment
(9) Gainsharing is considered a successful driver of biosimilar uptake
(10) Physician incentive policies are only sustainable if payers monitor physicians’ policy adherence
In 2018, pharmaceutical consultancy IQVIA published the multi-stakeholder assessment Advancing Biosimilar Sustainability in Europe [13], which was commissioned and funded by Pfizer. In this assessment IQVIA identified five key areas that have significant influence on the sustainability in the biosimilar marketplace: the current regulatory environment, clinical guidelines for biosimilars, product and supply features, incentives for biosimilar use and competitive market pressures such as pricing. Based on an analysis of policies in seven EEA countries (France, Germany, Italy, Norway, Poland, Spain, UK), the following key sustainability elements emerged:
(1) Regulatory environment and clinical guidelines favourable to biosimilar approval and uptake
(2) Guidelines and policies supporting a physician-guided therapy switch
(3) Prescription freedom for physicians enabling them to select therapy for patients, i.e. no automatic substitution
(4) Availability of multiple products, enabling physicians’ choice of approved therapies
(5) Well-designed incentives that foster biosimilar uptake, while safeguarding physician choice and patient input into treatment decisions, such as treatment switch
(6) Consideration of the long-term sustainability of the market as well as physician needs when designing incentives to facilitate budget release
(7) Incentivize biological manufacturers towards continued patient-friendly innovation
(8) Balance price pressures of payer purchasing mechanisms with requirements for long-term market sustainability; implement policies that address the needs of all market stakeholders
(9) Sustain healthy competition with multi-winner tenders as compared with single-winner tenders
(10) Sustain healthy product supply by enabling access for both originator and biosimilar products
In 2019, boutique consultancy Patch Consilium published the study Towards a sustainable European market for off-patent biologics [14] which was commissioned and funded by EFPIA. The objective of this study was to assess how policy and governance frameworks should be designed and implemented to ensure long-term sustainable off-patent biological markets through an analysis of the current policy ecosystem in 15 EEA countries (Belgium, Czech Republic, France, Germany, Greece, Hungary, Italy, Netherlands, Norway, Poland, Romania, Slovakia, Spain, Sweden, UK) regarding the pricing environment, procurement practices and the degree of physician autonomy and patient choice and through a survey among different stakeholders in these countries regarding their perceptions of the off-patent biologicals. The core findings are summarized below:
(1) Pricing environment
(2) Procurement practices
(3) Physician autonomy and patient choice
(4) Stakeholder* perception
*Stakeholders interviewed were: Healthcare Professionals, Patients/Patient Group Representatives, Government Institution Representatives and Industry/Trade Association Representatives; due to the limited number of responses (76) and uneven distribution between stakeholders, the study states that they may not reflect the common view of these stakeholder groups.
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/policy-recommendations-for-a-sustainable-biosimilars-market-lessons-from-europe.html
Author byline as per print journal: Stephen P Murby, FRSA; Michael S Reilly, Esq
|
Introduction: As the number of biosimilar approvals in Australia increases, it is important to build on the existing regulatory framework to continue to bring high quality, safe and efficacious biosimilars to the widest number of patients most cost-effectively. As new policies regarding the regulation, reimbursement and uptake of biosimilars are being considered, the Alliance for Safe Biologic Medicines (ASBM) has asked Australian prescribers for their views on the naming, substitution and prescribing of biologicals and biosimilars. Currently, biologicals and biosimilars are approved by Australia’s Therapeutic Goods Administration (TGA). The country’s Pharmaceutical Benefits Advisory Committee (PBAC) has indicated it will consider pharmacy-level substitution of biosimilars for reference biological medicines on a ‘case-by-case basis’. |
Submitted: 9 May 2017; Revised: 13 July 2017; Accepted: 19 July 2017; Published online first: 1 August 2017
In Australia, biologicals and biosimilars are approved nationally by the Therapeutic Goods Administration (TGA). Aczicrit and Grandicrit (epoetin lambda) were the first products approved in Australia as biosimilars in 2010 [1]. To date, TGA has approved 13 biosimilars within the product classes of human growth hormone, granulocyte colony-stimulating factor (G-CSF), insulin, erythropoietin, follicle stimulating hormone (FSH) and tumour necrosis factor-inhibitor [1].
With an increasing number of biosimilars seeking entry to the Australian market, it is important to build on the existing regulatory framework established to continue to bring high quality, safe and efficacious biosimilars to the widest number of patients most cost-effectively. As new policies regarding the regulation, reimbursement and uptake of biosimilars are being considered, the Alliance for Safe Biologic Medicines (ASBM) has asked Australian prescribers for their views on the naming, substitution and prescribing of biologicals and biosimilars.
Australia’s Pharmaceutical Benefits Advisory Committee (PBAC), an independent expert body appointed by the Australian Government to recommend new medicines for listing on the PBS (Pharmaceutical Benefits Scheme), has indicated that it will consider pharmacy-level substitution of biosimilars for reference biological medicines on a ‘case-by-case basis’.
Some clinicians hold concerns with pharmacy-level substitution due to the current paucity of data on such practices, and also matters associated with tracking which product is dispensed. PBAC has stated that its ‘default position’ would be to advise that a biosimilar is suitable for substitution by a pharmacist ‘where the data are supportive of this conclusion’ and that a relevant consideration is ‘the absence of data to suggest significant differences in clinical effectiveness or safety compared with the originator product’ [2].
Biologicals are used in the prevention, diagnosis, or treatment of a range of chronic diseases. Since biologicals have large, complex, inherently diverse molecular structures made, or derived, from living organisms, they are always heterogeneous. Unlike non-biological medicines, there is a degree of natural variability in biologicals, and there are generally some differences between the reference and biosimilar products. Current methods to analyse physicochemical and structural differences are extremely sensitive. Analysis of different batches of reference products following a change in the manufacturing process has revealed differences between the pre- and post-change batches [3]. This molecular heterogenity within the originator biological is distinct from molecular differences between the originator and biosimilar.
A biosimilar is a version of the active substance of an already authorized original (or reference) biological, see Box 1.
Biosimilars introduce competition, which has the effect of lowering prices of both originator and biosimilar and increasing patients’ access to these therapies [3].
With a growing number of reference biologicals and biosimilars, regulatory authorities across the globe are in discussion over how biosimilar medicines should be named and labelled [4, 5]. Distinct names will be crucial in order to facilitate post-market safety monitoring and help minimize the potential for medication errors.
There is a clear need for sufficiently detailed and transparent labelling and product information to enable informed decision-making by physicians and patients, ensuring appropriate safe and effective use of these medicines [6].
International Nonproprietary Names (INNs) are intended for use in drug regulation, prescribing, dispensing, pharmacopoeias, labelling, pharmacovigilance and in scientific literature [7]. However, biologicals, due to their increased molecular complexity and structural micro-heterogeneity, are not categorized by the INN alone [7]. An INN Expert Group recommended that the World Health Organization (WHO) develop and implement a system for assignment of Biological Qualifiers (BQs) to similar biotherapeutic products (SBPs) [8]. WHO has proposed the development of a global BQ for biological medicines that will provide a unique identifier for all biological active substances that are assigned an INN [6]. While the INN is a common and public non-proprietary name for a given active substance, the BQ would be applied to a particular manufacturer’s active substance. The BQ would not be part of the INN and it is envisaged that it would enhance identification, prescribing, dispensing and pharmacovigilance of biological medicines.
The US Food and Drug Administration (FDA) issued its guidance for the non-proprietary naming of biological products in January 2017 [9] following the release of two draft guidance documents outlining proposed methods for biological product naming and biosimilar product labelling [10]. According to its latest guidance, FDA will assign a non-proprietary name for all reference biologicals, related biologicals and biosimilars that will include an ‘FDA-designated suffix’. The ‘proper name’ will consist of a combination of the ‘core name’ and distinguishable suffix, which will be ‘devoid of meaning’ and be ‘composed of four lower case letters’. A survey of prescribers of biologicals in the US, carried out before the release of this guidance, found that two thirds (66%) of the prescribers surveyed supported the introduction of distinct names. Of those, the majority would prefer a suffix that indicated the manufacturer [11]. The WHO and FDA emphasis stands in contrast to the EMA approach to biosimilar naming, under which an originator biological and all biosimilars to that product will share the same non-proprietary name.
In Australia, biologicals and biosimilars are approved nationally by TGA. TGA issued a biosimilars guidance in 2013 that includes a section on naming conventions for biosimilars [12–14]. This guidance required the name of a biosimilar in Australia should be made up of the reference product Australian Biological Name (ABN), thus identifying the reference product with which the biosimilar has demonstrable comparability; and a biosimilar identifier, consisting of: the prefix sim(a)– and a three-letter code issued by the WHO INN Committee, according to its draft policy. This guidance was subsequently revoked and recently approved biosimilars to etanercept and infliximab have been given the same non-proprietary name as their reference products.
The country’s PBAC has indicated that it will consider pharmacy level substitution of biosimilars for reference biological medicines on a case-by-case basis, see Box 2. Since the introduction of this policy, two biosimilars to infliximab have been ‘a’ flagged, as has one biosimilar to etanercept.
In June 2016, 160 prescribers in Australia completed a survey sponsored by the ASBM about their knowledge of, attitudes toward and beliefs regarding biosimilars. They were asked their opinions on the naming of biologicals and biosimilars and how these medicines are identified in the patient record and in adverse event reporting. They were also asked for their views on substitution.
Sample characteristics and methodology
In June 2016, 605 prescribers in Australia were invited to complete a 15-minute web-based survey on biologicals and biosimilars. Potential respondents were identified in, and recruited from, a large, global, commercial database of healthcare professionals. A high response rate was expected because prescribers in this database had previously indicated a willingness to participate in market research.
A total of 451 prescribers responded. Respondents were screened as follows: they had to specialize in one of seven therapeutic specialties, including dermatology, endocrinology, gastrointestinal, nephrology, neurology, oncology or rheumatology; they had to have been in practice for one year or more; and they had to have prescribed biological medicines in their practice. A total of 174 respondents were screened out because they did not meet these criteria. A further 80 prescribers did not qualify because they specialized in a therapeutic specialty for which data collection had closed. In addition, 37 started but failed to complete the survey. Any data they contributed are not included in the analysis and report.
A total of 160 prescribers completed the survey. All data collected refer only to those who completed the survey. Participants received a standard cash stipend of US$76 for their time.
Prescribers practised in public hospitals (46%); private hospital/private practice (42%); academic medical centre (11%); and other (1%). They had spent between one and 30 years in practice, see Figure 1.
A quarter (25%) of the prescribers were rheumatologists, 25% were oncologists, and 25% were gastroenterologists. The remaining 25% prescribers were divided equally among dermatologists, neurologists, nephrologists and endocrinologists, see Figure 2.
Regarding responses from participants to the question of how often they used different information sources to learn about the details of a medicine for prescribing and monitoring, 46% of prescribers said they always used published literature, whilst 43% said they never used published literature. Only 19% of prescribers said they always used information from TGA, and 27% said they always used information from PBAC. Only a fifth (19%) of those surveyed said they learnt about the details of a medicine by reading the product information label, and 13% from hospital formulary, see Figure 3.
Reporting and naming of biologicals and biosimilars
Participants were asked how they identified biological medicines when they were prescribed or entered in patients’ records. Similar numbers of prescribers identified medicines by brand name or non-proprietary scientific name (39% and 38%, respectively). A smaller proportion identified medicines by brand name and non-proprietary scientific name (21%), and 2% identified them by Australian Register of Therapeutic Goods (ARTG) number. When adverse events (AEs) were reported, medicines were identified by brand name by 39% of prescribers, by brand name and non-proprietary scientific name by 34% of prescribers, by non-proprietary scientific name by 25% of prescribers, and by ARTG number by just 2% of prescribers, see Table 1.
Respondents were asked how often they included batch numbers when reporting AEs. A total of 23% of prescribers said they never used batch numbers, almost a third of prescribers (30%) said they rarely included batch numbers, 20% sometimes used batch numbers and 28% said they always included batch numbers.
When prescribers were asked why a batch number was not always reported, they replied that the batch number was not available at time of reporting (41%); or they were not sure where to find the information (36%); or they had forgotten to include the information (19%).
Respondents were also asked whether a loss of efficacy should be reportable as an AE. Most prescribers (65%) said it should not, while 19% of prescribers thought it should, and 16% of prescribers had no opinion about whether a loss of efficacy should be a reportable AE.
Asked whether TGA should insist on a distinct non-proprietary scientific name for every biological or biosimilar medicine that it approves, three quarters of prescribers (76%) said yes, 18% of prescribers said no, and 7% of prescribers had no opinion.
Asked what the best way was for TGA to differentiate a biosimilar medicine from its reference product, a large proportion of physicians responded that it would be best if biosimilars had the same non-proprietary scientific name as their reference medicines but with either a differentiating prefix (38%) or suffix (29%). 30% of participants opted for entirely different non-proprietary scientific names for the biosimilar and its reference product.
Substitution attitudes and beliefs
Prescribers were asked what level of evidence would be supportive of pharmacy-level substitution, see Table 2. They were also asked what role they thought TGA should play in advising PBAC on the suitability of a product for pharmacy-level substitution, see Figure 4, and whether TGA or PBAC should be responsible for providing the primary advice to the Australian Government that a product is suitable for pharmacy-level substitution, see Figure 5.
Prescribers completing the survey were asked how important it was for them to have the sole authority to decide, together with their patient, the most suitable biological medicine to be dispensed to the patient. Over half of prescribers (54%) thought it was very important and over a third (36%) thought it was critically important, see Figure 6. When the prescribing physicians were asked whether their prescription software/documentation included a box marked ‘brand substitution not permitted’, 61% responded that it did, 21% responded that it did not, and 18% were unsure.
Respondents were asked about switching between biological medicines for patients with chronic disease. Over half (51%) of prescribers said that pharmacy-level substitution was not acceptable for these patients, 9% thought it was totally acceptable, while 40% thought it was acceptable providing they were notified in advance.
Biosimilars familiarity and knowledge
In response to the question of how familiar survey respondents were with biosimilar medicines, 21% said they were very familiar and had a complete understanding, 73% said they had a basic understanding, 6% said that they could not define them (although they had heard of them), and 1% said they had never heard of them.
Prescribers were asked a series of questions to gauge their understanding of biosimilars, their awareness of and comfort with the biosimilars approval process, and their opinions on switching. Responses to these questions are given in Table 3.
Respondents were asked how comfortable they would be prescribing a biosimilar medicine that had been approved for several or all of the indications of its reference medicine on the basis of clinical trials in only one of those indications, or in fewer indications than for which the biosimilar is approved, 73% would have some concerns, depending on data and indications; 16% would be comfortable; and 11% would not be comfortable.
Of the prescribers who completed the survey, over three quarters (76%) agreed that TGA should insist on distinct non-proprietary scientific names for all biosimilars and reference products. In addition, well over half (61%) of respondents believed TGA should be responsible for recommendations on pharmacy-level substitution, while only a third (33%) thought that PBAC should be responsible.
Nearly all the prescribers in this survey (98%) use either brand name or non-proprietary scientific name for recording and prescribing biosimilars and their reference biologicals. Most prescribers (61%) want TGA to play a major role in naming biosimilars.
It was clear that the reporting of biosimilars use via brand name and batch number varied between respondents. Respondents indicated that robust data are needed to support substitution, and the vast majority of prescribers (94%) said that the final decision over which biological to prescribe should rest with the prescriber and the patient, and strongly supported clinically supervised switching over pharmacy-level substitution.
Respondents used different information sources to learn about the details of a medicine for prescribing and monitoring, but each source was used by surprisingly few prescribers. Less than half of prescribers said they always used published literature, and a similar proportion said they never used published literature. Clearly there were gaps in how the regulatory process is understood since about half of those surveyed thought, incorrectly, that biosimilars and originators are approved through the same regulatory process.
Despite a spread of responses from the prescribers surveyed, there was general agreement that biosimilars should be distinguished from originators with either the same non-proprietary scientific name and a differing prefix or suffix, or with a completely unique name. The vast majority of prescribers thought that they and their patients should decide which biological is dispensed and that they should be notified of any substitution by the pharmacist.
|
Key points
|
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
The Australia 2016 prescribers and biosimilars survey was sponsored by ASBM.
This paper is funded by ASBM.
Competing interests: Mr Stephen P Murby, FRSA, is a member of the International Advisory Board of Alliance for Safe Biologic Medicines (ASBM). Mr Michael S Reilly, Esq, is the Executive Director of and employed by ASBM. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Stephen P Murby, FRSA, former Head of Consumers Health Forum of Australia; International Advisory Board Member of ASBM
Michael S Reilly, Esq, Executive Director, ASBM
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Australia [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Australia
2. GaBI Online – Generics and Biosimilars Initiative. Australia’s PBAC recommends substitution of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Biosimilars/General/Australia-s-PBAC-recommends-substitution-of-biosimilars
3. Kurki P. Biosimilars for prescribers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):33-5. doi:10.5639/gabij.2015.0401.008
4. Alexander EA. The biosimilar name debate: what’s at stake for public health. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):10-2. doi:10.5639/gabij.2014.0301.005
5. Dolinar RO, Reilly MS. A survey of Australian prescribers’ views on the naming and substitution of biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018
6. European Biopharmaceutical Enterprises. Tell me the whole story: the role of product labelling in building user confidence in biosimilars in Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):188-92. doi:10.5639/gabij.2014.0304.043
7. Robertson JS. The challenges of nomenclature – INN, biosimilars and biological qualifiers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):110-2. doi:10.5639/gabij.2015.0403.025
8. Shaw B. Biosimilars naming and prescribing policy in Australia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):168-9. doi:10.5639/gabij.2013.0204.048
9. GaBI Online – Generics and Biosimilars Initiative. FDA issues final guidance on naming biologicals [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Guidelines/FDA-issues-final-guidance-on-naming-biologicals
10. US FDA proposals for naming of biologicals and labelling of biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):140-3. doi:10.5639/gabij.2016.0503.036
11. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
12. GaBI Online – Generics and Biosimilars Initiative. Australia issues new biosimilars guidance [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Guidelines/Australia-issues-new-biosimilars-guidance
13. GaBI Online – Generics and Biosimilars Initiative. Naming requirements in Australian biosimilars guidance [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Guidelines/Naming-requirements-in-Australian-biosimilars-guidance
14. World Health Organization. Biological Qualifier [homepage on the Internet]. [cited 2017 Jul 13]. Available from: http://www.who.int/medicines/services/inn/inn_bio_bq/en/
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-survey-of-australian-prescribers-views-on-the-naming-and-substitution-of-biologicals.html
|
Introduction: The US Food and Drug Association (FDA) released its requirements for the non-proprietary naming of biological products in January 2017. Before the FDA’s release, the Alliance for Safe Biologic Medicines (ASBM) asked physicians for their views on the labelling and naming of biosimilar medicines. |
Submitted: 9 January 2017; Revised: 12 February 2017; Accepted: 13 February 2017; Published online first: 27 February 2017
Biological medicines are therapeutic proteins produced using living cells. A copy of an original biological made by a different manufacturer is referred to as a biosimilar or follow-on biological rather than a generic drug because it will be similar, not identical, to the product it copies. Biosimilars are also referred to as subsequent entry biologics (SEBs) in Canada. As a result of the abbreviated biosimilar approval pathway [1], biosimilar medicines are now available in the US market.
The market uptake of biosimilars in the US will depend on regulatory policies [2], for which an agreed naming and labelling system will be key. A survey of the views of European physicians on familiarity of biosimilar medicines has demonstrated the need for distinguishable non-proprietary names to be given to all biologicals [3]. There have been calls for clear regulation in this area from Latin America [4], Malaysia [5] and beyond.
Since FDA has only distributed draft guidance on the naming of biosimilar medicines [6] at time of the survey, feedback from the physicians who prescribe biologicals may well be helpful in determining how these drugs are to be regulated. The Alliance for Safe Biologic Medicines (ASBM) invited 5,423 physicians in the US to complete a study on the naming of biologicals. A total of 433 physicians responded, of which 400 prescribers of biologicals qualified and completed the study. Prescribers were asked for their feedback on the non-proprietary biologicals naming proposal issued by FDA in August 2015 [6].
FDA has proposed a new policy that would require every biological – whether originator or biosimilar – to have a distinct non-proprietary scientific name. Prescribers concluded that FDA was right to require a distinct non-proprietary scientific name for every biological product – originator or biosimilar – that FDA had approved.
Product labelling is seen at the heart of building user confidence in biosimilars [7]. In a subsequent study, the ASBM invited 9,813 prescribers to complete a study on the labelling of biologicals. 624 of these responded, of which 400 qualified and completed the study. Physicians who completed the study were asked what information they would like to see in a biological product label in order to choose between multiple biosimilars and their reference products. Physicians were asked what information could be included in a label, such as what clinical data should be present; whether or not the product was a biosimilar; and whether or not it was interchangeable.
All the information included in the labelling survey was considered important by the physicians surveyed. The greatest importance was accorded to an indication that the drug was a biosimilar. Physicians responded that including information on interchangeability was slightly less important than this.
Physician biosimilars labelling survey
Four hundred physicians were recruited in the US to complete a 15-minute web-based questionnaire on biosimilar labelling. In a separate, independent study, 400 prescribers were recruited in the US to complete a 15-minute web-based questionnaire on biosimilar naming. Participants in both surveys received a standard cash stipend of US$25 for their time to complete the survey.
All participants in the labelling survey were located in the US. They were recruited from a large, reputable panel of physicians and were all board certified in one of the following six specialities: dermatology, endocrinology, nephrology, neurology, oncology, or rheumatology. All participants prescribed biological medicine.
Of the 400 physicians who completed the labelling survey, 23% specialized in dermatology, 15% in endocrinology, 16% in nephrology, 15% in neurology, 16% in oncology, and 16% in rheumatology.
Prescribers completing the labelling survey worked in different settings. Twenty-six per cent of respondents worked in a community setting, 24% worked in an academic medical centre, 22% worked in a multi-speciality clinic, 17% worked in a private or family practice, 8% worked in a hospital, see Figure 1.
A total of 7% of participants in the labelling survey had spent 1–5 years in practice, 26% had spent 6–10 years, 41% had spent 11–20 years, 22% had spent 21–30 years, and 4% had spent more than 30 years in practice.
Physician biosimilars naming survey
The 400 participants in the naming survey were also based in the US. They were recruited from a large, global panel of healthcare professionals. Participants specialized in one of the following seven therapeutic specialities: dermatology, endocrinology, gastrointestinal, nephrology, neurology, oncology or rheumatology.
Among physicians completing the naming survey, 13% specialized in dermatology, 15% in endocrinology, 14% in gastrointestinal, 14% in nephrology, 14% in neurology, 16% in oncology, and 14% in rheumatology.
Prescribers completing the naming survey worked in different settings. 26% of respondents worked in an academic medical centre, 25% in a community setting, 20% in a private or family practice, 18% in a multi-specialist clinic, 7% in a hospital, and 2% in a military/veterans affairs hospital, see Figure 2.
A total of 7% of the physicians completing the naming survey had spent 1–5 years in practice, 33% had spent 6–10 years in practice, 32% had spent 11–20 years, 21% had spent 21–30 years, and 6% had spent more than 30 years in practice.
Participants in the naming survey were asked about their familiarity with FDA’s ‘Orange Book’ [8], the resource for Approved Drug Products with Therapeutic Equivalence Evaluations. The ‘Orange Book’ is a reference that identifies drug products approved on the basis of safety and effectiveness by FDA. Only 13% considered themselves very familiar with the book, 33% somewhat familiar, 26% vaguely familiar, and over a quarter (28%) had never heard of it. Only 4% used the Orange Book on a daily basis, 17% used it weekly, 14% used it monthly, 28% used it rarely, and 37% never used it.
Asked about their familiarity with FDA’s ‘Purple Book’ [9], that is, the resources for Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations, 8% were very familiar, 23% were somewhat familiar, 28% were vaguely familiar, and 41% had never heard of it. Only 4% used the Purple Book daily, 11% used it weekly, 12% used it monthly, 23% used it rarely, and over half (51%) never used it. Information contained in the Purple Book is designed to help enable a user to see whether a particular biological product has been determined by FDA to be biosimilar to or interchangeable with a reference biological product.
All physicians questioned in the naming survey claimed that they identified all medicines that they prescribed (biological and chemical) in the medical record.
Asked how they identified a medicine in the patient record, 25% said by scientific name, 34% by brand name, and 39% said it varied by medicine. Asked whether they would report an adverse event by using a drug’s product name or National Drug Code (NDC) number, 47% said they would use the brand name, 38% said they would use the scientific name, 2% would use the NDC number, and 13% had no preference.
Participants in the naming survey were asked for their attitudes and beliefs on product naming for originator and biosimilar products. 72% of respondents thought that if medicines had the same non-proprietary scientific name then they were probably structurally identical, 16% thought they would not be structurally identical, and 12% had no opinion. If two products have the same non-proprietary scientific name then 68% of respondents thought that a patient could safely receive either product and expect the same result. Twenty-one per cent of respondents would not expect the same result, and 11% had no opinion.
Asked about switching (when a patient is switched from one medicine to another), 60% of respondents thought that patients could safely be switched between two medicines that had the same non-proprietary scientific name and expect the same results. A quarter of respondents (25%) thought that patients could not safely be switched between two medicines with the same non-proprietary scientific name, 14% of respondents had no opinion.
Physician responses to biosimilar labelling survey
On a scale of 1–5, where 1 is not important at all and 5 is very important, 90% of the physicians questioned rated the importance of whether a product label for a biosimilar should clearly indicate that it is a biosimilar as either a 4 or 5, see Figure 3. On the question of post-marketing data, 79% of respondents rated the importance of including this data on the biosimilar label as either a 4 or 5, see Figure 4. On the question of interchangeability, 79% of respondents rated the importance of including whether or not a biosimilar is interchangeable as either a 4 or 5, see Figure 5. The responses of physicians to questions on biosimilar labelling are shown in Table 1.
Physician responses to biosimilar naming survey
In the naming survey, prescribers were asked whether FDA should require a distinct non-proprietary scientific name for every biological product FDA had approved – whether originator or biosimilar. FDA has recently proposed a distinct non-proprietary scientific name for all products, whether originator or biosimilar. This is intended to aid the process of pharmacovigilance and accurate prescribing and dispensing of medicines.
Respondents were asked for their views on the information that should be included in a product name and for their attitudes and beliefs on substitution.
Responses to questions in the physicians’ naming survey are shown in Table 2.
The spread of prescribers’ responses to questions related to the information contained in the representative suffix are given in Figure 6.
Generally speaking, all issues raised in the labelling survey were considered by the physicians surveyed to be very important for label inclusion. The inclusion of an indication of interchangeability was considered very important by 54% of the physicians. In fact, the physicians concluded that interchangeability was – of all the features included in the survey questions – the least important feature, with an average score of 4.1 out of 5. On average, the physicians gave the highest score, 4.4, to indicating that the drug was a biosimilar.
Segment differences were examined for all issues included in the survey. Segments among physicians included specialty, time spent working in health care, and practice setting. Very few specialty differences were noted and no differences for practice setting were noted. In general, the longer a physician had been in practice, the more important they thought it was to include the features mentioned in the survey on the biosimilar product label.
FDA proposal for a random suffix on a product’s name that does not indicate the manufacturer was not broadly welcomed, with only 9% of respondents agreeing with FDA. Instead, most physicians (60%) would prefer a suffix on the non-proprietary scientific name that is indicative of the product’s manufacturer. A third of respondents (32%) had no opinion.
In the naming survey, it was clear that respondents were not in complete agreement on how biological medicines, whether originators or biosimilars, are named. Reaching an agreement on the naming of these medicines will be key in building user confidence in biosimilars. The data presented here provide important feedback from a wide range of physicians who prescribe biologicals in the US. The findings will be helpful in determining how these biosimilar medicines are regulated in future.
|
Key points of the 2015 physicians naming and labelling survey
|
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Harry L Gewanter, MD, FAAP, FACR, Chairman of the Alliance for Safe Biologic Medicines (ASBM), and Mr Michael S Reilly, Esq, Executive Director; are employed by ASBM.
This paper is funded by ASBM.
Provenance and peer review: Not commissioned; externally peer reviewed.
Harry L Gewanter, MD, FAAP, FACR, Chairman
Michael S Reilly, Esq, Executive Director
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Biologics Price Competition and Innovation Act of 2009, Public Law 111-148, Sec. 7001–7003, 124 Stat. 119. Mar. 23, 2010.
2. Cohen JP, Felix AE, Riggs K, Gupta A. Barriers to market uptake of biosimilars in the US. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):108-15. doi:10.5639/gabij.2014.0303.028
3. O Dolinar R, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018
4. Feijó Azevedo V, Mysler E, Aceituno Álvarez A, Hughes J, Javier Flores-Murrieta F, Ruiz de Castilla EM. Recommendations for the regulation of biosimilars and their implementation in Latin America. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):143-8. doi:10.5639/gabij.2014.0303.032
5. Abas A, Siew Khoon Khoo Y. Regulation of biologicals in Malaysia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):193-8. doi:10.5639/gabij.2014.0304.044
6. GaBI Online – Generics and Biosimilars Initiative. FDA issues draft guidance on biosimilars labelling [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Feb 12]. Available from: www.gabionline.net/Guidelines/FDA-issues-draft-guidance-on-biosimilars-labelling
7. European Biopharmaceutical Enterprises. Tell me the whole story: the role of product labelling in building user confidence in biosimilars in Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):188-92. doi:10.5639/gabij.2014.0304.043
8. U.S. Food and Drug Administration. Orange Book: approved drug products with therapeutic equivalence evaluations [homepage on the Internet]. [cited 2017 Feb 12]. Available from: http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm
9. U.S. Food and Drug Administration. Purple Book: lists of licensed biological products with reference product exclusivity and biosimilarity or interchangeability evaluations [homepage on the Internet]. [cited 2017 Feb 12]. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/naming-and-labelling-of-biologicals-a-survey-of-us-physicians-perspectives.html
Author byline as per print journal: Professor Philip J Schneider, MS, FASHP; Michael S Reilly, Esq
|
Introduction: To date, the US Food and Drug Administration (FDA) has offered only draft guidance on the naming of biosimilar medicines. The Alliance for Safe Biologic Medicines (ASBM) has asked pharmacists for their views on the labelling and naming of biosimilar medicines. |
Submitted: 28 September 2016; Revised: 28 October 2016; Accepted: 31 October 2016; Published online first: 14 November 2016
The market uptake of biosimilars in the US and worldwide will depend on regulatory policies [1], for which an agreed naming and labelling system will be important [2]. A survey of the views of European physicians on familiarity of biosimilar medicines demonstrated the need for distinguishable non-proprietary names to be given to all biologicals [3]. This has been supported by a number of discussions surrounding the development of clear regulation in this area [4–7], and a number of countries across the globe from Latin America [8, 9], Australia [10] and beyond; have called for specific nomenclature to be developed. The results of these surveys reinforce the value of a global naming policy for biologicals and the importance of the World Health Organization (WHO) moving forward with its biological qualifier proposal.
Since the US Food and Drug Administration (FDA) has only distributed draft guidance on the naming of biological medicines [11] and biosimilar labelling [12, 13], feedback from the pharmacists who prepare and dispense them is also important in determining how these drugs are regulated. In Europe, product labelling is seen as important to build user confidence in biosimilars [14]. In response to concerns in Europe, the Alliance for Safe Biologic Medicines (ASBM) invited 3,525 pharmacists in the US to complete a survey that included questions related to the information that could be included in a label, such as whether or not the product was a biosimilar; what analytical/clinical data and clinical similarity data should be present; post-marketing data; approved and non-approved indications; data source; and whether or not it was interchangeable/substitutable.
FDA has proposed a new policy that would require every biological – whether originator or biosimilar – to have a distinct non-proprietary scientific name. Both pharmacists and prescribers concluded that FDA was right to require a distinct non-proprietary scientific name for every biological product – originator or biosimilar – that FDA had approved.
In 2015, the ASBM invited 3,525 pharmacists in the US to complete a survey on the naming of biological medicines and biosimilar labelling [15], including feedback on FDA draft guidance on non-proprietary biologicals naming [11]. A total of 849 pharmacists replied (a response rate of 24%). Of these, 448 pharmacists were screened out predominately for their lack of knowledge on biologicals or did not complete the survey. A total of 401 pharmacists (11.4% of all those invited) completed the survey, and are collectively termed ‘respondents’. Pharmacists were reimbursed US$22 for completing the survey.
Pharmacists were recruited from a large, global panel of healthcare professionals and were either employed in a hospital/health system pharmacy (60%) or retail pharmacy setting (40%). All 401 pharmacists that completed the study had dispensed biological medicines and had been in practice as a pharmacist for one year or more, see Figure 1.
Pharmacists were asked what information they would like to see included in a biological product label in order to choose between multiple biosimilars and their reference products. Data were analysed using MS Excel, and checked manually.
Use of biological product reference material by pharmacists
The majority (64%) of pharmacist respondents were very familiar with the FDA ‘Orange Book’ [16], that is, the resource for Approved Drug Products, with Therapeutic Equivalence Evaluations. The ‘Orange Book’ is a reference that identifies drug products approved on the basis of safety and effectiveness by FDA. One third (29%) of pharmacists refer to this at least weekly, 24% monthly, 6% daily, with the rest referring to it less frequently. In contrast, 28% of respondents had never heard of the FDA ‘Purple Book’ [17], that is, the resource for Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations, and almost 80% of respondents never or infrequently used or referred to it. Information contained in the Purple Book is designed to help enable a user to see whether a particular biological product has been determined by FDA to be biosimilar to or interchangeable with a reference biological product. The results from the survey are outlined below and presented in Tables 1 and 2. Only 2% of pharmacists used the Purple Book daily, 7% of pharmacists used it weekly, 12% of pharmacists used it monthly, 30% of pharmacists used it rarely, and 49% of pharmacists never used the Purple Book at all.
Knowledge of biosimilars
Survey participants were asked how familiar they were with biosimilar medicines with the following question:
‘Biosimilar medicines are intended to be copies of already approved biological medicines. They are referred to as “biosimilar” rather than “generic” because they will be similar, but not identical to the product they copy. How familiar are you with biosimilar medicines?’
Of the pharmacists who completed the survey, 57% of respondents said that they were familiar with biosimilars having a basic understanding and 35% said they had a complete understanding. Hospital pharmacists included in the survey reported being the most familiar with biosimilars, with 44% saying they had a complete understanding, while only 23% of retail pharmacists reported having a complete understanding.
Pharmacists were asked about their knowledge of the approval process for biosimilars using the following question:
‘Originator medicines are approved by the US Food and Drug Administration based on an evaluation of clinical data that demonstrates a medicine is safe and effective for the specified indication and data must be provided for every indication. The approval pathway for biosimilars is different than for originator medicines. Are you aware a biosimilar medicine may be approved for several or all indications of the reference product on the basis of clinical trials in only one of those indications?’
The responses suggest that overall knowledge among respondents was good with 86% answering yes (91% hospital pharmacists are more likely to respond ‘Yes’ versus 78% retail pharmacists). When asked if they thought that their understanding of the biosimilar approvals process was acceptable, there was a consensus that this was ‘acceptable’ (27%) or at least ‘somewhat acceptable’ (51%).
Naming knowledge
The survey participants were asked questions about their knowledge of the naming of biosimilars and what this meant about how these products could be used. Most respondents (63%) indicated that if biological medicines have the same non-proprietary scientific name, this would imply that the products are identical, with 68% hospital pharmacists are more likely to answer ‘Yes’ versus 57% retail pharmacists. Those respondents would expect the same results from biological medicines with the same non-proprietary scientific name. The majority of respondents (58%) believed that products sharing non-proprietary scientific names could be safely switched from a reference biological medicine to its biosimilar during a course of treatment and the same result would be expected with either of the products. When the same non-proprietary scientific name is used in two biological medicines, 55% of respondents would also assume that the medicines had both been approved for the same indications.
Naming requirements
When questioned about how biological naming should be regulated, the majority (68%) of the respondents thought that each product – originator or biosimilar – should have a distinct/unique non-proprietary scientific name. Furthermore, 77% of respondents thought the suffix on a biological medicine name should indicate its manufacturer, rather than a random suffix in future product approvals.
Biosimilar labelling
A total of 58% of respondents thought that it was very important that a product label for a biosimilar clearly indicates that it is a biosimilar. They also agreed that labels should include an explanation as to what a biosimilar is. Analytical information resulting from biosimilarity studies should be included, as should any clinical data submitted to FDA and any post-marketing data. There was also a consensus that the label should include reference to the brand name of the originator product.
Participants felt strongly that it should be clear and explicit on the label if a biosimilar product has not been approved for all indications approved for use of the reference product. The label should clearly distinguish data generated from the use of the biosimilar sponsor and that generated by the originator sponsor, and that it is clear which indications were studied with the biosimilar sponsor and which indications were approved based on extrapolation from studies of the reference product. Respondents also felt that labels should clearly include all relevant data used to establish similarity and clearly indicate if the biosimilar is interchangeable with a reference product.
The results of this survey of pharmacists are consistent with the results from a survey of physicians in Europe [3]. In both surveys, respondents believed that products sharing a non-proprietary name could be considered identical, could be expected to produce the same results, could be used interchangeably and would be approved for all the indications of the reference product.
Many of the findings in this study support a recent survey sponsored by the Academy of Managed Care Pharmacy (AMCP) [18]. In the AMCP study, more than 60% of participants (62.3%) reported preferring a biosimilar naming convention that uses either a designated suffix (48.1% of all participants) or prefix (14.2% of all participants). Of the 48.1% who would prefer a designated suffix, the vast majority (83.4%) wanted a suffix that was based on the name of the manufacturer. In this survey, 77% of pharmacists expressed a preference for a suffix based on the manufacturer.
To date, FDA is yet to finalize its guidance for the naming of biologicals and labelling of biosimilars. This is a fast-evolving area – following publication of its draft guidance in 2015 and 2016 [11, 12], FDA issued a request for comments on expanding the number of suffixes that biosimilars makers could propose. The request was swiftly withdrawn; following what FDA said was an administrative error [19]. Some have speculated that the agency wanted to extend the comment period, which was originally in July 2016.
The results from surveys like the one described here will aid in the development of a clear and comprehensive system to promote the safe and effective use of biologicals and biosimilars, and as a result facilitate consumer confidence and market uptake in the US.
The pharmacists responding to this survey reported having a good overall knowledge of biosimilars and their approval process. The results suggest that when a biosimilar and the reference product share the same non-proprietary scientific name, it could lead to confusion among pharmacists. This is because two products sharing a non-proprietary name could be considered identical, be expected to produce the same results from both drugs, be used interchangeably, and be approved for all of indications of the reference product. As these are assumptions that cannot be made with biosimilars, pharmacist respondents agreed that all biological products should have unique names and that clear and explanatory labelling of these products should be required.
In general, all issues raised with regards to labelling were considered by pharmacist respondents to be very important for label inclusion. This means that they are supportive of biosimilar products being labelled specifically as such, with the clear inclusion of what a biosimilar product is. There should also be clear information about the analytical studies and clinical studies used for the approval of the product. In cases where the biosimilar is not approved for all indications of the reference product, this should be clearly indicated where these indications are based on extrapolation of the indications approved for the reference product, and whether the biosimilar is interchangeable.
|
Key points of the 2015 pharmacists naming and labelling survey
|
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Disclosure of financial and competing interests: Mr Michael S Reilly, Esq, Executive Director, is employed by ASBM.
Professor Philip J Schneider is a member of the International Advisory Board of ASBM since 2012 without compensation. From September 2014, Professor Schneider has been the Chair of the International Advisory Board and is paid a small stipend for that role.
This paper is funded by ASBM and represents the policies of the organization.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor Philip J Schneider, MS, FASHP
Associate Dean, College of Pharmacy, University of Arizona, Phoenix Biomedical Campus 3384, 1295 N Martin, PO Box 210202, Tucson, AZ 85721, USA
Michael S Reilly, Esq
Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Cohen JP, Felix AE, Riggs K, Gupta A. Barriers to market uptake of biosimilars in the US. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):108-15. doi:10.5639/gabij.2014.0303.028
2. Fuhr JP, Chandra A, Romley J, et al. Product naming, pricing, and market uptake of biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(2):64-71. doi:10.5639/gabij.2015.0402.015
3. Dolinar RO, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018
4. Robertson JS. The challenges of nomenclature – INN, biosimilars and biological qualifiers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):110-2. doi:10.5639/gabij.2015.0403.025
5. Declerck PJ. Common or distinct INN for biosimilars? Only characteristics of the active substance prior to formulation should be considered. Generics and Biosimilars Initiative Journal (GaBI Journal).2014;3(1):8. doi:10.5639/gabij.2014.0301.003
6. Alexander EA. The biosimilar name debate: what’s at stake for public health. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):10-2. doi:10.5639/gabij.2014.0301.005
7. Maggio ET. Critical immunogenicity differences will be obscured by a common INN for biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):166. doi:10.5639/gabij.2013.0204.046
8. Feijó Azevedo V, Mysler E, Aceituno Álvarez A, et al. Recommendations for the regulation of biosimilars and their implementation in Latin America. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):143-8. doi:10.5639/gabij.2014.0303.032
9. Gewanter HL, Reilly MS. Prescribing practices for biosimilars: questionnaire survey findings from physicians in Argentina, Brazil, Colombia and Mexico. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):161-6. doi:10.5639/gabij.2015.0404.036
10. Shaw B. Biosimilars naming and prescribing policy in Australia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):168-9. doi:10.5639/gabij.2013.0204.048
11. U.S. Food and Drug Administration. Nonproprietary naming of biological products. August 2015 [homepage on the Internet]. [cited 2016 Oct 28]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM459987.pdf
12. GaBI Online – Generics and Biosimilars Initiative. FDA issues draft guidance on biosimilars labelling [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Oct 28]. Available from: http://www.gabionline.net/Guidelines/FDA-issues-draft-guidance-on-biosimilars-labelling
13. U.S. Food and Drug Administration. Labeling for biosimilar products. March 2016 [homepage on the Internet]. [cited 2016 Oct 28]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm493439.pdf
14. Tell me the whole story: the role of product labelling in building user confidence in biosimilars in Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):188-92. doi:10.5639/gabij.2014.0304.043
15. Safe Biologics. Olson K. Biosimilars naming and labelling. A study of U.S. pharmacists. October 2015 [homepage on the Internet]. [cited 2016 Oct 28]. Available from: https://safebiologics.org/wp-content/uploads/2015/10/2015-US-Pharmacists-Survey.pdf
16. U.S. Food and Drug Administration. Orange Book: Approved drug products with therapeutic equivalence evaluations [homepage on the Internet]. [cited 2016 Oct 28]. Available from: http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm
17. U.S. Food and Drug Administration. Purple Book: Lists of licensed biological products with reference product exclusivity and biosimilarity or interchangeability evaluations [homepage on the Internet]. [cited 2016 Oct 28]. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/Therapeutic
BiologicApplications/Biosimilars/ucm411418.htm
18. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-26.
19. GaBI Online – Generics and Biosimilars Initiative. FDA withdraws biosimilar suffix proposal aplasia [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Oct 28]. Available from: www.gabionline.net/Guidelines/FDA-withdraws-biosimilar-suffix-proposal
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/naming-and-labelling-of-biologicals-the-perspective-of-hospital-and-retail-pharmacists.html
Copyright ©2025 GaBI Journal unless otherwise noted.
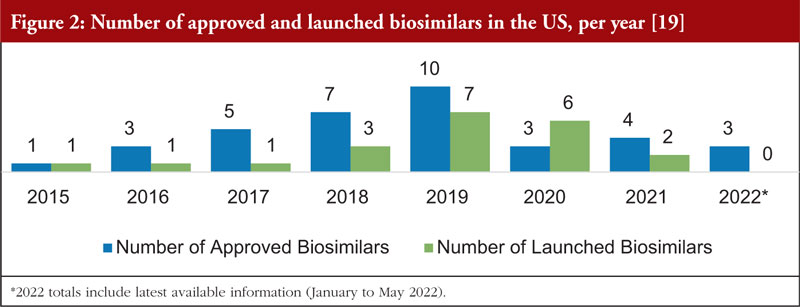
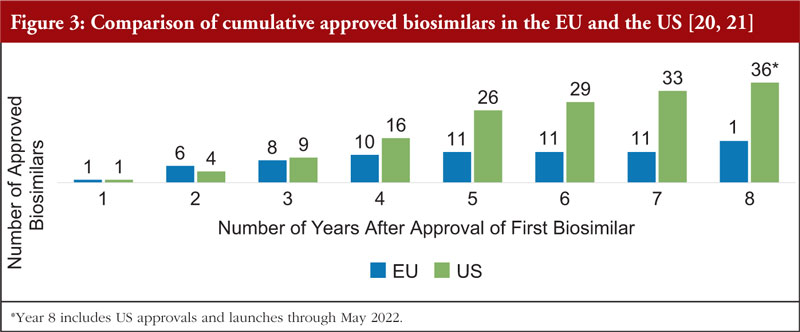













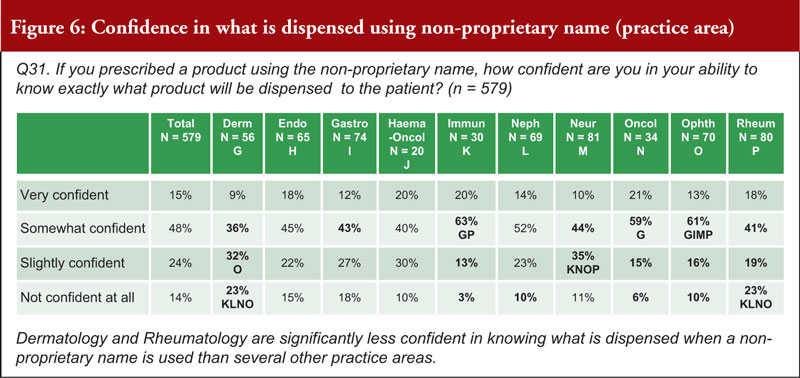

















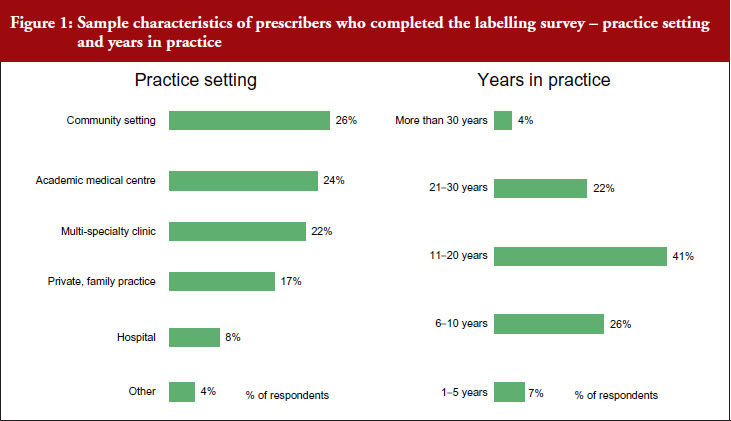
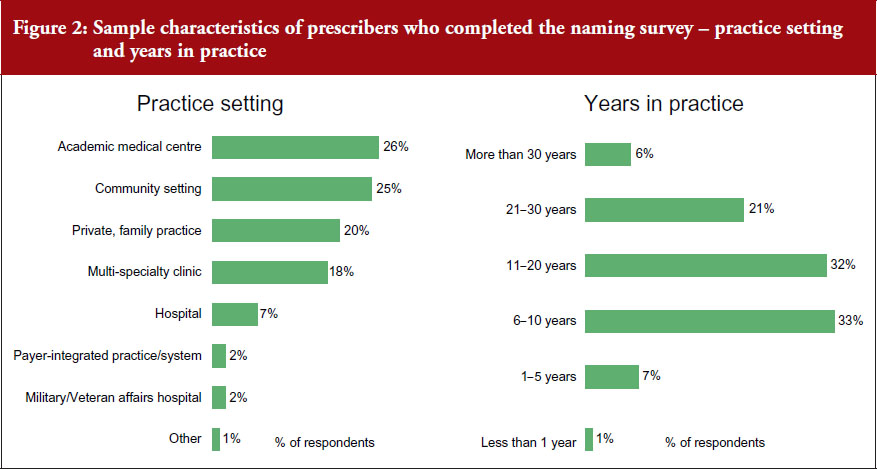
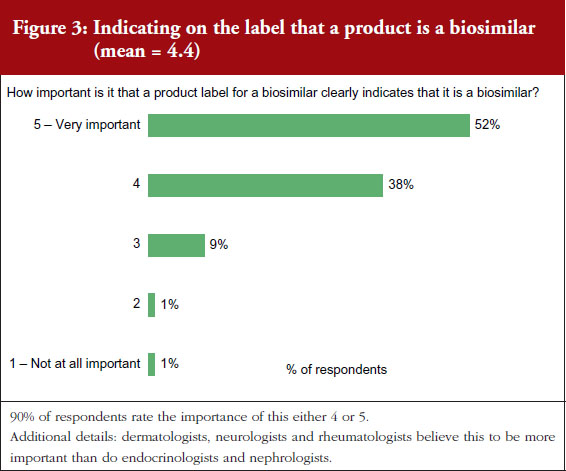
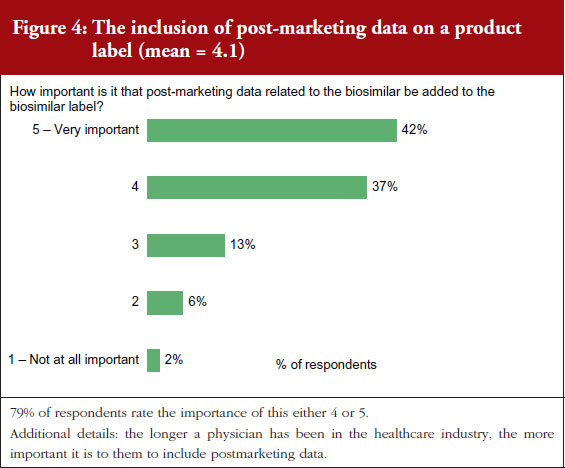
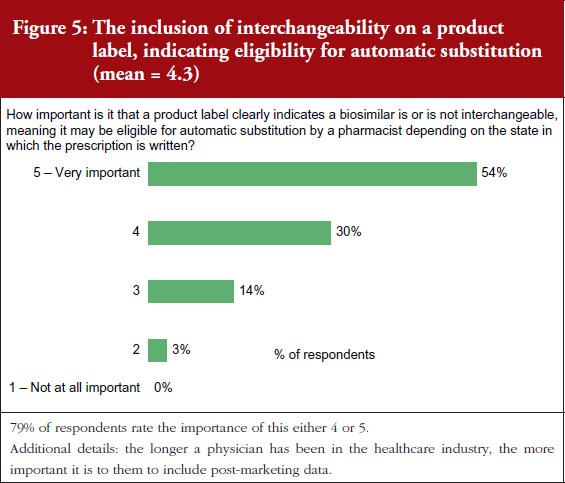
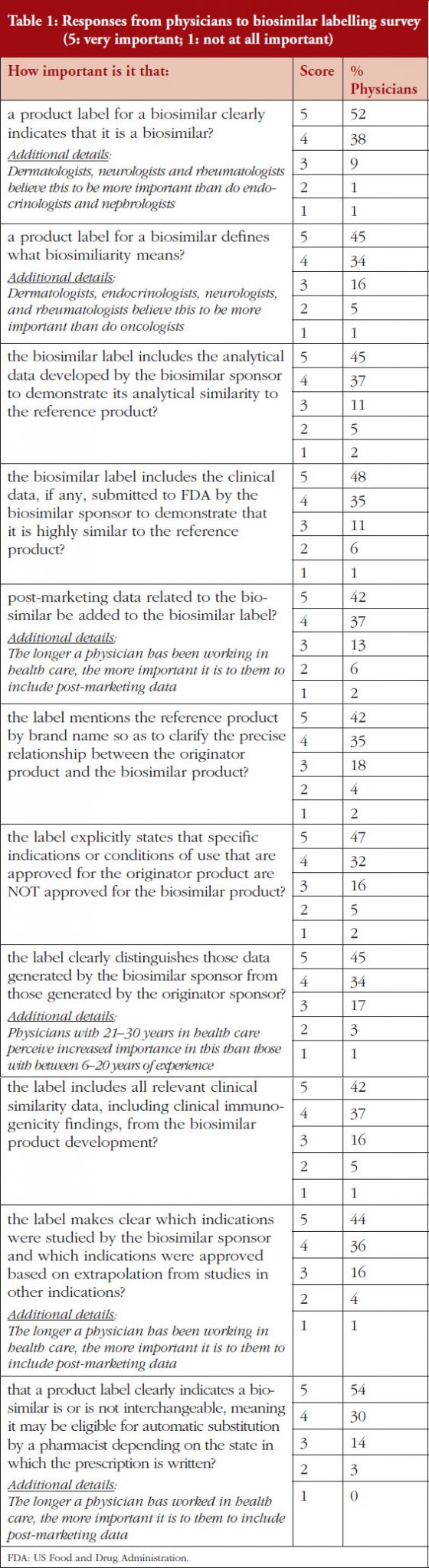
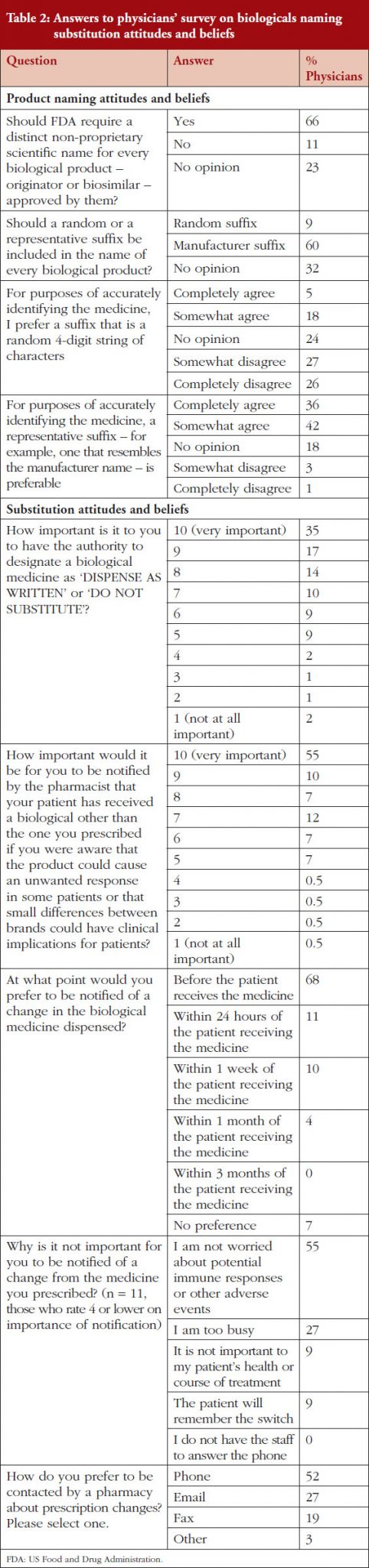
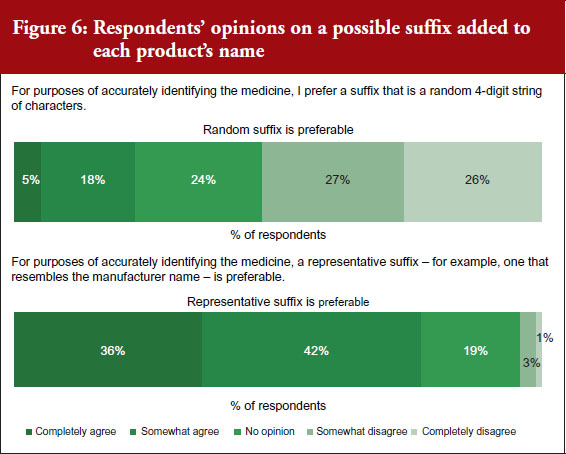



A white paper: US biosimilars market on pace with Europe
Abstract:
In the US, 28 biosimilars have been approved, with 10 in the last two years. The US is keeping pace with the EU who pioneered biosimilars approvals a decade earlier. Herein, current FDA regulations and hurdles encountered for US biosimilar approval and uptake are discussed.
Submitted: 12 October 2020; Revised: 28 October 2020; Accepted: 29 October 2020; Published online first: 9 November 2020
While many sources claim that Europe is winning the race when it comes to biosimilars, a broader assessment of the landscape reveals a more encouraging story for the US. Although the European Medicines Agency (EMA) pioneered the framework for biosimilar regulation, the US Food and Drug Administration (FDA) is moving at approximately the same pace as EMA based on the number of approvals at the same time after implementation of its regulatory pathway [1].
The biosimilar regulatory framework in Europe was implemented in 2004. Within this framework, the Committee for Medicinal Products for Human Use (CHMP) provides initial assessments for marketing authorization of new medicines that are ultimately approved centrally by EMA [2]. In the US, the Biologics Price Competition and Innovation Act of 2009 (BPCIA Act) created an abbreviated licensure pathway for biosimilars and granted FDA the authority to approve these near-identical biologicals [3]. This legislation was implemented in 2010 as part of the Patient Protection and Affordable Care Act signed by President Barack Obama.
As of September 2020, approximately 10 years after implementation of the biosimilar approval pathway, 28 biosimilars have been approved by FDA, with 18 of those approvals granted in the last two years [4]. In the 10-year time period following the creation of Europe’s biosimilar regulatory pathway, EMA approved 13 biosimilar products (some of which were marketed under several different brands) [5]. From this perspective, the US appears to be on a faster pace than the EU in terms of biosimilar approvals. Currently, there are 45 biosimilars approved in Europe; however, these estimates fall to 35 when products approved in the US as follow-on biologicals via the 505(b)(2) pathway, e.g. somatropin, insulin, teriparatide, or abbreviated new drug application (ANDA) are excluded, see Table 1. Furthermore, Europe’s filgrastim biosimilar Tevagrastim®/Ratiograstim® was approved as Granix® (tbo-filgrastim) in the US via a Biologic License Application (BLA) prior to the implementation of a biosimilar approval pathway and is not included in the US biosimilar count.
Biosimilars have been developed in various therapeutic areas, including oncology and rheumatology, and are based on nine reference products. For some reference products, numerous biosimilars have been developed; for example, five biosimilars of Genentech’s Herceptin® (trastuzumab) and Amgen’s Neulasta® (pegfilgrastim) have been approved in the US and Europe. A summary of the biosimilars landscape in the US and EU is presented in Table 1.
There are currently nine biosimilar applications under CHMP evaluation, including biosimilar candidates for adalimumab (2), bevacizumab (5), pegfilgrastim (1), and trastuzumab (1) [6]. Unlike the transparency on biosimilar filings in Europe, FDA does not provide information on biosimilar candidates under review until they are approved.
Since the creation of a regulatory approval pathway, numerous guidance documents have been developed by both EMA and FDA to guide manufacturers in their development of biosimilar candidates. At least 10 documents have been released by FDA that provide guidance on scientific and quality considerations in the demonstration of biosimilarity and interchangeability [7]. EMA has published three overarching guidelines and nine product-specific guidelines for biosimilars manufacturers that address both non-clinical and clinical issues as well as quality-related issues [8].
Because EMA pioneered biosimilar regulations, the question has commonly been raised: To what extent does the European biosimilar experience translate to the US? Although the US and the EU are on a similar trajectory in terms of the number of biosimilar products approved, these markets differ in several respects. A critical point of divergence between the US and EU biosimilars terrain is the concept of interchangeability. Unique to the US, FDA may designate a product ‘interchangeable’ if it meets additional requirements beyond being biosimilar, which translates to more clinical development that includes switching studies and increased cost from a manufacturer’s perspective. To date, no biosimilar products have interchangeability status. Having a separate designation of interchangeability for an approved biological has been said to give the impression that interchangeable biosimilars are superior in quality to non-interchangeable biologicals ‒ which is not the case. A major focus area for the Association for Accessible Medicines’ Biosimilars Council has been educating physicians and payers on interchangeability designations as they are ‘waiting for interchangeability to really get on board with biosimilars’ [9].
Perceptions of prescribing physicians about the safety of biosimilars is an ongoing issue. There is a need for increased confidence, particularly around switching from a reference product to a biosimilar. Concerns are particularly heightened in the oncology therapeutic area, where treatments can be curative and clinicians fear the loss of efficacy and increased immunogenicity [10]. One of the key concerns around switching to a biosimilar is the negative counterpart of the placebo effect: the nocebo effect, a phenomenon in which negative expectations lead to worsening of symptoms [11]. Some physicians continue to have doubts regarding the rigorous approval process for biosimilars and switching studies that have been performed thus far and transfer these negative concerns to patients through body language or tone of voice when discussing treatment options. Raising awareness about the nocebo effect by educating healthcare professionals on this phenomenon may mitigate its effect in patients receiving biosimilar products [12, 13].
The biosimilars marketplace
While the European biosimilars market has been credited with higher uptake compared to the US market, rates of uptake differ from country to country in Europe and can vary significantly by product class. A report by KPMG commissioned by Medicines for Europe to analyse the procurement of medicines in hospitals in eight European countries highlighted the variability in biosimilar sales against originator in these different Member States [14]. An average of hospital biosimilar volume in March 2019 showed that Denmark achieved 63% overall biosimilar volume, with the UK coming in second at 45%. Germany had 40% biosimilar volume, France had 34%, and Belgium tied with Switzerland for last place among the countries studied at 14%. In a recent assessment of the impact of biosimilar competition in Europe, 16 European countries were reported to have achieved > 90% biosimilar utilization for filgrastim and pegfilgrastim in 2018, while utilization in Ireland was just 27%. Among antitumour necrosis factor biosimilars (adalimumab, etanercept and infliximab), Norway and Denmark had 81% and 96% biosimilar uptake, respectively, while every other country’s utilization was less than 50% [15]. Variations in adoption rates among individual European countries as well as across therapeutic areas are influenced by government involvement, reimbursement structures and tender procurement policies.
In the US, biosimilars have gained significant share in the majority of therapeutic areas in which they have been introduced, ranging on average from 20% to 25% within the first year of launch, with some projected to reach greater than 50% within the first two years [16, 17]. As expected, first-to-market biosimilars tend to capture a greater portion of the segment compared to later entrants. Filgrastim biosimilars have been on the market the longest at five years and have achieved a 72% share, while bevacizumab and trastuzumab biosimilars have approximately 40% share. Rituximab and infliximab have had the most limited adoption, with approximately 20% market share [16].
Biosimilar competition has a significant potential to impact overall drug spending, with the amount of savings per country based on the volume and list price of products prior to biosimilar entry [15, 17]. Manufacturers are reducing healthcare costs by launching biosimilars at a wholesale acquisition cost (WAC) that is generally 15% to 37% lower than the reference product WAC and 3% to 40% below the reference product average sales price (ASP). Manufacturers of originator products have had to adapt their strategies in order to stay competitive, whether it be launching a second-generation product, or a new formulation aimed at reducing side effects. Biosimilar competition and originator manufacturer responses are translating into significant savings. The annualized savings reached US$6.5 billion in the second quarter of 2020, where reference molecules sold US$20.8 billion (annualized) pre-biosimilar [18]. Savings over the next five years as a result of biosimilar alternatives are projected to exceed US$100 billion [17].
Biosimilars include both self-administered drugs obtained at retail pharmacies as well as provider-administrated drugs in inpatient and outpatient settings. In the US, the balance of cost savings to healthcare payers, providers and patients is different for each of these due to differences in payment and cost-sharing arrangements [19]. Most self-administered pharmacy-dispensed biologicals are paid for on a fee-for-service basis and the final amount paid by insurers reflects several transactions, including a confidential rebate payment. On the other hand, the costs associated with provider-administered biologicals are purchased directly from manufacturers and wholesalers or through group purchasing organizations either incorporated into prospective, bundled payments or under fee-for-service arrangements. Access to these biologicals (reference or biosimilar) is determined by formularies that may reflect the highest price concession rather than the lowest list price.
To date, there have been no self-administered biosimilars launched in the US. This is in stark contrast to Europe, where biosimilars administered via self-injection at home are available, including a recently approved subcutaneous form of infliximab [20]. It remains to be seen how self-administered biosimilars will fare in the US system where higher list price drugs may be ‘preferred’ on the formulary based on their profitability for those constructing the formulary.
Overcoming the hurdles
Despite having approved 28 biosimilars in 10 years, the US faces one of its biggest biosimilar hurdles when it comes to launching these products. So far, 18 of 28 biosimilars have been made available to patients, while the others have not been commercialized due to patent-related issues. In an effort to protect their originator products, brand manufacturers block competition by filing numerous follow-on patents. The expense of challenging these patents can serve as a deterrent to potential biosimilar competitors [21]. Brand manufacturers also disincentivize biosimilar utilization by leveraging a ‘rebate trap’ wherein brand manufacturers offer significant rebates for their products that disable competition from biosimilars coming on the market [22].
While there is work to be done to accelerate commercialization of biosimilars, FDA has taken active steps to mitigate some of these barriers to biosimilar utilization. In July 2018, Health and Human Services Secretary Alex Azar announced a Biosimilars Action Plan (BAP) to advance policies that facilitate the development of the biosimilars market and increase competition for biological drugs [23].
Key strategies of FDA’s BAP of 2018:
FDA’s most recent actions addressed biosimilar market competition in a joint statement issued by FDA and the Federal Trade Commission (FTC) that identified four goals to help combat anti-competitive practices [24]. In particular, the agencies noted their concern with false or misleading statements comparing biological reference products and biosimilars, which may be hampering biosimilar uptake by creating negative misperceptions about the safety and efficacy of biosimilar therapies. To further clarify how data and information about biosimilars should be presented in a truthful and non-misleading manner in regulated promotional materials, FDA announced the release of the ‘Draft guidance for industry: promotional labeling and advertising considerations for prescription biological reference and biosimilar products ‒ questions and answers’ and invited comment by stakeholders in the docket. As part of their efforts to promote greater competition, FDA and FTC held a public workshop in March 2020 to discuss their ‘collaborative efforts to support appropriate adoption of biosimilars, discourage false or misleading communications about biosimilars, and deter anticompetitive behaviors in the biological product marketplace’.
Targeting faster reviews, FDA issued new draft guidance in February 2020 detailing how it will speed its review of biosimilar or interchangeable application supplements, which can be used to update the initial biosimilar approval when it is for fewer than all the reference product’s licensed conditions of use [25]. At the time of submission, applicants may decide not to seek licensure of a proposed biosimilar for conditions of use that are protected by patent for the innovator. FDA had previously committed to reviewing and acting on original 351(k) BLA supplements with clinical data within 10 months of receipt; however, the agency now says such supplements will be reviewed and acted upon in a 6-month timeframe.
The US Centers for Medicare and Medicaid Services (CMS) have also revised a number of their payment policies in an effort to promote biosimilar competition. Three policy changes made under Medicare Part B to incentivize biosimilar uptake were: 1) BPCIA required Medicare Part B to pay for biosimilars at the ASP of the biosimilar plus 6% of the innovator product’s ASP [26]; 2) unique payment codes were provided for each biosimilar (reversing policy that grouped drugs under single codes) [27]; and 3) biosimilars were allowed ‘pass-through’ status under the 340B Drug Pricing Program, which meant they could be paid at ASP plus 6% rather than being heavily discounted (–22.5%) [28]. Furthermore, under Medicare Part D, the Bipartisan Budget Act of 2018 changed the treatment of biosimilars in the coverage gap (doughnut hole) discount programme, requiring manufacturers to give discounts for biosimilars [29]. Furthermore, in 2019, there was a lowering of the maximum copay amount on biosimilars to rates commensurate with generic copay amounts for lower income beneficiaries.
In parallel with updated regulatory policies that encourage development of the biosimilars market, a growing body of evidence has amassed to support the clinical safety of biosimilars. In a systematic review of primary data from 90 studies that enrolled 14,225 unique individuals, the nature and intensity of safety signals reported after switching from reference medicines to biosimilars were the same as those already known from continued use of the reference biological [30]. Similar results were observed in a systematic review of 57 switching studies evaluating the efficacy, safety and immunogenicity risk of transitioning between an originator biological and a biosimilar [31]. These results should increase confidence of patients, healthcare professionals and the public in biosimilars, leading to increased acceptance of these safe and effective medicines.
Conclusion
Although it has been suggested that biosimilars are not living up to their promise in the US, the current landscape suggests otherwise. FDA has approved 18 biosimilars in the past two years alone and a total of 28 since the implementation of a regulatory pathway just 10 years ago. Although getting these drugs into the hands of patients has hit some stumbling blocks due to anti-competitive behaviours from brand manufacturers, FDA has been proactive in addressing these ‘shenanigans’ and has created a BAP as well as additional guidances to mitigate such barriers to uptake and ensure the biosimilars market is poised for success. Clinical studies on the impact of switching between reference medicines and biosimilars have underscored the safety of biosimilars and impart confidence in their utilization for both physicians and patients.
In short, the overall outlook on biosimilars is positive. Pharmaceutical Research and Manufacturers of America (PhRMA), an organization representing biopharmaceutical manufacturers, believes that biosimilars are helping to bring down the cost of drugs in the US market and are positioned to achieve further savings. Katie Verb, Director of Policy and Research for PhRMA, says that federal policies are having a positive effect. These sentiments were echoed by Dr Leah Christl, Amgen’s Executive Director for Global Regulatory and Research and Development Policy, who believes the US biosimilars market is showing healthy vital signs.
Funding sources
This paper is funded by the Alliance for Safe Biologic Medicines (ASBM).
The ASBM is an organization composed of diverse healthcare groups and individuals ‒ from patients to physicians, innovative medical biotechnology companies and others ‒ who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Competing interests: Dr Madelaine Feldman is the Chairperson of the Alliance for Safe Biologic Medicines. She has participated in advisory boards for Gilead, Lilly, Pfizer and Samsung. Mr Michael S Reilly, Esq is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002 to 2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Madelaine Feldman, MD, FACR
Michael S Reilly, Esq
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Mehr SR, Brook RA. Biosimilars in the USA: will new efforts to spur approvals and access spur uptake and cost savings? Pharmaceut Med. 2019;33(1):1-8.
2. European Commission. Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending 2001/83/EC on the Community code relating to medicinal products for human use [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2004_27/dir_2004_27_en.pdf
3. Carver KH, Elikan J, Lietzan E. An unofficial legislative history of the Biologics Price Competition and Innovation Act of 2009. Food Drug Law J. 2010;65(4):671-818, ii.
4. U.S. Food and Drug Administration. Biosimilar product information. FDA-approved biosimilar products [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
5. European medicines Agency. Centrally authorized biosimilar medicines [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_bio-similar.
6. European Medicines Agency. EMA/535780/2020. Applications for new human medicines under evaluation by the Committee for Medicinal Products for Human Use. 8 October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/documents/report/applications-new-human-medicines-under-evaluation-chmp-october-2020_en.pdf
7. U.S. Food and Drug Administration. Biosimilar guidances. Current as of 21 June 2019 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances.
8. European Medicines Agency. Scientific guidelines on biosimilar medicinal products. 2020 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/ multidisciplinary/multidisciplinary-biosimilar
9. The Center for Biosimilars. AAM’s Christine Simmon on Biosimilars Council Initiatives and educating around interchangeability. 16 February 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.centerforbiosimilars.com/news/aams-christine-simmon-on-biosimilars-council-initiatives-and-educating-around-interchangeability
10. Konstantinidou S, Papaspiliou A, Kokkotou E. Current and future roles of biosimilars in oncology practice. Oncol Lett. 2020;19(1):45-51.
11. Planès S, Villier C, Mallaret M. The nocebo effect of drugs. Pharmacol Res Perspect. 2016;4(2):e00208.
12. Ebbers HC, Schellekens H. Are we ready to close the discussion on the interchangeability of biosimilars? Drug Discov Today. 2019;24(10):1963-7.
13. Rezk MF, Pieper B. To see or NOsee: the debate on the nocebo effect and optimizing the use of biosimilars. Adv Ther. 2018;35(6):749-53.
14. KPMG. Improving healthcare delivery in hospitals by optimized utilization of medicines: A study into 8 European countries. Commissioned by Medicines for Europe. 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2019/10/20190903_Hospital-Reform-Study_final.pdf
15. IQVIA. White Paper. The impact of biosimilar competition in Europe. Oct 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://ec.europa.eu/docsroom/documents/ 31642/attachments/1/translations/en/renditions/native
16. Amgen. 2020 Biosimilar trends report [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.amgenbiosimilars.com/-/media/Themes/Amgen/amgenbiosimilars-com/Amgenbiosimilars-com/spandf/USA-CBU-80723-2020-Amgen-Biosimilar-Trends-Report.pdf
17. IQVIA. Biosimilars in the United States 2020-2024. Competition, savings and sustainability. October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.iqvia.com/-/media/iqvia/spandfs/institute-reports/iqvia-institute-biosimilars-in-the-united-states.pdf
18. Fein AJ. Drug Channels Institute®; The Booming biosimilar market of 2020. 6 October 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.drugchannels.net/2020/10/the-booming-biosimilar-market-of-2020.html
19. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3.
20. GaBI Online – Generics and Biosimilars Initiative. Celltrion launches infliximab biosimilar Remsima SC in Europe. 20 March 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Oct 28]. Available from: www.gabionline.net/Biosimilars/News/Celltrion-launches-infliximab-biosimilar-Remsima-SC-in-Europe
21. Chen BK, Yang YT, Bennett CL. Why biologics and biosimilars remain so expensive: despite two wins for biosimilars, the Supreme Court’s recent rulings do not solve fundamental barriers to competition. Drugs. 2018;78(17):1777-81.
22. U.S. Food and Drug Administration. Speech by Scott Gottlieb: Capturing the benefits of competition for patients.7 March 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/news-events/speeches-fda-officials/capturing-benefits-competition-patients-03072018
23. U.S. Food and Drug Administration. Biosimilars action plan: balancing innovation and competition. July 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/114574/download
24. U.S. Food and Drug Administration. Joint statement of the Food & Drug Administration and the Federal Trade Commission regarding a collaboration to advance competition in the biologic marketplace. 3 February 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/134864/download
25. U.S. Food and Drug Administration. Draft guidance for industry. Biosimilars and interchangeable biosimilars: licensure for fewer than all conditions of use for which the reference product has been licensed. February 2020 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.fda.gov/media/134932/download
26. Centers for Medicare & Medicaid Services. MLN Matters® Number SE1509. 2014 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/Downloads/SE1509.pdf
27. Centers for Medicare & Medicaid Services. Part B Biosimilar biological product payment and required modifiers. Last modified 2 February 2018 [home-page on the Internet]. [cited 2020 Oct 28]. Available from: Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice/Part-B-Biosimilar-Biological-Product-Payment
28. Centers for Medicare & Medicaid Services. MLN Matters® Number MM11099 Revised. Update of the Hospital Outpatient Prospective Payment System (OPPS). Jan 2019 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM11099.pdf
29. Congress.Gov. 115th Congress. H.R.1892 ‒ Bipartisan Budget Act of 2018 [homepage on the Internet]. [cited 2020 Oct 28]. Available from: https://www.congress.gov/bill/115th-congress/house-bill/1892
30. Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463-78.
31. McKinnon RA, Cook M, Liauw W, Marabani M, Marschner IC, Packer NH, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32(1):27-52.
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-white-paper-us-biosimilars-market-on-pace-with-europe.html