|
Abstract: |
Submitted: 27 June 2023; Revised: 1 August 2023; Accepted: 18 September 2023; Published online first: 2 October 2023
Antibody drug conjugates (ADCs) are antibody-based anticancer therapeutics consisting of monoclonal antibodies attached to a cytotoxic drug via a linker of some kind. These highly targeted delivery systems offer the promise of lower cytotoxic drug levels in the patient and, as a result, potentially reduced drug side-effects [1].
The first globally approved ADC was Pfizer and Wyeth’s Mylotarg (gemtuzumab ozogamicin) in 2000, however, it was not until 2011 that the second ADC approval came in the form of Seagen and Takeda’s Adcetris (brentuximab vedotin). These early ADCs came with ‘black box’ warnings making clear that although the benefits outweighed the risks, there remained issues with toxicity. Over the next decade companies explored improved conjugation chemistries to link their non-proteinaceous drug to the antibody vector of choice with 14 ADCs in total receiving US Food and Drug Administration (FDA) approval to date, with many reaching blockbuster statuses.
The question is though, are ADCs a class of drug that may be almost immune to biosimilar competition given their complexity? Undoubtedly the development challenges are far greater which may limit the number of biosimilar sponsors willing to take them on. In 2021, the Drugs Controller General of India approved Zydus Cadila’s ‘similar biological’ of Genentech/Roche’s Kadcyla (trastuzumab emtansine) marketed as Ujvira for treating both early and advanced human epidermal growth factor receptor 2 (HER2) positive breast cancer. To date, neither FDA nor the European Medicines Agency (EMA) have yet approved a biosimilar ADC under their respective regulatory pathways.
The requirements for structural characterization of novel ADCs and potential biosimilar versions are the same as for other biomolecules in that the precepts of ICH Q6B [2] should be followed to achieve a full and detailed overall picture of primary and higher order structure (HOS). However, it must be noted that the requirement for conjugation and the nature of the conjugation chemistry itself mean that investigations need to be performed to:
Careful analysis of conjugation products must be carried out to ensure that the conjugation itself has proceeded as expected. This includes determining that the antibody has not been adversely affected through, for example, oxidation, deamidation, non-specific drug binding or any other side reactions that are chemically feasible under the conjugation conditions used.
Many of the structural analyses performed will involve an analytical assessment of the primary structure, i.e. amino acid sequence, but it is important to remember that the attachment of drug groups with their own characteristics of size, charge distribution and hydrophobicity/hydrophilicity at various points could also influence secondary and tertiary structure, as well as creating the potential for aggregation.
For these reasons, it is important that the HOS is also investigated to fully understand the impact of the conjugation process on the monoclonal antibody (mAb) and the resultant ADC. It must be pointed out that alteration of the HOS between the parent mAb and the ADC is not necessarily in itself problematic, after all the ADC is not being marketed as a biosimilar to the parent mAb. Rather, the data are generated to give a better understanding of the product following conjugation. Furthermore, assessment of ADC HOS across different batches of product provides part of the structural characterization evidence for batch-to-batch consistency (or otherwise).
Alteration of the HOS of an ADC when compared to its native mAb may lead to questions regarding potential immunogenicity or could be a cause of increased aggregation, if observed. Furthermore, from a functional point of view, HOS changes may impact mAb target binding and therefore affect the ability of the mAb to deliver the drug to the site of action.
Secondary and tertiary structural techniques are used routinely in protein analysis, the most frequently used being circular dichroism (CD), fourier transform-infra-red (FT-IR), fluorescence analysis and nuclear magnetic resonance (NMR; both 1D (1 H) and 2D (1 H- 13 C) [3, 4]. These techniques are all equally applicable to ADCs since the protein structural features are preserved in ADCs, as would be expected. Each of these techniques works on a different principle and therefore will probe HOS in slightly different ways, resulting in unique outputs from each technique.
Circular dichroism
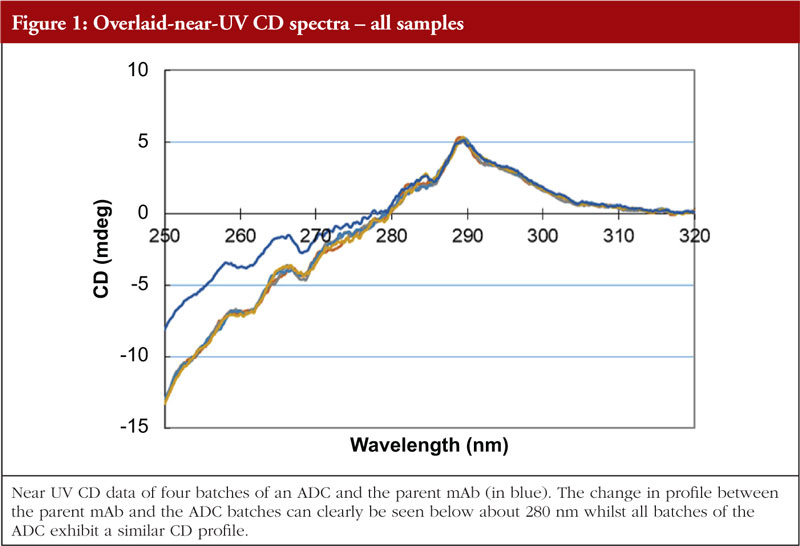
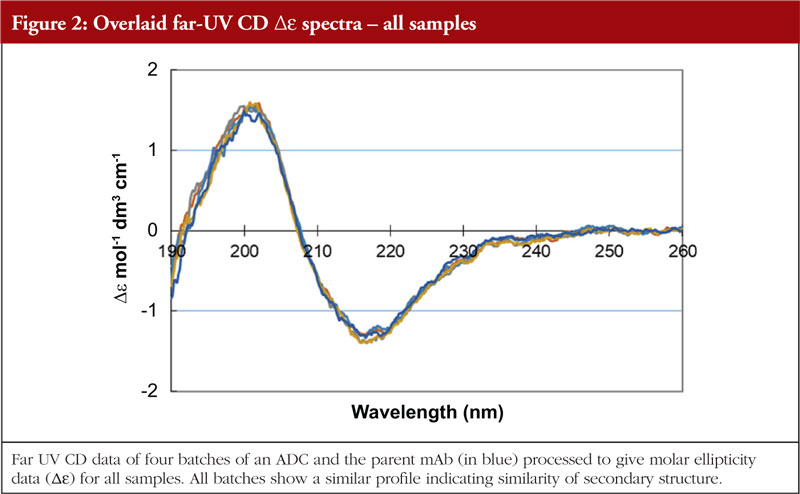
Circular dichroism measures the differences in absorbance by the sample of right and left hand circularly polarized light. This technique works well for chiral molecules and, since proteins exhibit chirality, has proven effective for their analysis. Wavelengths used are in the far and near UV region, giving information on secondary and tertiary structure, respectively. The far UV data are computer processed and, with database searching (data obtained from analysis of proteins with known secondary structures), provide a breakdown of secondary structural features. The protein component of ADCs is just as amenable to CD analysis as the parent monoclonal antibody; thus, CD data can be generated allowing an investigation into the similarity, or otherwise, of the HOS structural profiles. The data shown in Figures 1 and 2 show the stacked near and far UV data, respectively, for several ADC batches and the parent mAb [unpublished results, 5]. All data images presented in this article are from these same ADC batches and parent mAb, analysed by different HOS techniques. The difference in the spectra in the region below 280 nm in Figure 1 is consistent with the difference in absorbance between the disulphides and the pair of free/conjugated thiols as a result of the conjugation chemistry (pers. comm).
FT-IR
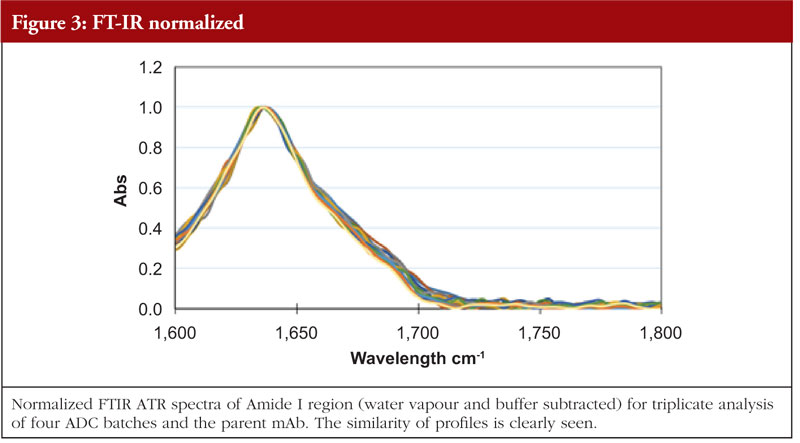
FT-IR uses infra-red light to investigate various bond flexures in the protein such as C=O, C-N, C-H, C-C and N-H. The chemical environments produced by the HOS features will have an impact on the precise wavelengths at which these flexures are seen. The absorption profile generated is subject to Fourier Transform mathematical interpretation and database interrogation to provide an output of relative abundances of secondary structural features. Figure 3 shows the FT-IR profiles of the same four ADCs and the parent mAb as shown in the previous figures [5].
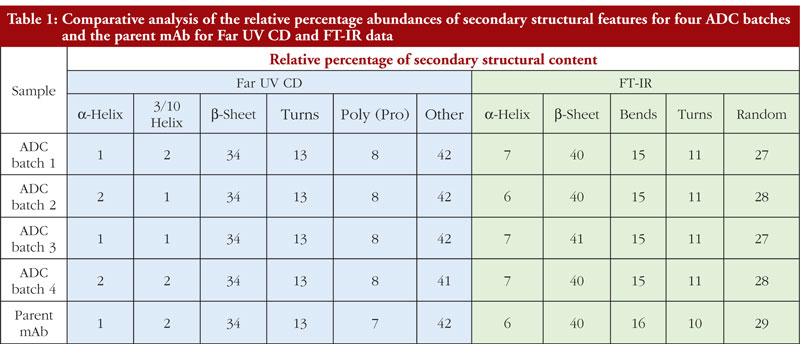
It is very important to bear in mind that secondary structure analysis by different techniques will provide data that will reflect the sensitivity of that technique for particular secondary structural features. In other words, Far UV CD and FT-IR data, when processed, are unlikely to provide the same relative ratios of secondary structural features. This can be seen in Table 1 where the Far UV CD and FT-IR data for the four ADCs and parent mAb analysed in this study are compared. The fact that different values are obtained between these techniques is not in itself a problem since what matters is that batches compare to one another within the same technique and data set. The data are not being used to give a precise structural definition for the ADCs but are aiming to demonstrate similarity of result and therefore similarity of structure between batches.
Fluorescence
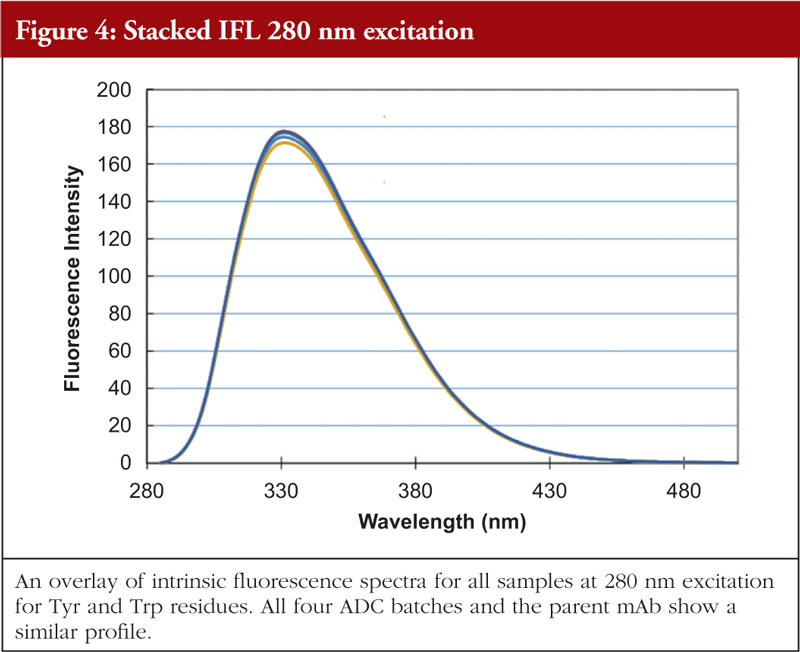
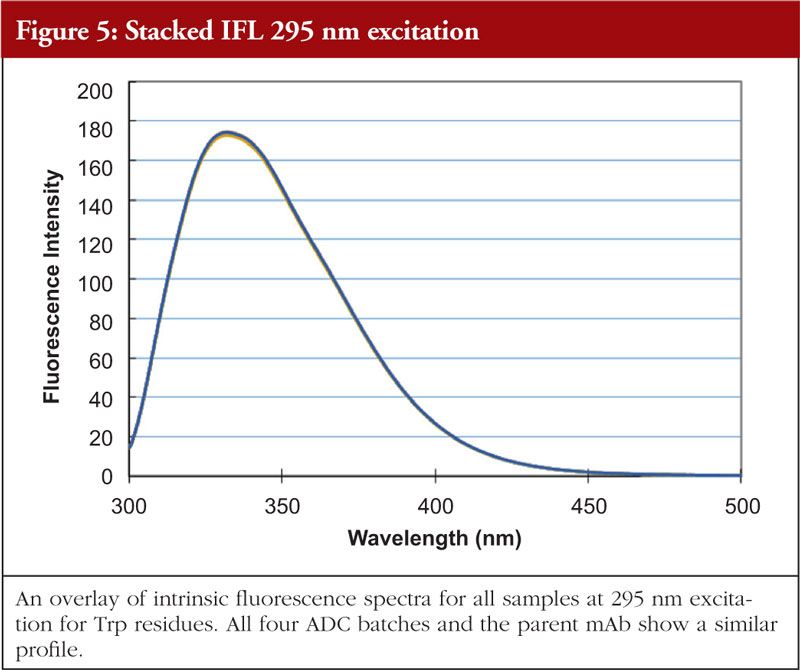
Fluorescence analysis can be both intrinsic to the molecule and extrinsic by means of fluorophore addition to the sample. In the intrinsic sense, fluorescent analysis involves measuring the fluorescence profile of tyrosine and tryptophan residues in the molecule using a spectrophotometer. The profile generated is related to the chemical environments that these amino acids find themselves in and thus the fluorescence profile gives information about the overall nature of the Tyr and Trp chemical environments. Relative amounts of secondary structural features are not determined with this technique. An example of intrinsic fluorescence is given in Figures 4 and 5 below for the same ADC batches and parent mAb as shown in the previous figures [5].
1D and 2D NMR
NMR relies on measuring the response of magnetic nuclei in the sample (most commonly 1 H and 13 C) to a radio frequency pulse applied perpendicular to a constant magnetic field. As the magnetic nuclei relax back to their ground state following the RF pulse they emit electromagnetic radiation, giving rise to the NMR signal. Again, the nature of the chemical environment that these atomic nuclei find themselves in will influence their relaxation profiles and this in turn influences the NMR profile.
A larger amount of structural information can be obtained by monitoring 2 magnetic nuclei in one experiment &ndash a so-called 2D NMR analysis. Most commonly 1 H and 13 C are measured but 1 H and 15 N measurements can also be used (although the relative abundance of 15 N is lower than 13 C). This gives rise to an intricate pattern of signals that are a function of the local environments the 1 H and 13 C nuclei find themselves in.
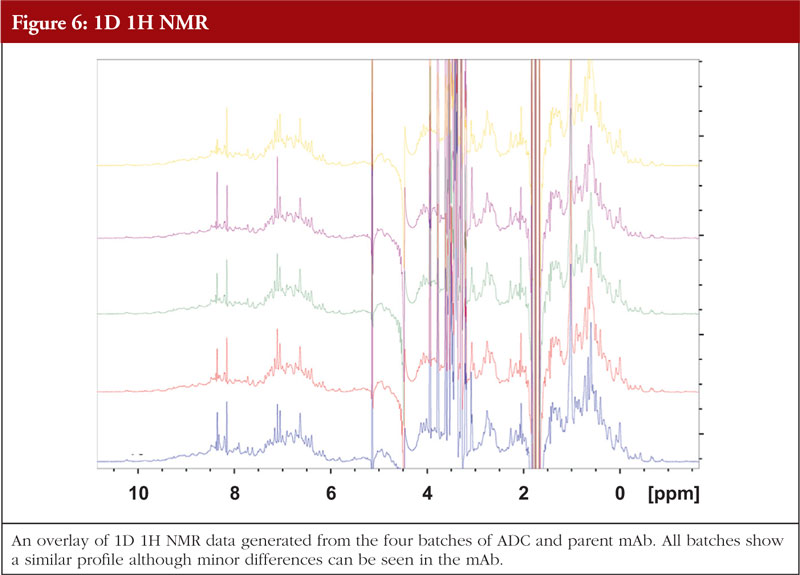
For ADCs and monoclonal antibodies, NMR particularly in the 2D sense, can be challenging due to the large size of the molecule, structural flexibility and long correlation time. Nonetheless, there are techniques available that allow data to be generated on such large proteins. Figure 6 gives an example of 1D NMR data generated for the same four batches of ADCs and the parent mAb as shown in previous figures (unpublished data).
Since these techniques are used to assess the HOS of a molecule, it is critical to analyse the unconjugated antibody alongside the ADC to demonstrate what to expect from the data from the mAb’s native state and to investigate if any changes have occurred in the HOS as a result of the conjugation process. One other point to consider is that each of these techniques described above will interact with the sample as the physics of the technique demands. Thus, since each technique works from different physical principles it follows that the outputs will be different between the techniques. This is perfectly fine from a structural characterization point of view and is a very good example of orthogonality, where data from different analytical approaches are used to support conclusions from the various techniques, forming a more self-sustaining whole.
The idea of orthogonality in HOS analysis is just as applicable for ADCs as it is for other proteins, since each individual technique will have its own sensitivities for different structural features such as CD having greater sensitivity for alpha helices compared to FT-IR. Any perturbation of the HOS in the ADC may disrupt these motifs to a greater or lesser degree both locally to the site(s) of conjugation or more globally across the molecule, therefore techniques need to be as all-encompassing as possible.
It is important to bear in mind that the ADC drug and linker will also have their own spectroscopic profile. Depending on the drug, it may have absorption characteristics in the spectral range of some of the instrumentation (CD in particular). A spectroscopic profile of the native drug is therefore absolutely necessary when assessing HOS data from an ADC to help assess any impact the drug is having on the overall ADC profile. Thus, comparison of the drug, ADC and native mAb can explain features seen in the ADC absorbance profile.
The chemical processes used to produce ADCs could result in enhanced aggregation as a result of structural disruption of the mAb and this needs to be investigated. Again, a sample of the native mAb should be analysed alongside the ADCs to provide baseline aggregate levels.
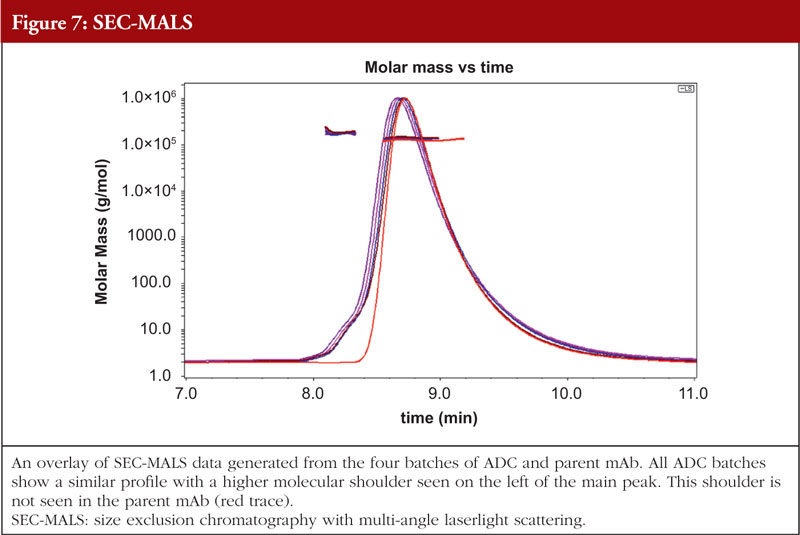
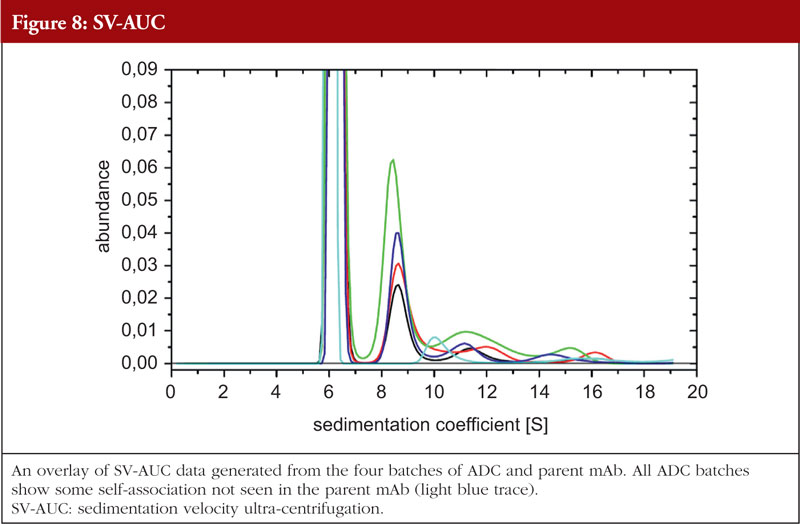
Just as with HOS analyses, orthogonality is very important in aggregation studies. This comes out of the general principle of the value of orthogonal investigations, as recognized by the regulatory agencies, stemming from the fact that different aggregation techniques will give different values for the level of aggregation due to the nature of the techniques themselves. A combination of sedimentation velocity ultra-centrifugation (SV-AUC) and size exclusion chromatography with multi-angle laserlight scattering (SEC-MALS) for aggregation studies have been shown to be applicable to ADCs. Data from the SEC-MALS and SV-AUC analyses of the same four ADC batches and the parent mAb as shown in previous Figures are shown in Figures 7 and 8, respectively (unpublished results). The parent mAb is likely to have slightly different hydrodynamic properties to the ADCs and this will result in slightly different sedimentation coefficients.
The range of HOS techniques available and applicable to structural analysis of biopharmaceutical products are equally applicable, and indeed vital, for the structural characterization of ADCs in order to gain a full structural understanding of the product. The fact that these techniques all have different biophysical attributes and thus analyse HOS from different perspectives is of great significance for building up a strong picture of biopharmaceutical HOS. When these techniques are used in an orthogonal manner, the various different features of individual techniques such as CD, FT-IR and NMR, with their unique biophysical properties, serve as meaningful molecular probes across different aspects of structure. Together with comparative data for the parent mAb, evaluations can be made not only with regard to batch-to-batch comparability for the ADC or in an ADC biosimilarity study but also for assessment of change in HOS compared to the parent mAb. This knowledge can be relevant for understanding any impact in terms of function or possibly aggregation that may be seen with the ADC. Furthermore, investigations of the extent of structural change between parent mAb and ADC can lead to the investigation of new conjugation chemistries that have less impact on HOS, thus producing mAb drug scaffolds more closely aligned structurally to the parent mAb.
Orthogonality of protein analytical aggregation techniques can also be successfully applied to ADCs with the data being used to either confirm satisfactory levels of aggregation or suggest possible modifications to process/purification procedures if levels are outside an expected or acceptable range. Thus, aggregation studies of ADCs are inextricably linked to HOS investigations and product development.
In summary, if ADCs are to become targets for biosimilar development, then, as with all biosimilar structural studies, their HOS and propensity to aggregate needs to be investigated using appropriate techniques such as those described here.
This paper is funded by BioPharmaSpec Ltd (www.biopharmaspec.com).
Competing interests: Dr Richard L Easton joined BioPharmaSpec in 2016 as Technical Director for Structural Analysis and is responsible for management of all aspects of carbohydrate and glycoprotein characterization at the primary structure level.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327-44.
2. European Medicines Agency. ICH Q6B Specifications: test procedures and acceptance criteria for biotechnological/biological products – Scientific guideline [homepage on the Internet]. [cited 2023 Aug 1]. Available from: https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products
3. Astier A. Importance of the determination of the higher order structure in the in-use stability studies of biopharmaceuticals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):49-51. doi: 10.5639/gabij.2020.0902.009
4. Vieillard V, Astier A, Paul M. Extended stability of a biosimilar of trastuzumab (CT-P6) after reconstitution in vials, dilution in polyolefin bags and storage at various temperatures. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):101-10. doi: 10.5639/gabij.2018.0703.022
5. McKee C, Chapman C, Bayley C. SDE-100 a stochastic cysteine linked vedotin ADC: assessing comparability of higher order structure using multiple orthogonal analytical approaches. Results presented as poster at 13th World ADC; 13&ndash16 March 2023; London.
|
Author: Richard L Easton, BSC (Hons), DIC, PhD, Technical Director – Structural Analysis, BioPharmaSpec Ltd, Suite 3.1 Lido Medical Centre, St Saviour, Jersey, JE2 7LA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2023 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/biosimilar-antibody-drug-conjugates-considerations-of-higher-order-structure-and-aggregation.html
|
Abstract: |
Submitted: 18 June 2022 Revised: 19 July 2022; Accepted: 20 July 2022; Published online first: 29 July 2022
Since the legal frameworks for biosimilar approvals were established in Europe in 2003 [1] and the US in 2009 [2], the use and uptake of biosimilars has increased across the globe. There have now been many years of biosimilars development, and the process of getting this class of drugs to market is being continually examined.
Multiple global regulatory guidelines require in-depth analysis to demonstrate that the biosimilar is similar enough to the originator in terms of structure, composition, and in-vitro activity, in addition to clinical trials to compare pharmacokinetics (PK) and examine possible clinical inequivalence. Questions are now being asked regarding the need for extensive clinical trials and whether it is more appropriate to front-load biosimilar drug investigations with risk-based analytics [3, 4].
An online roundtable discussion entitled ‘Front-loading biosimilar development with analytical characterization’ was recently organized and held by BioPharmaSpec.
BioPharmaSpec is a global contract research organization (CRO), led by industry experts in the field of mass spectrometry, specializing in the structural, physicochemical, and biophysical characterization of biopharmaceuticals such as biosimilars.
The roundtable was chaired by Dr Fiona Greer, an independent consultant with over 35 years’ experience in glycoprotein analysis and previously a founding Director of M-Scan and Global Director of Biopharma Services Development, SGS. Dr Greer was joined by three panel members, Dr Richard Easton, Technical Director for Structural Analysis at BioPharmaSpec, Dr Dan Mamelak, Founder and President of Custom Biologics, and Dr Marius Schmid Chief Executive Officer of Zentriforce Pharma. The panel discussed how detailed structural and functional investigations, as part of a comparability exercise, can be front-loaded in a biosimilar development programme to improve confidence levels in the nature of the biosimilar, reduce development costs and, in some circumstances if guidelines permit, reduce the requirement for full clinical trials. The premise of the discussion was that technological advances in hardware and software mean that detailed investigations can now take place in all aspects of primary and higher order molecular structure and the subsequent impact that any modifications can have on functional biological activity.
During the roundtable, the following key issues were discussed:
The panel members gave their opinions through their answers to the three questions listed below.
Certain regulatory authorities are taking a different view on the necessity of comparative clinical efficacy trials. How do you think this impacts the analytical and functional work performed as part of drug development?
Dr Easton was the first to answer the question. He highlighted the fact that traditionally, for a biosimilar to be approved, it is compared to an innovator reference product. However, now that many biosimilars have been approved, there is a large body of historical data on multiple biosimilars themselves. He noted that, in a post-Brexit world, the UK’s health authority is reconsidering biosimilar evaluation and the potential to reduce reliance on clinical trial data in favour of knowledge of structural and functional properties. To do this, structural and functional information must be very robust.
Dr Schmid followed on by noting that, when it comes to removing clinical studies, the question is: who will take the risk and do this first? Although the UK is considering removing the need for clinical trials of biosimilars, the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) are not at present, so clinical trials will still need to be done for products to be approved outside the UK.
In the UK, the biosimilar guideline provides a clear outline of the requirements and needs for clinical studies. The European Medicines Agency (EMA) follows a similar approach to the UK. Although the US regulatory stance on biosimilars may differ from that of the UK and the EMA, the US FDA can be flexible in its decisions. Nevertheless, clinical trials remain a crucial requirement for the approval of biosimilar products in both Europe and the US unless their omission can be justified by the applicant.
Dr Schmid also stated that, to expand biosimilarity study data, it could be possible to shift everything to a good manufacturing practice (GMP) environment, or to get larger statistical samples. However, he highlighted that neither seem to be completely necessary, but they might be an option to increase the trust in data. He also made the point that we have a lot of historical data on the biosimilars themselves and it might be interesting to compare new with old biosimilars as well as to reference products.
Dr Mamelak concluded the answers to this question by noting that regulatory agencies want to make it less expensive to produce biosimilars, thus reaching a wider market. With the changing approaches of some regulatory agencies, it is likely that the decision-making process will rely more on robust structure/function packages. This will help remove the need for animal and PK testing that are costly to perform and, it could be argued, unethical. Thus, moving forward, it will be important to rely on historical and empirical data.
What is the best way to develop a front-loaded characterization study? How can you build orthogonality into your approach?
Dr Schmid highlighted that, when carrying out structural assessments of biosimilars, accuracy is the most difficult aspect to assess and the need for orthogonal methods is key. He noted that, ‘aggregation analytics are often needed and it is common to perform three different techniques’. In many cases, orthogonal information can be obtained, but accuracy can be harder to achieve. He concluded by noting that one way to ensure accuracy is to run positive control samples.
Dr Mamelak noted that, although orthogonal tests are key for structural testing, there isn’t always the same need for orthogonal approaches in functional analyses. He noted, ‘with functional testing, molecules are received that are known to be structurally sound and the mechanism of action is what needs to be measured’. At this point in the analysis of biosimilars, orthogonal testing is no longer needed due to the emergence of very sophisticated recombinant cell lines that are used as the target cell line for these biological molecules. In reference to these assays, he concluded by noting, ‘if the molecule works the way it’s proposed to, the cell line glows and the glow is measured’.
He also highlighted that, although it is not important to carry out orthogonal tests within functional testing, it is important to qualify the assay against a reference product to ‘show that the biosimilar is similar’. When it comes to quality, it is important to run assays under good laboratory practice (GLP), or GLP-like, conditions. In summary, Dr Mamelak noted that the emergence of sophisticated recombinant cell lines and inclusion of a reference standard means orthogonal approaches to measure mechanism of action are not critical, which streamlines the functional characterisation plan and, in turn, reduces costs.
Dr Easton noted that orthogonality is crucially important for the structural and physicochemical types of analyses carried out by BioPharmaSpec. He highlighted that such tests are needed to produce a self-supporting dataset so that all aspects of the data can be relied upon. There are various orthogonal techniques employed at BioPharmaSpec, see Table 1. He noted, ‘to design an orthogonal experiment, it is key to understand what each technique can and can’t do so that you can deduce solid conclusions, a concrete package and in-depth knowledge of the molecule’.
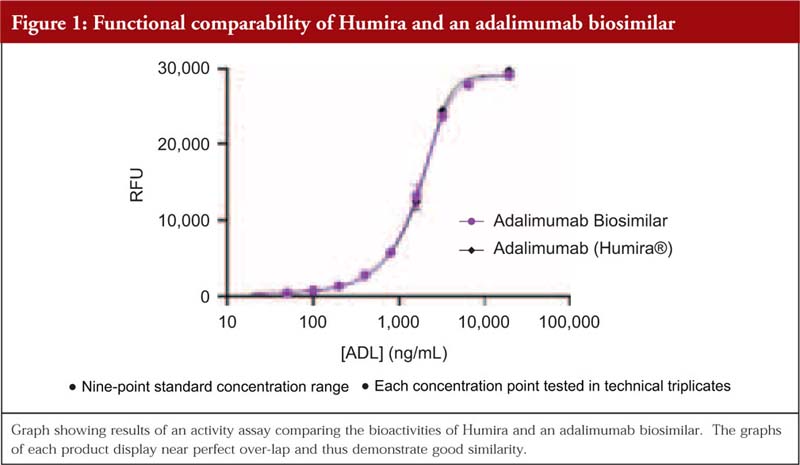
Dr Mamelak then added that, when it comes to functional testing, it is advantageous if the product’s structure has already been well-defined. When the comparability study is carried out within a cell-based assay, a curve is often produced to demonstrate how similar a biosimilar is to the reference. He shared the example of a comparability study of adalimumab biosimilar/originator behaving similarly, here the graphs overlap and thus demonstrate that they are behaving similarly. In addition, he remarked that the graph also showed that the structural analysis carried out to determine potential differences prior to the functional testing had been done very well.
Lastly, Dr Mamelak noted that in some cases, the buffer of the biosimilar can be different to that of the reference product and this can cause differences. Here, a cell-based functional assay is a useful tool to determine if there are any effects on functional properties, see Figure 1.
What is your experience of linking analytical characterization to function, i.e. an integrated strategy? What about the challenges of performing these correlations between the originator and biosimilars? Can you illustrate with examples?
Dr Mamelak highlighted that this question touches on the ‘holy grail’ of developing a biosimilar package. The structure and function analyses are the driving force that will bring the cost of developing biosimilars down. A good structure-function package with orthogonal approaches at the structural phases and well-designed functional characterization of the molecule is what the regulatory agencies want to see.
From a functional perspective, the main challenge is that the assay has to show that it can pick up subtle differences and must be able to confirm differences and similarities. An assay must be sensitive enough to show that differences can be captured, and any differences must be explained. He noted that it is important to ask: ‘Can the assay pick up differences in functional activity that are likely due to differences in structural properties?’ He then demonstrated this by giving some examples of cell-based bioassays. Overall, he stressed that a solid structure-function package is the key driving force to help bring the cost of biosimilar development down.
Dr Schmid provided a case study from aggregation analyses where a generic drug (cyclosporine) performed differently to the originator and they found, by examining oil drop distribution, that the issue was in the micellar structure of the Tween.
Dr Easton concluded the answer to the final question by noting that, to get a good understanding of the structure-function relationship, you have to know your assays and your product well enough to be able to understand any differences you might see and put them into the context of your development programme. Developing good lines of communication between the scientists carrying out the structure and the functional analyses is crucial to front-loading analytics and truly defining biosimilarity.
Dr Greer concluded that it is clear that detailed structure-function investigations as part of the comparability exercise can give great confidence in the nature of the biosimilar, providing information from which risk-based assessments can be made. Results from orthogonal methods can provide a meaningful insight into how similar the biosimilar is to the originator and potentially reduce the requirement for full confirmatory clinical trials. Overall, structural-functional data should be viewed in tandem for full understanding in integrated studies to enable a reliable assessment of biosimilarity.
This paper is funded by BioPharmaSpec Ltd (www.biopharmaspec.com).
The roundtable discussed how biosimilar manufacturers might be able to decrease the number of clinical trials required for registration now that the UK is no longer part of the EU. It was noted that until/unless the FDA and/or EMA change their current approach the need for clinical PK, safety and efficacy studies is unlikely to be decreased by what has been proposed in the UK.
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Jul 19]. Available from: www.gabionline.net/biosimilars/general/biosimilars-approved-in-europe
2. GaBI Online – Generics and Biosimilars Initiative. FDA approves its first biosimilar [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2022 Jul 19]. Available from: http://www.gabionline.net/biosimilars/news/FDA-approves-its-first-biosimilar
3. Bielsky M, Cook A, Wallington A, Exley A, Kauser S, Hay JL, et al. Streamlined approval of biosimilars: moving on from the confirmatory efficacy trial. Drug Discov Today. 2020;S1359-6446(20)30343-3. doi: 10.1016/j.drudis.2020.09.006
4. BioPharmaSpec. Biosimilars roundtable discussion with analytical characterization experts [homepage on the Internet]. [cited 2022 Jul 15]. Available from: https://biopharmaspec.com/biosimilars-roundtable-analytical-characterization/
|
Author: Richard L Easton, BSC (Hons), DIC, PhD, Technical Director, BioPharmaSpec Ltd, Suite 3.1 Lido Medical Centre, St Saviours Road, St Saviour, Jersey JE2 7LA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2022 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/front-loading-biosimilar-development-with-analytical-characterization.html
|
Abstract: |
Submitted: 15 February 2022; Revised: 16 March 2022; Accepted: 22 March 2022; Published online first: 28 March 2022
Structural analysis of a biopharmaceutical product, be it a biosimilar or new drug, inevitably requires the use of a wide range of techniques and technologies. The ICH Q6B [1] guidelines for structural investigation, which are invoked by the regulatory authorities as the document detailing their expectations for structural characterization of biosimilars [2-4], state that all areas of biomolecular structure must be investigated. This requirement covers primary amino acid sequence and post-translational modifications including glycosylation (if present), through to secondary and tertiary structure and aggregation assessments, as well as an assessment and characterization of product related impurities (forms of the product that are not structurally consistent with the product specification). Structural analysis is therefore the starting point in the development of a drug product. It goes hand in hand with developing the product itself, as well as the manufacturing process, to produce the expected end product suitable for use in pharmacokinetics (PK), pharmacodynamics (PD) and clinical trials as necessary.
Structural investigations are performed at the drug development stage to examine and indeed confirm that the product has been made correctly, and also to assess the nature and levels of product related impurities prior to any clinical-based work. It is therefore understandable that analytical methods are liable to evolve as structural data are generated and interpreted, and any further specifically targeted analytical studies are carried out. To this end, there needs to be a degree of flexibility in the details of the analytical methodologies involved, since the precise conditions required for optimal data generation within a specific technique may not yet be defined.
At this point in the development of the product there is therefore no requirement for the application of specifically qualified or validated methods for analysis since the methods, product structure and therefore any product specifications or parameters are not yet defined. This does not mean to say of course that the methods used should not be demonstrated as being fit for purpose and indeed, prior to analysis, investigations need to be performed to optimize a method for application to that particular product. This may include the demonstrated requirement for excipient removal if that is likely to interfere with analysis, e.g. high levels of sugar or amino acid excipients which would interfere with monosaccharide and amino acid analysis, respectively, or assessment of incubation times for a proteolytic digest associated with a peptide mapping study. Purification may be carried out so that both biosimilar and innovator drugs are subsequently analysed from within the same buffer system, which precludes the influence of buffer effects on results. If any up-front purification is required for excipient removal, then an assessment needs to be made as to any possible impact on the active ingredient being assessed [2].
When we consider biosimilars, the requirement for the application of such a broad range of characterization techniques, with their inherent need for a degree of flexibility to suit the molecule under investigation, is recognized by the regulatory agencies as falling outside the area of ‘Quality Control’ since these procedures are used as part of product development and ultimately product definition, see Table 1 for comparison of qualification and validation features.
To this end the methods are not therefore required to be validated in accordance with ICH Q2(R1) [5] but; as stated by the US Food and Drug Administration (FDA) in their Biosimilar guidance document (Quality), methods ‘…should be scientifically sound, fit for their intended use, and provide results that are reproducible and reliable’ [3]. The European Medicines Agency (EMA), in their Biosimilar guidance document (Quality) state ‘Methods used in the characterization studies form an integral part of the quality package and should be appropriately qualified for the purpose of comparability. If applicable, standards and reference materials, e.g. from European Pharmacopoeia (Ph. Eur.), World Health Organization (WHO), should be used for method qualification and standardization’ [2]. The UK Medicines and Healthcare products Regulatory Agency (MHRA) Biosimilars guidance document echoes this sentiment, stating ‘Analytical methods need to be sensitive, qualified and sufficiently discriminatory to detect possible differences. Robust data require the application of suitable orthogonal methods’, [4].
The idea of the suitability of methods for their analytical application, i.e. being fit for purpose, is clearly recognized in these regulatory documents but at the same time the need for release method type controls of methodologies is also understood to be unnecessary at this characterization stage. Strength of data and the subsequent conclusions drawn for structural characterization purposes are derived from the use of orthogonal techniques where possible, a point emphasized in the guidelines.
For biosimilar analysis, qualification is useful for the determination of the expected experimental range or error of the method being employed. With this knowledge in hand, a comparison can be made between innovator and biosimilar-derived values, allowing a better assessment of similarity or otherwise between samples for the parameter in question.
There is no explicit mention of an expected procedure for qualification in the quoted biosimilar guidelines, and no request drawing upon any principles of the ICH Q2(R1) document on validation of analytical methods. However, when qualifying any method, the application of the principles in this document for generation of a qualified (as opposed to ‘validated’) method in a broad sense would seem wise. Use of appropriate standards to qualify a procedure, in terms of both instrumentation and methodology, can demonstrate its general applicability to a particular analysis with due regard given to whether the method is assessing a particular aspect of the sample in a qualitative or quantitative sense (through considerations of specificity, repeatability, accuracy, linearity, LOD, LOQ, intermediate precision and robustness as appropriate). The method then becomes suitable for use in characterization protocols where the same tenets hold true of the sample as for the standard used in the qualification.
This general method qualification should be repeated on a cycle of one or two years where the same process is repeated to demonstrate that the system (i.e. instrument and methodology) is still performing within the defined parameter range. Where consideration may need to be given to unique features of any biosimilar that requires specific assessment in a method application, qualification may be performed once a method has been developed specifically for that product and structural aspect. An example of this could be a method used to assess the presence of a unique or uncommon post-translational modification.
It should be noted that the basic ‘go-to’ analytical techniques used during product characterization have usually been applied across the analyses of many molecules, including biosimilars, over time and thus have been demonstrated to have basic applicability within the area of analytical structure determination. Therefore, specific qualification of these methods may not be appropriate.
Wherever possible, appropriate checks of the method should be used during the course of sample analysis regardless of whether the method is qualified or not. These checks normally comprise of system suitability tests (SSTs) run at the start and end of an instrument analytical session. The data obtained serve to demonstrate that detection parameters, such as sensitivity and resolution, are optimal and therefore sample results are valid and data can be processed and interpreted. If there are a high number of samples to run, SSTs can also be run at regular intervals throughout the sample runs as an ongoing check that the system has not deviated from the optimal run parameters during the course of analysis. This of course can also be supplemented by interspersed repeat runs of key preparations, such as standard mixtures or blanks, to ensure a consistent and optimal level of performance across the analytical session. The SST may well have its own set of acceptance criteria that must be met, these criteria having come from a qualification of the method and thus a knowledge of how the SST will behave.
One other point to consider is the use of the term ‘qualification’ itself. Qualification does not necessarily only cover the method itself. Qualification is also performed at the time of installation of analytical instruments, in order to demonstrate performance at the expected level for a variety of defined parameters, e.g. sensitivity and resolution. Instrument qualification procedures cover installation, operation and performance qualifications (often described as IQ, OQ and PQ, respectively). On this basis, the instrument itself is then qualified to operate as expected and the method can then be developed with confidence in the system it is being run on.
Extinction coefficient determination
One area where method qualification is important at an early stage of product development is in determining the extinction coefficient (e). The extinction coefficient is the measure of light absorption per unit amount of sample and is intimately related to absorbance (A), through the Beer-Lambert Law A=ecl, to the concentration of the sample (c) and pathlength (l) of the sample through which the light passes. Thus, in order to determine the concentration of a biopharmaceutical active ingredient from an absorbance measurement, one must determine the extinction coefficient.
For determination of the extinction coefficient, concentration of the protein/glycoprotein can be measured using amino acid analysis, see Figure 1, and the determined concentration assessed alongside the measured optical density and a value obtained. The significance of this value is that it can be used to measure the concentration of all prepared samples using an ultraviolet (UV) spectrophotometer for studies in subsequent biological analyses. It is critical that the amount of sample used in these studies is accurately known for dosing and safety reasons. Thus, a qualification of the methods used to determine the extinction coefficient (optical density (OD) spectroscopy and amino acid analysis) is important in demonstrating that the value obtained is coming from a reliable method and therefore can itself be analytically supported and relied upon.
Impurities
Another area that the ICH Q6B and biosimilarity guidelines highlight is that of assessing impurities present in the product. For biosimilar products it is not expected that the impurities will be the same as the innovator since the manufacturing process will not necessarily be the same. Impurities are defined as being either product or process related.
Product related impurities are the result of differential processing of the product either during biosynthesis or subsequent purification. These are considered part of product characterization itself and will thus naturally be identified and characterized during those analyses. Process related impurities cover all aspects of the drug production process where contaminants are likely to be derived both from cellular and production sources. Removal of these impurities to the extent possible, or at least to below set limits, is critical for product quality and safety.
Methods that assess impurities in the characterization phase can be used in the absence of qualification, however given the importance of impurity clearance as a key consideration in the drug production process it is worth considering qualification of residual testing methods for key process-related impurities, see Figure 2.
This will allow confidence in the method and thus support for the results generated, so that meaningful changes can be made to the production process, if necessary, on the basis of reliable data. This alleviates a potential area of risk that could be very costly and time-consuming to fix if issues with levels of impurities were discovered at a later date. These qualified methods can, if required, be subsequently validated for batch release purposes.
As mentioned above, structural analysis uses a wide variety of instrumentation and techniques to probe all aspects of biopharmaceutical/biosimilar structure. Once the structure is determined (using multiple batches of biosimilar and innovator, if the product is a biosimilar), and the manufacturing process has been tied down, batches can be manufactured for subsequent biological testing.
At this point, the methods that will be used for testing of particular key structural features, or critical quality attributes, should be considered. These methods are designed to demonstrate that the batch being tested is showing the expected structure with no anomalies or aberrations. Tests to consider will vary between different products but should provide unique information on the molecule and be responsive to a change in molecular structure such that the method should reliably detect any change in the product.
Example: Peptide mapping – MS or UV detection?
The online LC/ES-MS analysis used for the peptide mapping study during the characterization phase can potentially be reduced down to UV detection rather than MS detection, once structure and the manufacturing process are determined. This means that, with all other parameters, such as column and gradient conditions remaining constant, a simple fingerprint profile could be used, where the identities of the UV peaks are known from the initial MS characterization work. This method can be qualified/validated to demonstrate its applicability prior to use in batch release which would include an assessment of the sensitivity of the method to any structural changes that may occur, for example, variation in the levels of post-translational modifications. Other tests will need to be considered to monitor other aspects of molecular structure, such as intact mass analysis and glycan profiling, since peptide mapping is not a catch-all for assessment of all aspects of a molecule’s structure.
Ultimately, appropriately selected methods used to assess the quality of a product will need to be validated and performed in a GMP environment. The decision to qualify a method in no way affects the necessity to also validate the method for the later phases of drug development, with validation of methods expected by the regulatory authorities at these stages of drug development. So what good is qualification of a method as a sort of half-way house? This comes down to the need or desire to ensure that the method is producing reliable and meaningful data for the structural aspect of the product being measured, such that the quality of the product and the process is built in from an early stage. A method, qualified through the principles of ICH Q2(R1), should be straightforward to validate since that method will already have gone through the testing phase. It will therefore stand the validation process with less risk than an untried method that has come to be relied upon but whose characteristics have not been tested rigorously.
Method qualification is something that needs to be considered during product development based on the nature of the specific analysis being undertaken and the nature of the methodology being used. For example, qualifications of methods for residual testing and impurities, or other methods used to demonstrate the correct nature of a particular structural feature may be viewed differently compared to qualifications of structural characterization techniques using mass spectrometry. There is no hard and fast rule covering when to qualify a method or what methods should be qualified, but the qualification of selected methods early in product development can help to avoid problems with test processes that may be required for use later but which are shown to have features that preclude meeting validation criteria.
In summary, what is required at the early stage of product development, i.e. at the characterization and definition stage, is that methods are fit for purpose and scientifically accurate such that data derived from these methods are trustworthy and sound. Thus, in biosimilar analysis, the comparative data generated will allow meaningful conclusions to be drawn regarding structural similarities and differences.
This paper is funded by BioPharmaSpec Ltd (www.biopharmaspec.com).
About the author: Richard obtained his PhD in glycoprotein structural characterization using mass spectrometry from Imperial College of Science, Technology and Medicine. He subsequently spent several years there as a postdoctoral research scientist working in the field of glycoprotein structural characterization with emphasis on glycan elucidation. The projects he was involved in required detailed structural analysis of glycoproteins derived from animal, plant and fungal systems, very frequently expressing unusual glycosylation profiles. He moved to GlaxoSmithKline for a short time where he was head of mass spectrometry for the toxicoproteomics and safety assessment group. Richard joined M-Scan Limited (now part of SGS Life Sciences) in 2003 as a biochemist and became the Team Leader for Carbohydrate Analysis before being appointed Principal Scientist. Richard joined BioPharmaSpec in 2016 as Technical Director for Structural Analysis and is responsible for management of all aspects of carbohydrate and glycoprotein characterization at the primary structure level.
References
1. European Medicines Agency. ICH Topic Q6B specifications: test procedures and acceptance criteria for biotechnological/biological products. Guidance Document. August 1999 [homepage on the Internet]. [cited 2022 Mar 28]. Available from: https://www.ema.europa.eu/en/ich-q6b-specifications- test-procedures-acceptance-criteria-biotechnologicalbiological-products
2. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). EMA/CHMP/BWP/247713/2012. 22 May 2014 [homepage on the Internet]. [cited 2022 Mar 28]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf
3. U.S. Food and Drug Administration. Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations. Guidance for Industry. Draft Guidance. May 2019 [homepage on the Internet]. [cited 2022 Mar 28]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein- biosimilars-comparative-analytical-assessment-and-other-quality
4. Government UK. Guidance on the licensing of biosimilar products [homepage on the Internet]. [cited 2022 Mar 28]. Available from: https://www.gov.uk/government/publications/guidance-on-the- licensing-of-biosimilar-products
5. European Medicines Agency. ICH Q2(R1) validation of analytical procedures: text and methodology. June 1995 [homepage on the Internet]. [cited 2022 Mar 28]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf
|
Author: Richard L Easton, BSC (Hons), DIC, PhD, Technical Director, BioPharmaSpec Ltd, Suite 3.1 Lido Medical Centre, St Saviours Road, St Saviour, Jersey JE2 7LA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2022 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/structural-characterization-methods-for-biosimilars-fit-for-purpose-qualified-or-validated.html
Copyright ©2025 GaBI Journal unless otherwise noted.