|
Introduction: Launched in 2015, Zarxio™ (filgrastim-sndz) was the first biosimilar to gain US Food and Drug Administration (FDA) approval. In contrast, the first biosimilar was launched in Europe in 2006. |
Submitted: 14 June 2017; Revised: 7 November 2017; Accepted: 9 November 2017; Published online first: 22 November 2017
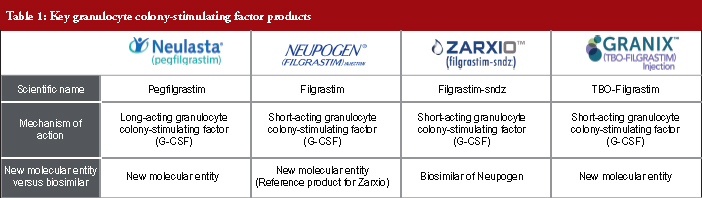
A biosimilar product is a biological product that is approved following demonstration that it is highly similar to an FDA (US Food and Drug Administration) approved biological product, known as a reference product, and has no clinically meaningful differences in terms of safety and effectiveness, from the reference product. The first biosimilar launched on the European market in 2006 and since then the US market has been anticipating the arrival of biosimilars and the potential cost savings they will bring to the healthcare system. Zarxio™ (filgrastim-sndz) was the first biosimilar to gain FDA approval in the US under the 351(k) regulatory pathway and was launched in September 2015. Zarxio™ is a short-acting granulocyte colony-stimulating factor product (G-CSF) which competes with Neupogen (its reference product), Granix and Neulasta, for a share of the neutropenia market, see Table 1. With over one year of data available, the biosimilar performance in the US short-acting (SA) filgrastim market was examined to identify key sales and marketing trends and their drivers. The analysis of sales performance, market share, pricing trends, branding strategies and coverage has yielded several key insights which may serve as a guide for biosimilar manufacturers looking to enter the US market or for reference product manufacturers who are trying to defend their market share against biosimilar entrants.
Using the first US biosimilar launch, Zarxio, as a testcase, a review and analysis of US sales performance, market share, pricing trends, branding strategies and coverage detail available through public and proprietary sources was conducted. The sources included (but were not limited to) pharmaceutical and promotional audits (evaluation of the expenditures companies spend on promoting their product), Centers for Medicare and Medicaid Services (CMS) data, market intelligence databases, news media, key academic literature and company annual/quarterly reports.
1. Market share and adoption
Initial uptake of biosimilars in the US market has been slower than anticipated, as healthcare providers are still getting comfortable with their use. However, as the first short-acting filgrastim biosimilar in the market, Zarxio’s adoption is tracking better in the US than it did in the EU at launch. This is possibly due to a combination of factors that include the improved familiarity amongst payers/physicians with biosimilars and the real-world outcomes data from their use in Europe.
Key insight: Biosimilar manufacturers are likely to see limited initial acceptance and uptake of their competing products in the near term.
2. Pricing and discounting
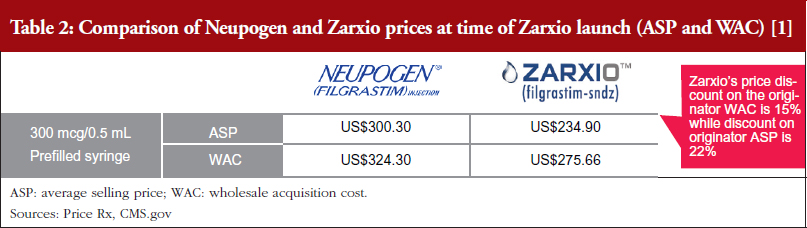
One biosimilar on the market – First-to-market biosimilar entrants will likely not need to employ aggressive discounting tactics to gain market share. Zarxio’s wholesale acquisition cost (WAC) was 15% lower than Neupogen, while Zarxio’s average selling price (ASP) was 22% lower as of the first quarter (Q1) of 2017 (note that WAC is typically referred to as the list price in the US, while ASP is the price an average purchaser would pay, typically less than the list price) [1, 2]. However, in general, US physicians lack a strong financial incentive to prescribe biosimilars in a buy and bill model (a model which involves the physician purchasing the product and getting reimbursed by an insurer), especially with only one biosimilar player on the market, as they do not stand to profit from any price differentials in the long run. In addition, reference product manufacturers are unlikely to respond with heavy price discounts via rebates unless a significant amount of the company’s business is at risk.
Multiple biosimilars on the market – Biosimilar manufacturers should expect more aggressive price discounts in markets with multiple biosimilar entrants for the same reference product, where physicians are incentivized to prescribe the lowest ASP biosimilar product. Additionally, if the pricing differential is significant, there is an incentive for early adopter physicians who are comfortable with biosimilars to switch to a new biosimilar with a lower ASP sooner rather than later, before the new lower ASP drags down the blended ASP rate.
Key insight: Given the infancy of the US biosimilar market, it is anticipated that biosimilar companies will modestly discount (20–30%) their products versus the reference product until there are multiple biosimilar competitors on the market.
3. Branding and promotion
Early biosimilar market entrants will need to invest in their brand to gain buy-in from key stakeholder groups – physicians, patients and payers through education and support via the appropriate channels. Sandoz spent a significant amount of money promoting Zarxio via Journal Advertising and Electronic Promotions in 2016 (46% of their money spent on promoting the product, categorized as ‘promotional spend’) with the rest spent on detailing (sales representatives promoting to healthcare providers) [3].
Key insight: The US biosimilar market is evolving to resemble more of a ‘branded’ market, which typically requires product promotion and advertisement in order to obtain buy-in from key stakeholders, rather than a ‘generics’ market, which typically requires little to no promotional activity.
4. Managed care and access
Payers have been slow to mandate Zarxio’s use versus the reference product; however, as Zarxio has increased its rebates (9.9% in Q2 2016, versus 15% in Q1 2017), Sandoz has been able to secure more favoured product coverage and/or exclusivity on product formulary lists [1, 2]. In Q2 of 2016, payers such as CVS Health Corporation and UnitedHealth Group announced their decision to favour Zarxio by dropping Neupogen coverage, starting in January 2017 in order to capture any additional savings [4, 5].
Key insight: Biosimilars will secure favoured tier coverage and/or formulary exclusivity from payers as biosimilar manufacturers increase their rebates (greater than a 20% price differential versus the reference product).
Market share and adoption
Zarxio’s adoption has been slow but steady since its launch in September 2015. The biosimilar has been gradually eroding Neupogen’s market share, as healthcare providers get more comfortable with prescribing biosimilars. A key reason behind healthcare providers getting more comfortable with biosimilars is the increased promotional activity and physician education by biosimilar manufacturers.
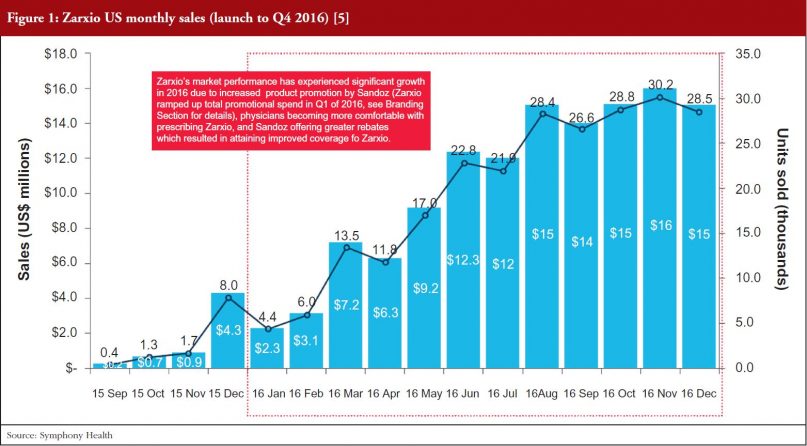
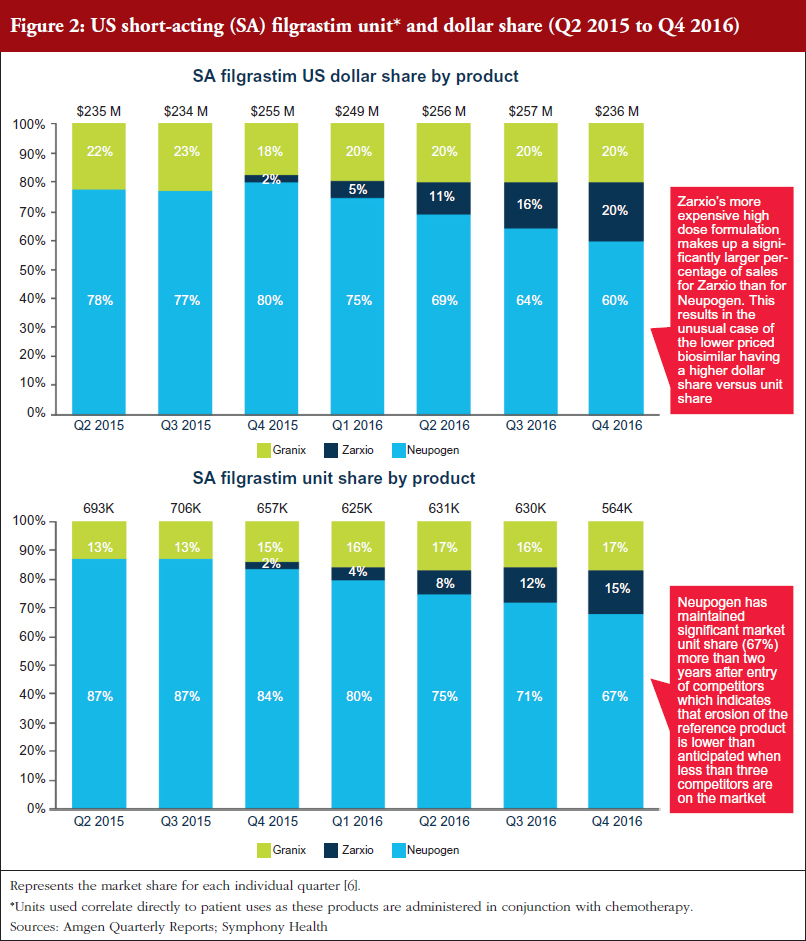
Zarxio only had 2% share (US$6 million in sales) of the SA G-CSF market (US$1.04 billion) at the end of December 2015. However, Zarxio’s share in 2016 grew to 15% by the end of the year (approximately US$129 million in 2016 sales, see Figure 1) [5]. As illustrated in Figure 2, Zarxio’s unit share doubled each quarter from launch to Q2 2016 [6]. The uptick in Zarxio sales in Q1 2016 (an increase from US$6 million in Q4 2015 to US$13 million in Q1 2016 and US$28 million in Q2 2016) is likely due to increased product promotion by Sandoz (Zarxio ramped up total promotional spend in Q1 2016, see Branding Section for details), physicians becoming more comfortable with prescribing Zarxio and Sandoz offering greater rebates which resulted in attaining improved coverage for Zarxio, see Figures 1 and 2.
Neupogen has been steadily losing its unit share to both Granix and Zarxio (87% unit share in Q2 2015 vs 67% in Q4 2016) [6]. However, Neupogen’s ability to maintain a significant market share two years after the entry of competitors indicates that erosion of the reference product is slower than some analysts had originally predicted when less than three competitors are on the market [7].
In comparison, one year after the first filgrastim biosimilar received market authorization in the EU, filgrastim biosimilars had gained only 5% volume share of the total filgrastim market. Neupogen had retained 52% share of the market, while Granocyte had 43% volume share [8]. This suggests that Zarxio adoption is tracking better in the US than in the EU at launch, possibly due to a combination of factors including: better familiarity amongst payers/physicians with biosimilars, the proven track record of biosimilar safety in Europe and Amgen’s shift in focus to Neulasta (which makes up about 80% of their G-CSF business, provides higher profit margins and lacks biosimilar competitors). However, it is also important to note that, while some countries in the EU can mandate use of biosimilars over the reference product, the US does not yet have any policies in place to do so given the relative infancy of the biosimilars market.
Pricing and discounting
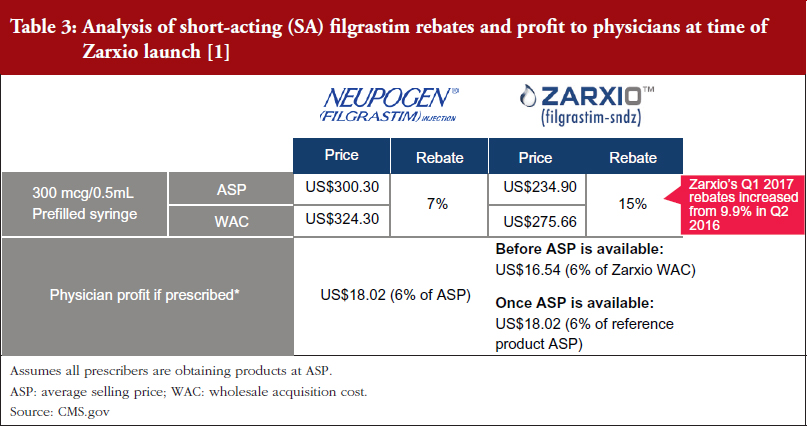
Zarxio was launched with a lower discount (15%) than the initial analyst consensus of 30%, see Tables 2 and 3 [1, 2], which suggests that first biosimilar entrants are expected to maintain minimal price differentiation with moderate discounts compared to the reference products. This discount is also less than what is currently observed outside the US, as the EU discount versus the reference product can range from 20% to 70%. As such, when there is only one biosimilar on the market it is expected that biosimilars will act as branded competitors by preserving the overall market value rather than eroding market value by acting as a typical small molecule generic would. However, larger price discounts are likely to be seen in situations where multiple competing biosimilars are on the market.
Pricing has a significant impact on prescribing patterns in the US. Prescribers have little to no incentive to drive biosimilar sales over the reference product in a buy and bill market (a market where US physicians are required to purchase a product to use in a procedure and are reimbursed for both the product and procedure by payers), as they get reimbursed the same amount for both the biosimilar and the reference product once the biosimilar ASP is established.
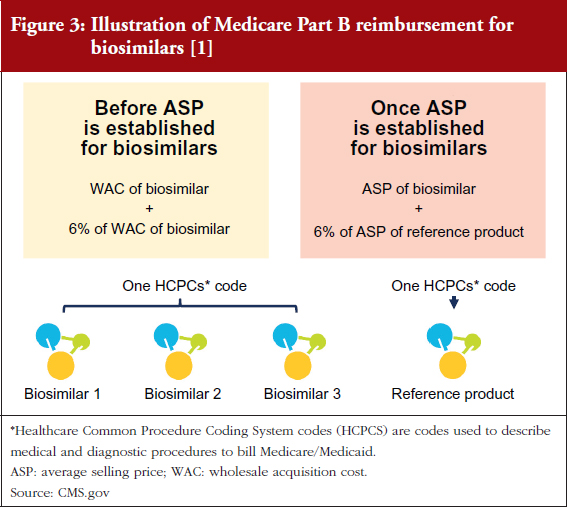
This is because Medicare Part B reimbursement from CMS for biosimilars is 106% of the biosimilar’s WAC immediately after launch, see Figure 3 [1, 2]. During this period, healthcare providers do not have an incentive to switch from Neupogen to Zarxio, where they would make a slightly lower profit. Approximately six months after launch, when the biosimilar ASP is available, reimbursement is 100% of the biosimilar’s ASP plus 6% of the ASP of the reference product. This formula is meant to prevent the financial incentive to prescribe the product with the higher ASP, which is typically the reference product (mandated by the Affordable Care Act).
As a result, there is no incentive for physicians to prescribe one product over the other in a buy and bill model as they do not stand to profit from any price differentials, see Table 4.
In contrast, in a market with multiple biosimilars, CMS – the US Government payer responsible for overseeing many federal healthcare programmes – will assign a single J-Code (a code used to identify injectable drugs for purposes of billing) to all biosimilar versions of a reference product biological, resulting in the biosimilar ASP being a blended rate of all the prices of the different biosimilar versions. This should provide an incentive to prescribe the lowest cost product and also means that a single manufacturer could lower the ASP with significant discounts/rebates. For early adopter physicians who are comfortable with biosimilars, this also presents an incentive to switch to a new biosimilar with a lower ASP sooner rather than later, before the new lower ASP drags down the blended ASP rate if the pricing differential is significant.
Furthermore, with a single biosimilar on the market, the innovator is unlikely to respond with heavy price discounts via rebates. Amgen has yet to increase rebates for Neupogen with CMS, perhaps due to their shift in focus to the Neulasta business. In addition, Sandoz only increased rebates from 9.9% in Q2 2016 to 15% in Q1 2017. However, reference product manufacturers may react more aggressively to competitors by offering large rebates if a significant amount of their business faces risk from biosimilars.
The analysis also suggests that the market entry of biosimilars could curb price increases of reference products by jeopardizing their competitive positioning on the market. In response to Zarxio’s market entry, Amgen halted price increases and has kept Neupogen’s price flat since July 2015 [6]. In contrast, Amgen has consistently increased Neulasta’s price by 5% every six months (Neulasta is yet to face competition from biosimilar products in the US). However, the market entry of several additional competitors is expected to result in a price war with steeper price discounts across the brands.
Branding and promotion
Competition between reference products and biosimilars looks more like competition between branded drugs, rather than between branded drugs and their generics. Sandoz is using a branded approach to promote their products with emphasis on physician education via journals/digital content and detailing to drive adoption.
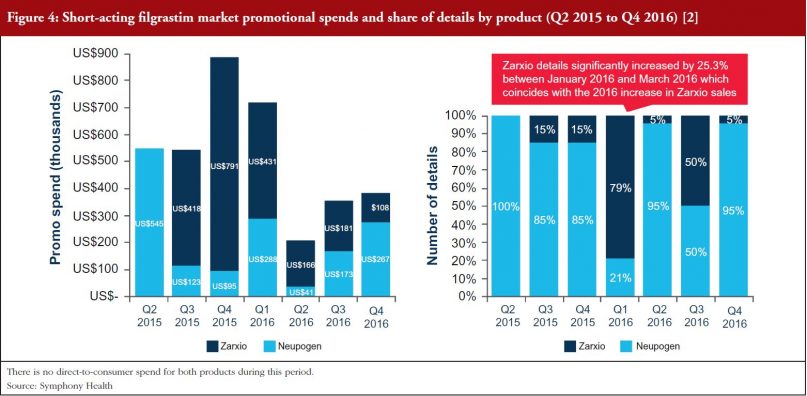
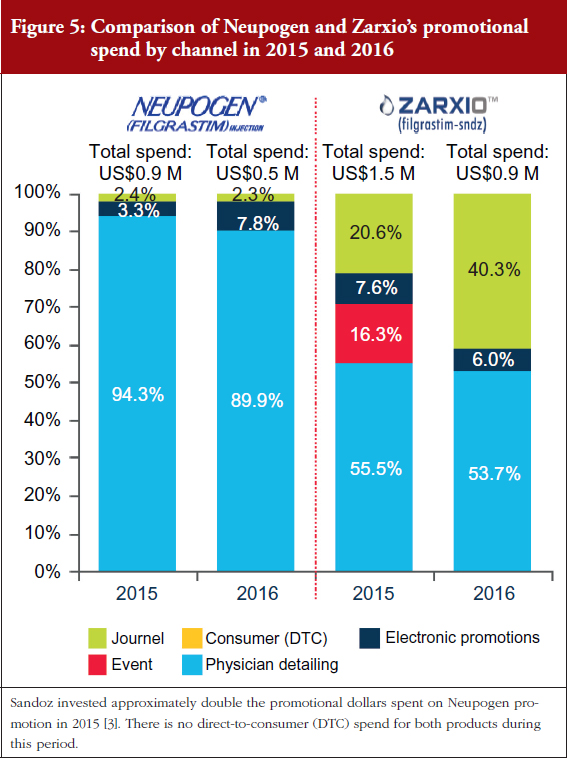
As can be seen in Figures 4 and 5, Sandoz’s total spend on promoting Zarxio was nearly twice that of Amgen on Neupogen in 2015 and 2016 respectively, which helped to drive Zarxio’s adoption. Zarxio’s promotional spend was US$1.5 million in 2015 and US$0.9 million in 2016, while Amgen is estimated to have spent less than US$1 million each year to promote Neupogen [3]. Zarxio’s total promotional spend in 2016 was focused on Detailing (54%), Journal Promotion (40%) and Digital/Electronic Promotions (6%) [3].
Zarxio product detailing by sales representatives increased significantly (by 253%) between January and March 2016, which coincides with the increase in 2016 Zarxio sales and unit share. Overall, Zarxio was detailed approximately four times more than Neupogen in Q1 2016 [3]. The emphasis on the physician here, demonstrates the importance of physician buy-in for biosimilars in the US as they have the freedom to prescribe to whomever they believe would be fit to receive the biosimilar versus the reference product. While this extra detailing could have helped with the uptick in sales, the potential changes in coverage could have contributed as well, as patient insurance could potentially dictate the prescription of Zarxio versus Neupogen.
Physician education also appears to have played a prominent role in the increase in Zarxio adoption in 2016. In Q1 2016, Sandoz spent a significant amount of promotional dollars (35% of their promotional spend) on Journal Advertising and Electronic Promotions, while the rest was spent on Detailing, see Figure 5 [3]. In contrast, Amgen spent only 10% of its entire 2016 promotional spend for Neupogen on Journal and Electronic Promotion [2]. In Q4 2015, Sandoz also promoted Zarxio significantly via Events (pharmaceutical company sponsored meetings and events), using 31% of the quarter’s promotional spend there, while spending the rest on Journal and Electronic Promotion (50%) and Detailing (19%) [3].
This is consistent with what QuantiaMD (an online physician platform and community) found in late 2015 when it surveyed US specialists and primary care physicians to gauge physician perceptions of biosimilars [9]. Only 17% of prescribing specialists (those who see patients with conditions commonly treated with biologicals) reported that they would be ‘very likely’ to prescribe biosimilars to eligible patients. The main concerns included safety/efficacy, drug substitution regulations and accurate evaluation of when to prescribe a biosimilar versus branded therapy. Specialty societies were prescribing specialists’ most trusted source of information about biosimilars (25%), followed by peers (19%) and key opinion leaders (18%). 80% of prescribing specialists said they would want to learn about biosimilars through expert-led digital content.
All of this data suggests that the acceptance and subsequent uptake of biosimilars is dependent on gaining buy-in from key stakeholder groups including physicians, patients and payers. This is especially important in the US, as opposed to the EU, as there is little real-world evidence of biosimilars given the number of products on the market. Biosimilar manufacturers should therefore plan to invest in promoting their products through channels such as journals and specialty societies in addition to detailing.
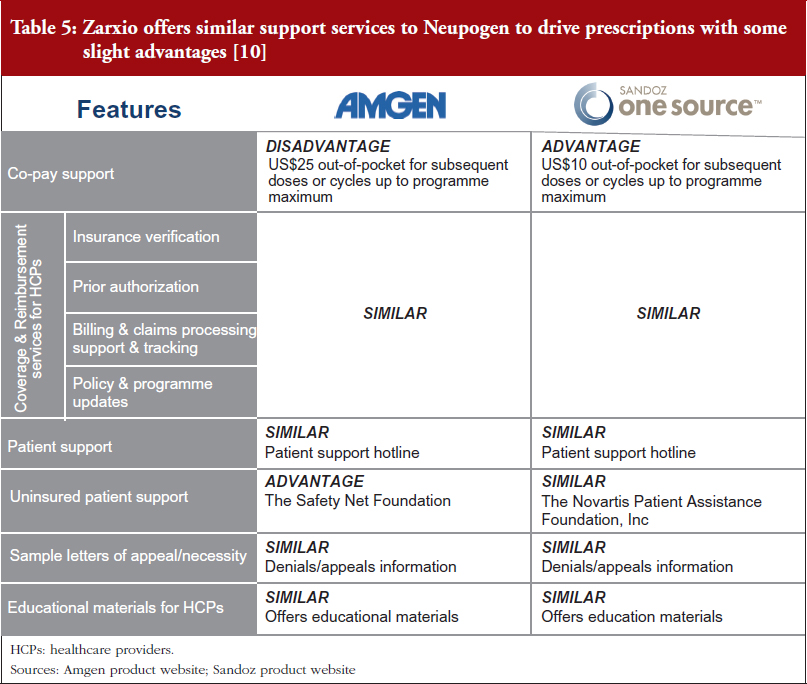
Biosimilar and reference product manufacturers are providing very similar patient support programmes. Zarxio offers similar support services to Neupogen to drive prescriptions, with slight advantages and disadvantages in some areas. In the absence of significant price discounts, biosimilar manufacturers (and reference product manufacturers) are likely to continue to invest in support services to ensure their products are differentiated in a sea of copycat versions.
For example, Zarxio’s One Source programme [9] (Sandoz’s patient support programme for Zarxio) offers similar co-pay support for patients and Coverage & Reimbursement services for healthcare providers (HCPs). However, Zarxio’s co-pay (US$10) is lower than Neupogen’s (US$25), while both have a maximum benefit of US$10,000 per patient annually. Nevertheless, Zarxio does not provide support for uninsured patients while Amgen does via the Safety Net Foundation, which helps qualifying uninsured patients to access Amgen medicines at no cost, see Table 5 [10].
Managed care and access
Payers are expected to be one of the major decision-makers affecting biosimilar adoption. Neupogen has generally had better coverage among the top payers which partially explains Zarxio’s slow adoption since launch. However, this trend will potentially reverse in the near future as payers increasingly favour biosimilars over their reference products to save on cost.
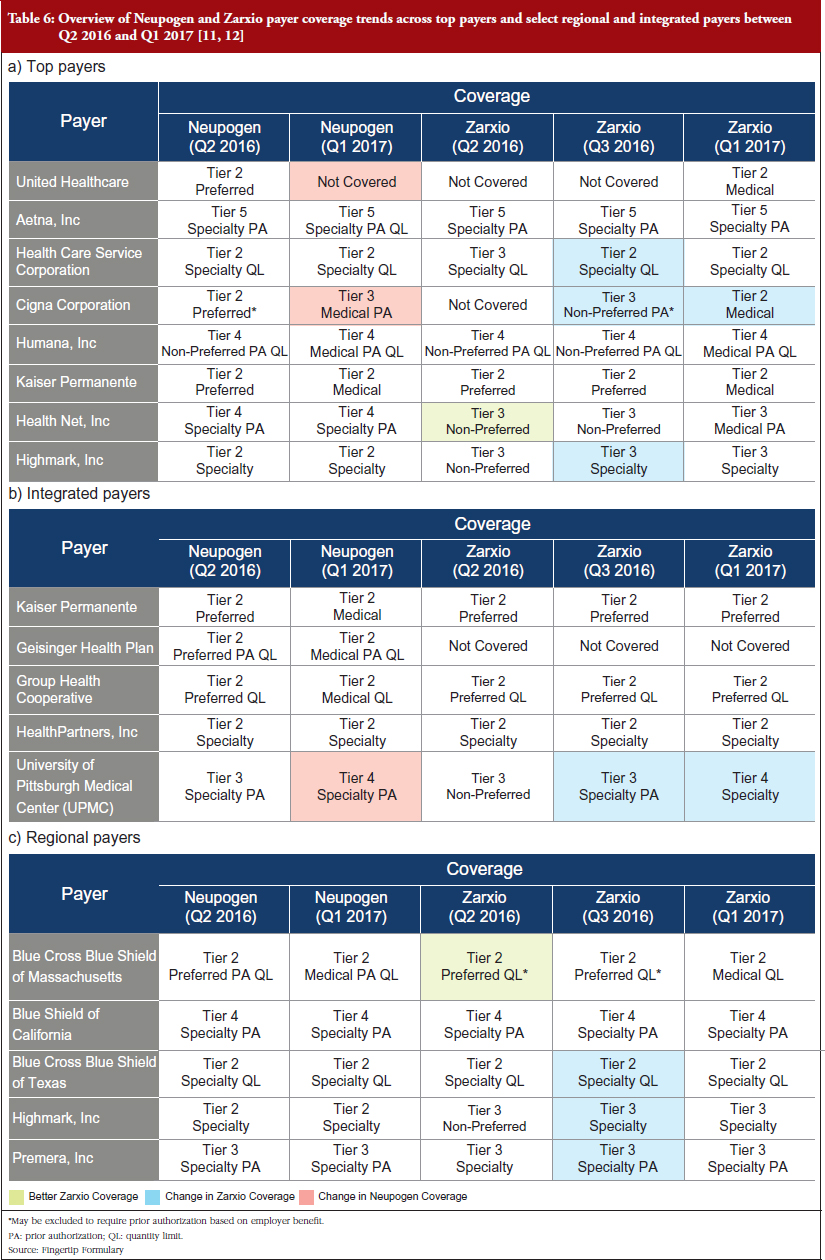
The trend observed among the payers reviewed in 2016, is that Neupogen generally has better coverage, meaning that it is covered by more plans or has a higher tier coverage equating to it being offered at a lower co-payment or co-insurance cost to patients, see Table 6 [10, 11]. The implications of Neupogen’s better coverage are that physicians will have fewer barriers to prescribe the product, whether that be prior authorizations (PAs) required before filling the prescription, quantity limits (QLs) on the prescription, or being a non-preferred brand. This could partially account for Zarxio’s slow adoption at launch. Subsequent payer reviews in 2017 revealed that Zarxio’s coverage has improved since launch as payers favour biosmilars over reference products to save on cost, which partially explains the uptick in Zarxio’s market share throughout 2016. Highlighted in Table 6 are some cases where Zarxio’s coverage improved between Q2 2016 and Q1 2017 [6]. National payers cover patients across the entire US, while Regional payers focus on specific regions or states. Integrated delivery networks (IDNs) or integrated payers, are payers who also act as providers and treat patients within their facilities or network of physicians.
Examples of changes across all payers and IDNs:
In comparison, Neupogen’s coverage was lowered in some cases:
Moving forward, Neupogen’s market share is likely to continue decreasing as payers/pharmacy benefit managers (PBMs) increase Zarxio coverage and restrict Neupogen coverage. Payers are beginning to favour biosimilar coverage, as evidenced by recent decisions by CVS Health Corporation and UnitedHealth to drop Neupogen coverage in favour of the biosimilar, effective as of January 2015. In August 2016, CVS Health corporation announced that it will drop coverage of Neupogen in favour of Zarxio, effective as of January 1, 2017 [4]. UnitedHealth soon followed CVS Health corporation’s lead by announcing their move to exclude Neupogen from their formulary in favour of biosimilars effective, as of 1 January 2017 [5].
Zarxio has paved the way for future biosimilars that are looking to enter the US market. Sandoz, Zarxio’s manufacturer, has refined its commercialization approach in the US by leveraging what it has learned from launching in the EU market first. The company’s primary strategies were to launch the product with a modest discounting strategy, target physicians in order to increase awareness, and to focus on improving insurance coverage in order to gain market share in the US. As reference product manufacturers become savvier and the market for biosimilars becomes saturated with multiple entrants in the same product class, the strategies of biosimilars manufacturers will need to evolve in order to gain adoption over time.
Disclosure of financial and competing interests: Mr Michael Sarshad worked with Amgen on Neupogen and Neulasta over three years ago, and none of the information disclosed in the paper is from that work. Everything is either public or has been accessible to inVentiv Health via the sources stated. He is currently not invested in or advising any of the companies described in the manuscript nor has he been for the past 24 months. Mr Sarshad is currently employed by Syneos Health.
This paper is funded by Syneos Health.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Evaluate Pharma [homepage on the Internet] [cited 2017 Nov 7]. Available from: http://evaluategroup.com/public/EvaluatePharma-Overview.aspx
2. Centers for Medicare and Medicaid Services [homepage on the Internet] [cited 2017 Nov 7]. Available from: www.cms.gov
3. inVentiv Health. Research Insights, Answer Suite™ Promotional Audits, Kantar Media. Proprietary, accessed in 2016 and 2017.
4. CVS drops coverage of 2 branded biotech drugs in favor of copies. The Wall Street Journal. 2016 Aug. 2.
5. Fierce Pharma. Updated: UnitedHealth adds to formulary pain for Sanofi, Amgen and Novartis. 2016 Sep 22.
6. Symphony Health. Pharmaceutical Audit Suite (PHASTTM) [homepage on the Internet]. [cited 2017 Nov 7]. Available from: https://symphonyhealth.com/product/pharmaceutical-audit-suite-phast/
7. Nasdaq. Amgen quarterly earnings calls and annual reports 2015, 2016 and 2017 [homepage on the Internet]. [cited 2017 Nov 7]. Available from: http://www.nasdaq.com/symbol/amgn/call-transcripts
8. Walsh K. Biosimilars’ utilization and the role payers do play in driving uptake in Europe: an industry perspective. Biosimilar Medicines 11th EGA International Symposium; April 2013.
9. New quantia report reveals physician attitudes toward biosimilars. Business Wire. 2015 Aug 18 [homepage on the Internet]. [cited 2017 Nov 7]. Available from: https://www.businesswire.com/news/home/20150818005839/en/New-Quantia-Report-Reveals-Physician-Attitudes-Biosimilars
10. Amgen First Step [homepage on the Internet]. [cited 2017 Nov 7]. Available from: https://www.amgenfirststep.com/
11. Sandoz One Source [homepage on the Internet]. [cited 2017 Nov 7]. Available from: https://www.sandozonesource.com/
12. Easy Counter. Analysis of Fingertip Formulary 2016 and 2017 [homepage on the Internet]. [cited 2017 Nov 7]. Available from: https://www.easycounter.com/report/fingertipformulary.com
|
Author: Michael Sarshad, BSc, MBA, Suite 270, 1315 Lincoln Boulevard, Santa Monica, CA 90401, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/major-lessons-learned-from-zarxios-us-launch-the-start-of-a-biosimilar-revolution.html
Author byline as per print journal: Michael Sarshad, BSc, MBA; Rosanne Campbell, BComm, PGdip, MSc; Peter J Pitts, BA; Jacqueline anderpuye- Orgle, MSc, PhD
|
Abstract: |
Submitted: 7 November 2018; Revised: 21 December 2018; Accepted: 27 December 2018; Published online first: 31 December 2018
The availability of biological drugs is an important advancement in treating complex diseases. However, the inherent complexities in developing and manufacturing biologicals have inevitably meant that their use is costly, which in turn places a high burden on the healthcare system. In the US, the per cent of pharmaceutical spending attributable to biologicals has increased from 13% in 2006 to 27% in 2016 [1]. In 2007, the average daily cost of a biological was 22 times that of a small molecule drug [2]. Biosimilars present the possibility of lowering these costs while offering additional treatment options for patients.
The pace of biosimilar approvals in the US has been accelerating, with the US Food and Drug Administration (FDA) approving five products in 2017, two of which were for the blockbuster originator biologicals Humira® and Remicade® [3]. Currently, there are 12 biosimilar products approved in the US and 22 distinct biosimilar products approved in the EU, see note. In fact, in the first eight and a half years after the regulatory pathway was established, FDA approved more biosimilars to more reference products (12 unique biosimilars to eight reference products) than the EU (five unique biosimilars to two reference products). As the number of biosimilar approvals continues to grow, it will become even more important to collect pharmacovigilance data that are accurate and attributable to the specific product. The World Health Organization (WHO) proposed an approach of assigning all biological products a 4-6 character alfa-numeric biological qualifier (BQ) code. The BQ would consist of four random consonants and an optional 2-digit checksum (calculated from the letters and their order in the BQ code). In October 2017, WHO announced that it would not proceed with BQ code system.
Different countries and regions have taken various approaches to ensuring accurate product traceability; the EU passed legislation requiring the recording of brand name, batch number and lot number, while the US and Japan pursued the use of distinguishable non-proprietary names. The latter has been criticized by some who believe it may negatively impact the uptake of biosimilars.
How biosimilars are named has become complex and inconsistent globally. FDA’s final guidance on the naming of biologicals (reference products and their biosimilars), with the addition of a distinct four-letter suffix to the non-proprietary name, was a major step forward in supporting more accurate pharmacovigilance. Those in favour of the suffix contend that the distinct nomenclature allows for better product-specific tracking of safety and real-world effectiveness. This in turn promotes prescriber and patient comfort with biosimilars, thus serving an important role in the overall adoption of biosimilars and enhancing a robust marketplace.
Those who argue against differential naming of biosimilars, including the US Federal Trade Commission (FTC), state that it causes confusion for prescribers, as distinguishable non-proprietary names will make physicians believe there is a difference between the originator and the biosimilar. This, they suggest, would create an artificial barrier by discouraging prescribing, with the consequence of slowing uptake and potentially of inhibiting cost savings.
The recent FDA guidance on biosimilar naming and an FTC statement in a recent Department of Health and Human Services (HHS) document, American Patients First: The Trump Administration blueprint to lower drug prices and reduce out-of-pocket cost, prompted a review of published evidence to examine the impact of a distinct four-letter suffix as part of a biological product and a global naming standard for biosimilars [4, 5].
An ad-hoc literature review was conducted between January and October 2018 to gather evidence related to the various stakeholders impacted by introducing a distinct suffix as part of biological product non-proprietary names. These stakeholders included regulatory agencies, physicians, payers, patients, government/affiliated agencies and advocacy groups. PubMed was searched for evidence related to differential nomenclature published in the last five years (2013–2018). Additional sources of evidence included ClinicalTrials.gov, pharmacovigilance databases, naming guidelines and surveys. Literature was reviewed to identify the link between differential nomenclature and better safety reporting, real-world effectiveness and prescriber comfort with biosimilars.
Better safety reporting and real-world effectiveness
In Europe, guidelines on good pharmacovigilance practices (GVP) have been developed for biological medicinal products and require that prescriptions for originator biologicals and biosimilars specify brand names [6]. This helps to reduce ambiguous adverse event (AE) reporting, i.e. an inability to link an AE to a specific product [originator or biosimilar]. A review of the European Eudravigilance database found that during the period March 2017 to February 2018, around 25% of the AEs reported for infliximab were ambiguous even after implementing GVP, with no link to originator or biosimilar, see Figure 1. While these guidelines are beneficial, adopting the current FDA system of a distinct four-letter suffix for biologicals can further reduce ambiguity.
However, the distinction between infliximab biosimilar products using differential scientific nomenclature has been sufficient to facilitate post-marketing studies, particularly in inflammatory bowel disease (IBD) indications, as limited data were available at the time of regulatory approval. The Clinicaltrials.gov site was reviewed to establish the number of ongoing, global real-world and observational studies conducted post-marketing for infliximab biosimilars, see Figure 2. The Clinicaltrials.gov database was selected as it includes research studies conducted in 205 countries. In addition, evidence from two published sources, Diena et al. (2017) and Ben-Horin et al. (2016), which reviewed Inflectra® and Remsima® post-marketing studies, see Figure 2 [7, 8], was incorporated.
Furthermore, a review of the Australian Therapeutic Goods Administration (TGA) Database of Adverse Event Notifications (DAEN) was conducted to determine if there was a difference in AE reporting between the biosimilars infliximab, filgrastim and epoetin. In Australia, infliximab and filgrastim biosimilars do not have distinct Australian Biological Names (ABN). However, the TGA has used a distinct ABN for epoetin lambda, a biosimilar of epoetin alpha, due to apparently different glycosylation patterns [9]. The data highlight that infliximab (25%) and filgrastim (36%) were associated with higher rates of ambiguous reporting compared to epoetin (3%), see Figure 3.
Prescriber comfort with biosimilars
It is relevant to understand physician perceptions of the safety of biosimilars. FDA can only approve a biosimilar if it is highly similar to, and has no clinically meaningful differences from, the originator biological. However, in a survey of US physicians across a variety of specialties, over 50% of respondents expressed safety concerns with FDA-approved biosimilars, see Figure 4 [10].
The Biosimilars Forum conducted a survey from November 2015 to January 2016 of US physicians who frequently prescribe biologicals (Reported in Cohen et al. [10]). The aim of the study was to investigate physician awareness, knowledge and perception of biosimilars. Figure 5 indicates that the vast majority of respondents (75%) considers it necessary to seek additional information on biosimilars prior to prescribing them.
In 2015, the Alliance for Safe Biologic Medicines (ASBM) conducted a survey of US Healthcare Professionals (HCPs) to establish their perspective on biosimilar naming. In this survey, 66% of respondents supported distinct non-proprietary naming for originator biologicals and biosimilars, see Figure 6 [11]. Similarly, a 2017 ASBM survey of Canadian healthcare providers found that 68% of respondents wanted distinct non-proprietary naming for all biologicals [12].
There are cases in which the biosimilar is approved in an indication based on the extrapolation of other clinical data. A 2016 survey of US physicians (Cohen et al. [10]) established that roughly 60%, see Figure 7, of respondents expressed concerns with using biosimilars in indications where approval was extrapolated from other clinical data [10]. By promoting effective pharmacovigilance, differential nomenclature may help to alleviate physician concerns with the concept of extrapolation.
Due to the inherent complexities associated with developing and manufacturing biosimilar products, differentiating them is essential to both product uptake and product-specific tracking. As highlighted in the introduction, those who argue against distinct biological naming maintain that it does not support biosimilar competition and creates an environment unconducive to a biosimilar manufacturer’s commercial success. However, based on the evidence presented, distinct nomenclature would seem to be essential for accurate pharmacovigilance and to increasing physician comfort with the use of biosimilars, thereby promoting biosimilar uptake and creating a viable competitive market.
Biosimilar uptake
Having identified the need for a global naming standard, different countries have taken varying approaches to implementation. Some may argue that the system used by FDA may hinder biosimilar uptake, however, market data have proven otherwise. While it is not currently possible to make any causal inferences in this paper, anecdotal evidence suggests that distinct non-proprietary names have not deterred uptake. One example of note is the launch of the biosimilar Zarxio®. Zarxio®, a biosimilar of filgrastim, was approved in Europe in 2009 and in the US in 2015. The FDA-designated suffix in the US is filgrastim-sndz, whereas in the EU, all biosimilars are identified by a new brand name with no suffix to the scientific name. A comparison of market share in the first two full calendar years after approval reveals that Zarxio® had slightly better uptake in the US versus in Europe, see Figure 8.
This suggests that the US differential naming approach of including a suffix does not hinder the possibility of a favourable biosimilar marketplace and the ability for a biosimilar sponsor to compete successfully.
As demonstrated in Figure 8, Zarxio® uptake more than doubled in the second full calendar year post-launch.
The recent US HHS blueprint (To Lower Drug Prices and Reduce Out-of-Pocked Costs) stated that physician and patient confidence in biosimilars is critical to increased acceptance of biosimilars [5]. The evidence suggests that additional information and data about biosimilars could help physicians become more comfortable with using them. In an ASBM (Alliance for Safe Biologic Medicines) survey of Canadian physicians, only 54% of respondents stated that should two biological medicines have the same INN, the implication is that they are identical [12]. In a survey of US physicians, 76% of respondents indicated that they had the greatest interest in learning more about the totality of evidence related to safety, efficacy and potency of biosimilars [10]. FDA’s current naming policy (a distinct four-letter suffix as part of a biological product) facilitates better pharmacovigilance and product traceability, which can promote physician comfort with prescribing biosimilars. Assigning distinct non-proprietary names for biosimilars also supports the development of real-world pharmacovigilance data for biosimilars and allows for that information to be disseminated to physicians who often request data to make an informed decision on product therapy choice. Increased comfort in prescribing biosimilars can contribute to a more robust and competitive marketplace if it impacts adoption positively.
Product-specific tracking and physician comfort
A lack of unique names across biological products complicates the reporting and tracking of AEs; allowing for better pharmacovigilance and safety tracking would help to alleviate some of these concerns. Unique non-proprietary names of biosimilars can facilitate access to information on individual products, which will lead to better physician understanding of biosimilars and ultimately comfort in prescribing them.
Biosimilar label extrapolation is an area of concern for some physicians. The data suggest that differential nomenclature may promote product-specific tracking in post-marketing studies for extrapolated indications, as described in Figure 2. Prescribers may be more comfortable with label extrapolation if they have confidence in the traceability of different biosimilar products and their specific studies.
The evidence suggests that differential nomenclature will facilitate traceability, helping to demonstrate the real-world safety and efficacy of biosimilar products. This supports the development of high quality products and continuous investment in manufacturing, as manufacturers can be held accountable for the products distributed. Establishing a reputation for high quality manufacturing eases entry into the biosimilar market, encourages adoption, and promotes marketplace competition. Developing high quality, traceable biosimilar products will contribute to formulary inclusion, enabling payers to better leverage competitive market forces to control the cost of care.
In summary, it is believed that findings outside of the US support the notion that a differential naming policy facilitates pharmacovigilance and better product traceability and will also help to minimize inadvertent substitution for drugs distributed via retail pharmacies. This supports manufacturer accountability and the development of high quality products in addition to more attributable measurement of efficacy and safety outcomes for biosimilars.
The number of available biosimilars will continue to grow, therefore, it is essential to have a system that allows these products to be differentiated and traced. The evidence collected in this review demonstrates that a distinct four-letter suffix as part of a biological product may reduce the ambiguity in AE reporting, facilitate post-marketing studies in indications with limited data at the time of regulatory approval and alleviate physician concerns related to biosimilar safety and extrapolation. Distinguishable suffixes for biologicals will allow better product tracking and awareness around product effectiveness/outcomes, which ultimately will strengthen physician and patient comfort with biosimilars. Thus, it will enable a competitive and sustainable biosimilar marketplace while promoting biosimilar uptake.
Competing interests: Michael Sarshad, Rosanne Campbell and Peter J Pitts have received funding for this research from Amgen.
Jacqueline Vanderpuye-Orgle was employed by Amgen and held Amgen stock during this research. She is currently employed by Parexel.
Provenance and peer review: Not commissioned; externally peer reviewed.
Michael Sarshad, BSc, MBA
Senior Engagement Manager
Commercial Strategy & Planning, Consulting Syneos Health
Rosanne Campbell, BComm, PGdip, MSc
Syneos Health
10 Bloomsbury Way, London WC1A 2SL, UK
Peter J Pitts, BA
Former Associate Commissioner, United States Food and Drug Administration
Visiting Professor, Université Paris Descartes Medical School President, Center for Medicine in the Public Interest 20/F, 757 Third Avenue, New York, NY 10017, USA
Jacqueline Vanderpuye-Orgle, MSc, PhD
Parexel International
Glendale Adventist Medical Center, Suite 140, 1560 E Chevy Chase Drive, Glendale, CA 91206, USA
Note: Distinct biosimilar products approved in EU include: (1) products in the EU that are considered ‘transition’ products in the US, i.e. biological products approved pursuant to new drug applications (NDAs), including section 505(b)(2) NDAs, e.g. growth hormones, insulins; and (2) products licensed in the EU under multiple brand authorizations.
References
1. Morton FS, Boller LT. Enabling competition in pharmaceutical markets. Hutchins Center Working Paper #30. May 2017.
2. Statement of Ed Weisbart, MD, Chief Medical Officer, Express Scripts in Hearing Before the Subcommittee on Health of the Committee on Energy and Commerce. May 2, 2017.
3. U.S. Food and Drug Administration. FDA-Approved Biosimilar Products [homepage on the Internet]. [cited 2018 Dec 21. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580432.htm
4. US Federal Trade Commission. Statement of the Federal Trade Commission to the Department of Health and Human Services Regarding the HHS blueprint to lower drug prices and reduce out-of-pocket costs [homepage on the Internet]. [cited 2018 Dec 21]. Available from: https://www.ftc.gov/policy/advocacy/advocacy-filings/2018/07/statement-federal-trade-commission-department-health-human
5. Federal Register. Notice by the US Health and Human Services Department. HHS blueprint to lower drug prices and reduce out-of-pocket costs. May 2018 [homepage on the Internet]. [cited 2018 Dec 21]. Available from: https://www.federalregister.gov/documents/2018/05/16/2018–10435/hhs-blueprint-to-lower-drug-prices-and-reduce-out-of-pocket-costs
6. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP). Product- or population-specific considerations II: biological medicinal products. 4 August 2016 [homepage on the Internet]. [cited 2018 Dec 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/08/WC500211728.pdf
7. Deiana S, Gabbani T, Annese V. Biosimilars in inflammatory bowel disease: A review of post-marketing experience. World J Gastroenterol. 2017;23(2):197-203.
8. Ben-Horin S, Vande Casteele N, Schreiber S, Lakatos PL. Biosimilars in inflammatory bowel disease: facts and fears of extrapolation. Clin Gastroenterol Hepatol. 2016;14(12):1685-96.
9. Power DA. Licensing and prescribing biosimilars in Australia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(3):152-4. doi:10.5639/gabij.2013.0203.030
10. Cohen H, Beydoun D, Chien D, Lessor T, McCabe D, Muenzberg M, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther, 2017;33(12):2160-72.
11. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
12. Industry Standard Research. ASBM Biosimilars. Canada prescribers and biosimilars. Biosimilars, familarity, knowledge, attitudes and beliefs. 2017.
|
Author for correspondence: Michael Sarshad, BSc, MBA, Syneos Health Consulting, Suite 270, 1315 Lincoln Boulevard, Santa Monica, CA 90401, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/the-need-for-distinct-nomenclature-for-originator-and-biosimilar-products.html
Copyright ©2025 GaBI Journal unless otherwise noted.