Author byline as per print journal: Stephen P Murby, FRSA; Michael S Reilly, Esq
|
Introduction: As the number of biosimilar approvals in Australia increases, it is important to build on the existing regulatory framework to continue to bring high quality, safe and efficacious biosimilars to the widest number of patients most cost-effectively. As new policies regarding the regulation, reimbursement and uptake of biosimilars are being considered, the Alliance for Safe Biologic Medicines (ASBM) has asked Australian prescribers for their views on the naming, substitution and prescribing of biologicals and biosimilars. Currently, biologicals and biosimilars are approved by Australia’s Therapeutic Goods Administration (TGA). The country’s Pharmaceutical Benefits Advisory Committee (PBAC) has indicated it will consider pharmacy-level substitution of biosimilars for reference biological medicines on a ‘case-by-case basis’. |
Submitted: 9 May 2017; Revised: 13 July 2017; Accepted: 19 July 2017; Published online first: 1 August 2017
In Australia, biologicals and biosimilars are approved nationally by the Therapeutic Goods Administration (TGA). Aczicrit and Grandicrit (epoetin lambda) were the first products approved in Australia as biosimilars in 2010 [1]. To date, TGA has approved 13 biosimilars within the product classes of human growth hormone, granulocyte colony-stimulating factor (G-CSF), insulin, erythropoietin, follicle stimulating hormone (FSH) and tumour necrosis factor-inhibitor [1].
With an increasing number of biosimilars seeking entry to the Australian market, it is important to build on the existing regulatory framework established to continue to bring high quality, safe and efficacious biosimilars to the widest number of patients most cost-effectively. As new policies regarding the regulation, reimbursement and uptake of biosimilars are being considered, the Alliance for Safe Biologic Medicines (ASBM) has asked Australian prescribers for their views on the naming, substitution and prescribing of biologicals and biosimilars.
Australia’s Pharmaceutical Benefits Advisory Committee (PBAC), an independent expert body appointed by the Australian Government to recommend new medicines for listing on the PBS (Pharmaceutical Benefits Scheme), has indicated that it will consider pharmacy-level substitution of biosimilars for reference biological medicines on a ‘case-by-case basis’.
Some clinicians hold concerns with pharmacy-level substitution due to the current paucity of data on such practices, and also matters associated with tracking which product is dispensed. PBAC has stated that its ‘default position’ would be to advise that a biosimilar is suitable for substitution by a pharmacist ‘where the data are supportive of this conclusion’ and that a relevant consideration is ‘the absence of data to suggest significant differences in clinical effectiveness or safety compared with the originator product’ [2].
Biologicals are used in the prevention, diagnosis, or treatment of a range of chronic diseases. Since biologicals have large, complex, inherently diverse molecular structures made, or derived, from living organisms, they are always heterogeneous. Unlike non-biological medicines, there is a degree of natural variability in biologicals, and there are generally some differences between the reference and biosimilar products. Current methods to analyse physicochemical and structural differences are extremely sensitive. Analysis of different batches of reference products following a change in the manufacturing process has revealed differences between the pre- and post-change batches [3]. This molecular heterogenity within the originator biological is distinct from molecular differences between the originator and biosimilar.
A biosimilar is a version of the active substance of an already authorized original (or reference) biological, see Box 1.
Biosimilars introduce competition, which has the effect of lowering prices of both originator and biosimilar and increasing patients’ access to these therapies [3].
With a growing number of reference biologicals and biosimilars, regulatory authorities across the globe are in discussion over how biosimilar medicines should be named and labelled [4, 5]. Distinct names will be crucial in order to facilitate post-market safety monitoring and help minimize the potential for medication errors.
There is a clear need for sufficiently detailed and transparent labelling and product information to enable informed decision-making by physicians and patients, ensuring appropriate safe and effective use of these medicines [6].
International Nonproprietary Names (INNs) are intended for use in drug regulation, prescribing, dispensing, pharmacopoeias, labelling, pharmacovigilance and in scientific literature [7]. However, biologicals, due to their increased molecular complexity and structural micro-heterogeneity, are not categorized by the INN alone [7]. An INN Expert Group recommended that the World Health Organization (WHO) develop and implement a system for assignment of Biological Qualifiers (BQs) to similar biotherapeutic products (SBPs) [8]. WHO has proposed the development of a global BQ for biological medicines that will provide a unique identifier for all biological active substances that are assigned an INN [6]. While the INN is a common and public non-proprietary name for a given active substance, the BQ would be applied to a particular manufacturer’s active substance. The BQ would not be part of the INN and it is envisaged that it would enhance identification, prescribing, dispensing and pharmacovigilance of biological medicines.
The US Food and Drug Administration (FDA) issued its guidance for the non-proprietary naming of biological products in January 2017 [9] following the release of two draft guidance documents outlining proposed methods for biological product naming and biosimilar product labelling [10]. According to its latest guidance, FDA will assign a non-proprietary name for all reference biologicals, related biologicals and biosimilars that will include an ‘FDA-designated suffix’. The ‘proper name’ will consist of a combination of the ‘core name’ and distinguishable suffix, which will be ‘devoid of meaning’ and be ‘composed of four lower case letters’. A survey of prescribers of biologicals in the US, carried out before the release of this guidance, found that two thirds (66%) of the prescribers surveyed supported the introduction of distinct names. Of those, the majority would prefer a suffix that indicated the manufacturer [11]. The WHO and FDA emphasis stands in contrast to the EMA approach to biosimilar naming, under which an originator biological and all biosimilars to that product will share the same non-proprietary name.
In Australia, biologicals and biosimilars are approved nationally by TGA. TGA issued a biosimilars guidance in 2013 that includes a section on naming conventions for biosimilars [12–14]. This guidance required the name of a biosimilar in Australia should be made up of the reference product Australian Biological Name (ABN), thus identifying the reference product with which the biosimilar has demonstrable comparability; and a biosimilar identifier, consisting of: the prefix sim(a)– and a three-letter code issued by the WHO INN Committee, according to its draft policy. This guidance was subsequently revoked and recently approved biosimilars to etanercept and infliximab have been given the same non-proprietary name as their reference products.
The country’s PBAC has indicated that it will consider pharmacy level substitution of biosimilars for reference biological medicines on a case-by-case basis, see Box 2. Since the introduction of this policy, two biosimilars to infliximab have been ‘a’ flagged, as has one biosimilar to etanercept.
In June 2016, 160 prescribers in Australia completed a survey sponsored by the ASBM about their knowledge of, attitudes toward and beliefs regarding biosimilars. They were asked their opinions on the naming of biologicals and biosimilars and how these medicines are identified in the patient record and in adverse event reporting. They were also asked for their views on substitution.
Sample characteristics and methodology
In June 2016, 605 prescribers in Australia were invited to complete a 15-minute web-based survey on biologicals and biosimilars. Potential respondents were identified in, and recruited from, a large, global, commercial database of healthcare professionals. A high response rate was expected because prescribers in this database had previously indicated a willingness to participate in market research.
A total of 451 prescribers responded. Respondents were screened as follows: they had to specialize in one of seven therapeutic specialties, including dermatology, endocrinology, gastrointestinal, nephrology, neurology, oncology or rheumatology; they had to have been in practice for one year or more; and they had to have prescribed biological medicines in their practice. A total of 174 respondents were screened out because they did not meet these criteria. A further 80 prescribers did not qualify because they specialized in a therapeutic specialty for which data collection had closed. In addition, 37 started but failed to complete the survey. Any data they contributed are not included in the analysis and report.
A total of 160 prescribers completed the survey. All data collected refer only to those who completed the survey. Participants received a standard cash stipend of US$76 for their time.
Prescribers practised in public hospitals (46%); private hospital/private practice (42%); academic medical centre (11%); and other (1%). They had spent between one and 30 years in practice, see Figure 1.
A quarter (25%) of the prescribers were rheumatologists, 25% were oncologists, and 25% were gastroenterologists. The remaining 25% prescribers were divided equally among dermatologists, neurologists, nephrologists and endocrinologists, see Figure 2.
Regarding responses from participants to the question of how often they used different information sources to learn about the details of a medicine for prescribing and monitoring, 46% of prescribers said they always used published literature, whilst 43% said they never used published literature. Only 19% of prescribers said they always used information from TGA, and 27% said they always used information from PBAC. Only a fifth (19%) of those surveyed said they learnt about the details of a medicine by reading the product information label, and 13% from hospital formulary, see Figure 3.
Reporting and naming of biologicals and biosimilars
Participants were asked how they identified biological medicines when they were prescribed or entered in patients’ records. Similar numbers of prescribers identified medicines by brand name or non-proprietary scientific name (39% and 38%, respectively). A smaller proportion identified medicines by brand name and non-proprietary scientific name (21%), and 2% identified them by Australian Register of Therapeutic Goods (ARTG) number. When adverse events (AEs) were reported, medicines were identified by brand name by 39% of prescribers, by brand name and non-proprietary scientific name by 34% of prescribers, by non-proprietary scientific name by 25% of prescribers, and by ARTG number by just 2% of prescribers, see Table 1.
Respondents were asked how often they included batch numbers when reporting AEs. A total of 23% of prescribers said they never used batch numbers, almost a third of prescribers (30%) said they rarely included batch numbers, 20% sometimes used batch numbers and 28% said they always included batch numbers.
When prescribers were asked why a batch number was not always reported, they replied that the batch number was not available at time of reporting (41%); or they were not sure where to find the information (36%); or they had forgotten to include the information (19%).
Respondents were also asked whether a loss of efficacy should be reportable as an AE. Most prescribers (65%) said it should not, while 19% of prescribers thought it should, and 16% of prescribers had no opinion about whether a loss of efficacy should be a reportable AE.
Asked whether TGA should insist on a distinct non-proprietary scientific name for every biological or biosimilar medicine that it approves, three quarters of prescribers (76%) said yes, 18% of prescribers said no, and 7% of prescribers had no opinion.
Asked what the best way was for TGA to differentiate a biosimilar medicine from its reference product, a large proportion of physicians responded that it would be best if biosimilars had the same non-proprietary scientific name as their reference medicines but with either a differentiating prefix (38%) or suffix (29%). 30% of participants opted for entirely different non-proprietary scientific names for the biosimilar and its reference product.
Substitution attitudes and beliefs
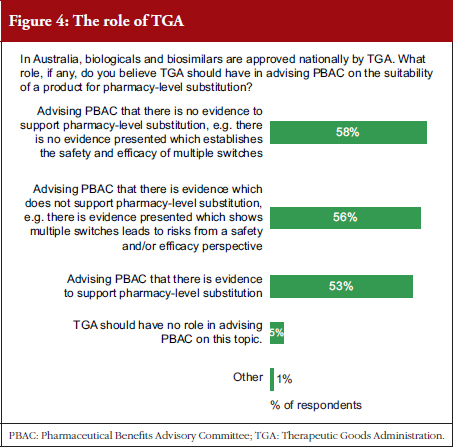
Prescribers were asked what level of evidence would be supportive of pharmacy-level substitution, see Table 2. They were also asked what role they thought TGA should play in advising PBAC on the suitability of a product for pharmacy-level substitution, see Figure 4, and whether TGA or PBAC should be responsible for providing the primary advice to the Australian Government that a product is suitable for pharmacy-level substitution, see Figure 5.
Prescribers completing the survey were asked how important it was for them to have the sole authority to decide, together with their patient, the most suitable biological medicine to be dispensed to the patient. Over half of prescribers (54%) thought it was very important and over a third (36%) thought it was critically important, see Figure 6. When the prescribing physicians were asked whether their prescription software/documentation included a box marked ‘brand substitution not permitted’, 61% responded that it did, 21% responded that it did not, and 18% were unsure.
Respondents were asked about switching between biological medicines for patients with chronic disease. Over half (51%) of prescribers said that pharmacy-level substitution was not acceptable for these patients, 9% thought it was totally acceptable, while 40% thought it was acceptable providing they were notified in advance.
Biosimilars familiarity and knowledge
In response to the question of how familiar survey respondents were with biosimilar medicines, 21% said they were very familiar and had a complete understanding, 73% said they had a basic understanding, 6% said that they could not define them (although they had heard of them), and 1% said they had never heard of them.
Prescribers were asked a series of questions to gauge their understanding of biosimilars, their awareness of and comfort with the biosimilars approval process, and their opinions on switching. Responses to these questions are given in Table 3.
Respondents were asked how comfortable they would be prescribing a biosimilar medicine that had been approved for several or all of the indications of its reference medicine on the basis of clinical trials in only one of those indications, or in fewer indications than for which the biosimilar is approved, 73% would have some concerns, depending on data and indications; 16% would be comfortable; and 11% would not be comfortable.
Of the prescribers who completed the survey, over three quarters (76%) agreed that TGA should insist on distinct non-proprietary scientific names for all biosimilars and reference products. In addition, well over half (61%) of respondents believed TGA should be responsible for recommendations on pharmacy-level substitution, while only a third (33%) thought that PBAC should be responsible.
Nearly all the prescribers in this survey (98%) use either brand name or non-proprietary scientific name for recording and prescribing biosimilars and their reference biologicals. Most prescribers (61%) want TGA to play a major role in naming biosimilars.
It was clear that the reporting of biosimilars use via brand name and batch number varied between respondents. Respondents indicated that robust data are needed to support substitution, and the vast majority of prescribers (94%) said that the final decision over which biological to prescribe should rest with the prescriber and the patient, and strongly supported clinically supervised switching over pharmacy-level substitution.
Respondents used different information sources to learn about the details of a medicine for prescribing and monitoring, but each source was used by surprisingly few prescribers. Less than half of prescribers said they always used published literature, and a similar proportion said they never used published literature. Clearly there were gaps in how the regulatory process is understood since about half of those surveyed thought, incorrectly, that biosimilars and originators are approved through the same regulatory process.
Despite a spread of responses from the prescribers surveyed, there was general agreement that biosimilars should be distinguished from originators with either the same non-proprietary scientific name and a differing prefix or suffix, or with a completely unique name. The vast majority of prescribers thought that they and their patients should decide which biological is dispensed and that they should be notified of any substitution by the pharmacist.
|
Key points
|
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others – who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
The Australia 2016 prescribers and biosimilars survey was sponsored by ASBM.
This paper is funded by ASBM.
Competing interests: Mr Stephen P Murby, FRSA, is a member of the International Advisory Board of Alliance for Safe Biologic Medicines (ASBM). Mr Michael S Reilly, Esq, is the Executive Director of and employed by ASBM. Mr Reilly served in the US Department of Health and Human Services from 2002–2008.
Provenance and peer review: Not commissioned; externally peer reviewed.
Stephen P Murby, FRSA, former Head of Consumers Health Forum of Australia; International Advisory Board Member of ASBM
Michael S Reilly, Esq, Executive Director, ASBM
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Australia [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Australia
2. GaBI Online – Generics and Biosimilars Initiative. Australia’s PBAC recommends substitution of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Biosimilars/General/Australia-s-PBAC-recommends-substitution-of-biosimilars
3. Kurki P. Biosimilars for prescribers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):33-5. doi:10.5639/gabij.2015.0401.008
4. Alexander EA. The biosimilar name debate: what’s at stake for public health. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):10-2. doi:10.5639/gabij.2014.0301.005
5. Dolinar RO, Reilly MS. A survey of Australian prescribers’ views on the naming and substitution of biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):58-62. doi:10.5639/gabij.2014.0302.018
6. European Biopharmaceutical Enterprises. Tell me the whole story: the role of product labelling in building user confidence in biosimilars in Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):188-92. doi:10.5639/gabij.2014.0304.043
7. Robertson JS. The challenges of nomenclature – INN, biosimilars and biological qualifiers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):110-2. doi:10.5639/gabij.2015.0403.025
8. Shaw B. Biosimilars naming and prescribing policy in Australia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):168-9. doi:10.5639/gabij.2013.0204.048
9. GaBI Online – Generics and Biosimilars Initiative. FDA issues final guidance on naming biologicals [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Guidelines/FDA-issues-final-guidance-on-naming-biologicals
10. US FDA proposals for naming of biologicals and labelling of biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):140-3. doi:10.5639/gabij.2016.0503.036
11. Gewanter HL, Reilly MS. Naming and labelling of biologicals – a survey of US physicians’ perspectives. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):7-12. doi:10.5639/gabij.2017.0601.003
12. GaBI Online – Generics and Biosimilars Initiative. Australia issues new biosimilars guidance [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Guidelines/Australia-issues-new-biosimilars-guidance
13. GaBI Online – Generics and Biosimilars Initiative. Naming requirements in Australian biosimilars guidance [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jul 13]. Available from: www.gabionline.net/Guidelines/Naming-requirements-in-Australian-biosimilars-guidance
14. World Health Organization. Biological Qualifier [homepage on the Internet]. [cited 2017 Jul 13]. Available from: http://www.who.int/medicines/services/inn/inn_bio_bq/en/
|
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-survey-of-australian-prescribers-views-on-the-naming-and-substitution-of-biologicals.html
Copyright ©2025 GaBI Journal unless otherwise noted.