Author byline as per print journal: Tobias Blank, PhD, Tilo Netzer, PhD, Wolfram Hildebrandt, MSc, MA, Angela Vogt-Eisele, PhD, Professor Marietta Kaszkin-Bettag, PhD
|
Background: As patents for biological drugs begin to expire, the need for scientific guidance on biosimilar drugs grows increasingly important. The European Medicines Agency provided the first guidelines to cover the approval of biosimilars in 2005. On 10 February 2012, the US Food and Drug Administration drafted three guidance documents relevant to approval of biosimilars in the US. |
Submitted: 22 February 2013; Revised: 16 July 2013; Accepted: 16 July 2013; Published online first: 29 July 2013
As the patent protection for many biological medicinal products begins to expire [1], the need for specific guidance regarding the development and approval of biosimilars grows increasingly important. The European Medicines Agency (EMA) was the first competent authority that issued guidelines for biosimilars in 2005 [2]. Other countries including Canada, Japan, and Korea followed with guidance documents for the approval of biosimilars, which mainly follow the approach of EMA. The World Health Organization (WHO) has also issued a guideline as a general framework, establishing regulatory requirements for the approval of biosimilars [3]. In February 2012, US Food and Drug Administration (FDA) released draft guidance documents for biosimilars; however, no biosimilar drug has yet been approved in the US.
The purpose of this review article is to compare the EU with the US regulatory approaches and to evaluate the potential differences with respect to their implications for characterization of the safety profile of a proposed biosimilar product. In this context, three main areas were identified as potentially relevant, namely non-clinical and clinical testing strategies as well as general regulatory requirements. For the comparison of the regulatory approaches, legal documents and guidelines from both regions were compared and the results are discussed on the background of publicly available literature.
Since the active substance of a biosimilar is not identical to the active substance of the reference product, the regulatory requirements for approval of generics are inadequate to demonstrate the quality, efficacy, and safety of biosimilars [4, 5].
Importantly, manufacturers of biosimilars do only have access to the finished reference product and have to conduct and establish their own strategies to develop the respective similar drug. Even minor changes to the original manufacturing process may result in alterations to the secondary, tertiary, and quaternary structure of the molecule, which may have an impact on the efficacy and safety of the drug. Thus, the demonstration of high similarity to the reference product regarding quality, safety and efficacy is necessary and has to be demonstrated using a set of comprehensive comparability exercises. Follow-on biologic products not authorized according to these standards of comparability cannot be referred to as biosimilars [6].
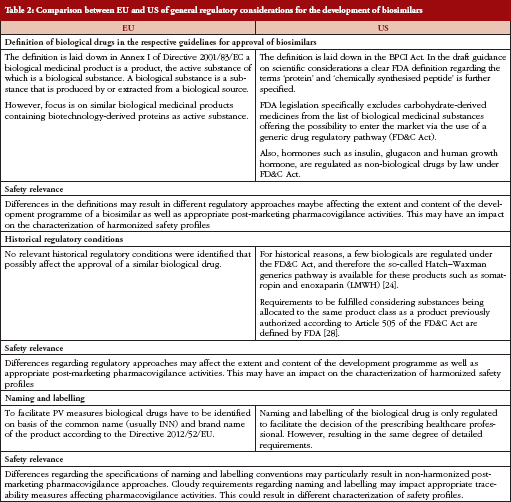
Since it is assumed that potential differences might lie not only in the current biosimilar guidelines on non-clinical and clinical aspects, differences in the definitions of biological products between EMA and FDA are outlined prior to the comparison of documents, see Table 1.
As most biological medicinal products, including biosimilars, are intended for long-term use to treat chronic diseases, safety concerns are likely to arise outside the time course of controlled clinical trials. A study of safety-related regulatory issues of biologicals approved in the EU and US investigated 174 approved products and identified 82 safety-related regulatory actions concerning 41 products [7].
Biologicals developed first had a higher risk for safety related regulatory actions compared with later approved products of the same class, indicating that the experience gained over time reduced safety concerns.
The most important general safety concern of biosimilars and follow-on biologics is their potential to trigger immunogenic responses in humans [8–11]. Therefore, the immunogenicity issue has become a focus area in their development and approval [12]. In the case of monoclonal antibodies (mAbs), these may cause safety issues, alter activity or have an impact on both parameters. For example, antibodies neutralizing endogenous erythropoietin (EPO) can result in a rare condition known as antibody-mediated pure red cell aplasia (PRCA) [8], which was observed inter alia after a manufacturing change initially considered to be minor to the originator product Eprex in 1998 [13].
Regarding biosimilar products currently approved by EMA, there are still some concerns in the long-term evaluation of these products, particularly the limited experience at the time of approval in terms of safety and their potential to trigger immunogenicity. Even if similar efficacy is proven in the comparability exercises conducted during biosimilar development, the safety profile might differ from that of the reference product. Therefore, the particular need for safety surveillance by rigorous pharmacovigilance (PV) is stressed also for biosimilars [8]. Only effective post-marketing PV including post-authorization safety studies (PASS) will provide sufficient evidence that a biosimilar is comparable to the reference product in terms of its safety profile [14]. Side effects related to use in daily practice, e.g. medication errors, off-label use, drug interactions or co-morbidities, can only be detected during post-approval exposure. A crucial point to consider in this context is the need for traceability of the products after launch and in daily practice [15].
The assessment of toxicity and safety of mAbs becomes more challenging due to the higher complexity and size of these products, giving rise to higher variability. The manufacture of biosimilar mAbs can lead to differences in glycosylation pattern, resulting in a high level of micro-heterogeneity. In addition, mAbs often have a complex mode of action as they comprise multifunctional molecules with biological properties involving both their Fab and Fc fragments. Many challenges are associated with the approval process of biosimilar mAbs [16].
Recently, EMA issued two guidelines dealing with non-clinical/clinical issues as well as with the immunogenicity assessment of biosimilar mAbs [17, 18]. In contrast, no specific requirements for the approval of biosimilar mAbs are addressed by FDA in their draft guidance documents so far. In June 2013, first approvals for biosimilar mABs have been granted in the EU for infliximab; however, it will be interesting to follow how future applications will be assessed by EMA and FDA (CHMP 2013 Summary of positive opinion for Remsima (infliximab). EMA/CHMP/363689/2013. released 27 June 2013; CHMP 2013 Summary of positive opinion for Inflectra (infliximab). EMA/CHMP/364710/2013. released 27 June 2013).
Overall, the regulatory requirements in both regions are comparable regarding their scientific content. However, the most obvious discrepancy is the structure and number of guidance documents. In contrast to the US, guidance documents in the EU are available for different product classes such as somatropins, EPO, granulocyte colony-stimulating factor (G-CSF) and insulin [5]. Content-related differences between the EU and US guidelines regarding distinct parameters with possible impact on toxicity and safety of biosimilar products are outlined below.
General requirements with a possible impact on toxicity and safety of biosimilars
General topics with a possible impact on toxicity and safety of the proposed biosimilar product include: the reference product; definitions of biological drugs and the resulting regulatory pathway; and naming and labelling.
Comparison of provisions determining the reference product.
In principle, only a reference product licensed in the EU or US, respectively, is accepted for any kind of testing during the approval process of a biosimilar drug. Furthermore, the same product has to be used throughout the entire development programme [12]. In certain cases, the use of non-EU or non-US reference products, respectively, is accepted for distinct non-clinical and clinical investigations and results may be bridged for final evaluation [19]. Under this approach, it is the applicant’s responsibility to establish that the batches sourced outside the European Economic Area (EEA) are representative of the reference medicinal product authorized in the EEA through an extensive analytical comparison.
According to the May 2013 EMA’s overarching guideline the use of foreign reference material to conduct the immunogenicity and clinical efficacy programme marketing is in principle possible in both regions which is considered a key advantage (CHMP 2013. Guideline on Similar Biological Medicinal Products. CHMP/437/04-Rev 1. Released 22 May 2013). This is considered by biosimilar developers as a key advantage since they can use the EU programme also for US filing [20]. However, FDA Q&A document (Q.1.8) states that ‘At this time, as a scientific matter, it is unlikely that clinical comparisons with a non-US licensed product would be an adequate basis to support the additional criteria required for a determination of interchangeability with the US licensed reference product’ [21]. In this context, interchangeability means that the biological product is biosimilar to the reference product, it can be expected to produce the same clinical result as the reference product in any given patient; and for a product administered more than once, the safety and reduced efficacy risks of alternating or switching are not greater than with repeated use of the reference product without alternating or switching [22]. This concept of interchangeability, however, is only applied in the US and not the EU [23]. Recently, it was reported that FDA accepts substitution of interchangeable biosimilars [24], while, for example, the Greek medicines agency released a document in March 2013 that it did not recommend automatic substitution or interchangeability of biological [25]. Automatic substitution will make post-marketing surveillance more difficult due to potential issues with traceability and identification of the products.
The reliable bridging of data derived from studies with a non-EU or non-US licensed product to the respective licensed reference product remains challenging. This has been followed in the May 2013 drafted revision of EMA’s Overarching guideline. As is stated in the response to Question 4 of the Q&A document of EMA [19], a close collaboration with FDA is foreseen in the upcoming revision of the biosimilar guideline CHMP/437/04 to ensure harmonization of requirements of reference products for the comparability exercise. It should be noted that bridging data between studies gathered by the use of different reference products may result in potential bias regarding safety data.
During the development programme, sponsors are encouraged to discuss with the authorities the adequacy of the scientific justification of such an approach, particularly the bridging to reference material authorized in another ICH region applying respective approval standards proving biosimilarity similar to that of the EU and the US [26].
Comparison of biological drug definitions for approval of biosimilars
In the US, for historical reasons and due to the definition of biological drugs, certain biologicals are regulated under the Federal Food, Drug, and Cosmetic (FD&C) Act and are therefore open to the so-called Hatch–Waxman generic drug pathway (The Hatch–Waxman Act, a 1984 amendment to the FD&C Act, created an abbreviated approval process for generic versions of conventional drugs while still protecting the patent rights of brand-name drug manufacturers). Such biological products include insulin, glucagon, somatropin, and low molecular weight heparins (LMWHs). Recently, FDA published requirements to be fulfilled by a biosimilar, thus offering the possibility of authorization by an abbreviated new drug application (ANDA) procedure [27].
In contrast, EU regulation does not allow for the use of classical generics application procedures for marketing authorization of biosimilars for these classes of biologicals [28].
Since there are different requirements to be fulfilled when the same drug is approved in either the US or the EU, e.g. the extent of non-clinical and clinical testing, the characterization of the safety profile of biosimilars may be affected. This applies, for example, to the product class of LMWHs. These are classified as generic drugs in the US and therefore become approved as generics under the FD&C Act using an ANDA [29]. The generic drug approval procedure in the US results in a less extensive development programme omitting studies evaluating non-clinical and clinical safety and efficacy. Only bioequivalence to the originator product has to be proven in company-sponsored studies [30]. In contrast, LMWHs are classified as biosimilars in the EU. Since heparins are derived from biological source material and are labelled according to potency rather than according to weight, these products are regulated as biologicals rather than generics. Consequently, mere physico-chemical similarity between a biosimilar such as LMWH and the originator product will be insufficient for a successful licensing application in the EU [31] and additional clinical studies assessing comparability with respect to pharmacokinetics (PK), efficacy and safety are required [32, 33]. Similarly, while insulins, glugacon, and human growth hormone (somatropin) are regulated under the FD&C Act (generics procedure) in the US, they are treated as biologicals or biosimilars in the EU.
In conclusion, when comparing the different regulatory approaches for distinct biological product classes in the EU and US, there is an obvious need for global harmonization of the available regulatory pathways.
Comparison of guidelines for biosimilar labelling
The impact of naming and labelling on the safety of already approved biosimilar products has been widely discussed because biosimilars are marketed under the same International Nonproprietary Names (INN) as the reference product. In this regard, it was outlined by the European Biopharmaceutical Enterprises (EBE) and the European Federation of Pharmaceutical Industries and Associations (EFPIA) that, for example, the labelling for Omnitrope is effectively indistinguishable to that of its reference product (Genotropin). It is not clearly highlighted that Omnitrope is a biosimilar, or which data quoted in the labelling were generated with Omnitrope and which data with Genotropin. EBE and EFPIA strongly recommended a consistent approach concerning the labelling of future biosimilars by providing the respective information about the origin of data in sections 4.8 and 5.1 of the product’s Summary of Product Characteristics [34].
The European Commission issued a directive by end of 2012 requiring biological products to be identified by brand name and not by INN [35]. Measures are described which should facilitate the recognition of medical prescriptions of biologicals and, thus, also of biosimilars issued in another Member State. However, FDA is less precise in this context only saying that naming and labelling of the drug should facilitate decision making by the prescribing healthcare professional [22].
Therefore, biologicals should be marketed with the brand name or at least with the INN together with the manufacturer’s name for a definite identification of the product by prescribers [35]. Unique, dissimilar names can help to prevent prescription mix-ups and can support reporting and tracking of adverse events. Identical or similar names may contribute to prescription mix-ups and complicated post-marketing surveillance [36].
Comparison of non-clinical testing strategies
There is no substantial difference in the general non-clinical approach between the EU and the US regarding the scientific content. Both regions require an extensive set of in vitro data which in principle are comparable. In the US draft guidances, residual uncertainty after every single study milestone is addressed within the scope of the next studies to be conducted. However, individual testing strategies are likewise applied in the EU approach providing product-class specific guidelines.
In the EU and US, in vivo studies are usually required in relevant species based on state-of-the-art technology. However, slight differences exist concerning the structural approach. In the US, some of the non-clinical tests are described in the quality guideline [37]. The step-wise procedure of the so-called ‘totality of the evidence’ approach is described in more detail in the US draft guidance documents compared with EMA’s general guideline on non-clinical and clinical issues, particularly regarding structural characterization of the product [12, 22]. Furthermore, consistent with the recommendations of EMA, non-clinical safety pharmacology, reproductive and developmental toxicity as well as carcinogenicity studies are not warranted in the US when the proposed product and reference product have been demonstrated to be highly similar by extensive structural and functional characterisation. In August 2012, FDA proposed a risk-based approach stating that animal repeated-dose toxicity studies may not be required, and may only be useful if safety uncertainties remain before first-in-man studies [38].
Both EMA and FDA agree that animal immunogenicity studies do not predict potential immunogenic responses to protein products in humans. However, if differences in manufacturing, e.g. the presence of impurities or excipients, between the proposed product and the reference product result in altered immunogenicity, measurement of anti-protein antibody responses in animals may provide useful information relevant to patient safety [26]. Overall, a step-wise testing approach before entering the clinical phase is required in both the US and EU.
Comparison of clinical testing strategies
Both, the EU guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues (EMEA/CHMP/42832/05) and the US draft guidance for industry, scientific considerations in demonstrating biosimilarity to a reference product [26], propose in general that the clinical comparability exercise is a stepwise procedure that should start with PK and pharmacodynamics (PD) studies followed by clinical efficacy and safety trials or, in certain cases, PK/PD studies. Both regions recommend selecting PD parameters on the basis of their relevance to demonstrate therapeutic efficacy of the product. Although reference-controlled clinical trials are usually required to demonstrate clinical comparability of safety, in certain cases PK/PD studies may be sufficient. For this purpose, there must be a high level of physico-chemical characterization of the biosimilar product in comparison with all the information collected with the originator during the development process or through post-marketing experience. If there are several potential indications, the most sensitive disease model to detect differences should be chosen in a homogeneous patient population. If efficacy and safety has been shown in this indication, the extrapolation to other indications is possible under certain circumstances [12, 26].
Overall, FDA and EMA recommendations for clinical testing of PK/PD and efficacy trials are highly similar.
Comparison of clinical safety and pharmacovigilance requirements
In terms of clinical safety and PV requirements there are no significant differences between FDA and EMA recommendations. In principle, study populations should be of sufficient size to detect possible safety concerns. Usually it is recommended to evaluate safety data within the scope of combined safety and efficacy trials. However, since the need for such clinical trials depends on the extent and relevance of data derived from non-clinical testing and PK/PD studies in humans, and particularly on the relevance of PD markers used, the size of the safety population to be evaluated may vary. Even if extensive phase III clinical trials are performed, the study population will be too small to allow the detection of rare side effects. Therefore, both FDA and EMA focus on post-marketing safety programmes according to the respective PV laws applicable in either region. To detect possible differences between reference and biosimilar products, it is crucially important to compare the frequency and severity of known side effects of the reference product with any differences or new side effects not yet observed with the reference product [12, 26].
Comparison of immunogenicity assessment
Both authorities, FDA and EMA, explicitly state the importance of the assessment of clinical immunogenicity in both pre- and post-approval settings. FDA and EMA agree that non-clinical data are not able to predict the immunogenic potential of a molecule. To date, current guidance documents focus on chronic administration in humans with a minimum follow-up period of one year being expected in both regions [12, 26]. However, FDA explains in more detail that the requirements depend on the extent and period of administration. EMA also addressed this aspect in a concept paper regarding non-clinical and clinical issues of biosimilars [39].
The regulatory approaches differ regarding extrapolation of immunogenicity data to other indications. While EMA clearly states that immunogenicity should be assessed for each indication separately [12], FDA in principle provides the possibility of extrapolating data to other indications [26]. However, caution is advised regarding the extrapolation of data to other indications and the applicant is recommended to discuss these aspects with the agency [20].
The possibility of extrapolating immunogenicity data depends on several factors, including understanding the mode of action, experience with the reference product, the extent of characterization of the reference and proposed product, and the amount and relevance of data collected so far. In principle, the most sensitive disease model with regard to immunogenicity should be used [26], and extrapolation is only possible from high-risk to low-risk patient populations and clinical settings [40]. However, testing for only one indication may not be sufficient to adequately explore the immunogenic potential of a biosimilar drug throughout several indications. This issue is of particular importance as biosimilar mAbs are already in the development pipeline, making the immunogenicity issue even more challenging [18, 41].
While the possibility of altered efficacy and safety of a product is outlined by both EMA and FDA, EMA provides examples of reactions possibly related to immunogenicity that should be closely monitored, such as hypersensitivity, infusion reactions, autoimmunity and loss of efficacy. Both agencies recommend defining certain PV measures depending on the specific characteristics of the product.
EMA was the first competent authority to issue guidelines for biosimilar approval in 2005 [2]. In February 2012, FDA issued draft guidance documents regarding the approval of biosimilars; however, no biosimilar drug has yet reached approval in the US. By March 2013, FDA’s drugs centre had received 51 requests for pre-Investigational New Drug (IND) meetings covering 12 different biological reference products. This is interesting to compare with Europe, where to date 14 biosimilars have been approved covering only three reference products [42].
The purpose of this review article was to compare EU with US regulatory approaches to biosimilar approval, evaluating the potential extent to which the safety profile of a proposed biosimilar product is characterized. Major issues comparing regulatory approaches with regard to a harmonized characterization of safety profiles of biosimilars in the two regions of interest are displayed in Table 2.
Overall, the requirements are similar; however, the language is different [20]. This fact makes it more complicated to identify relevant differences with respect to potential safety issues.
Despite differences in the structural approach and the language chosen, major differences possibly affecting understanding of the safety profile of the proposed products concern the general requirements, i.e. the choice of the reference product, the definition of biologicals and the resulting regulatory pathways, and to some extent the naming and labelling requirements. Regarding non-clinical and clinical testing strategies, substantial differences were identified concerning the extrapolation of immunogenicity data from one indication to another, which is precluded in the EU and in principle allowed in the US.
The naming and labelling of biosimilars is a topic under continuous discussion. With the new EC Directive 2012/52/EU, consensus was found in the EU regarding the naming of biologicals by brand name and not by INN. In contrast, until now no precise requirements are scheduled in the US. For PV in a global setting, harmonization in naming is crucial as different naming systems in different countries, particularly outside the scope of the EC directive and FDA regulations, may hamper clear data analysis for a specific biosimilar product.
Physicians need to be informed about the scientific principles of the biosimilar approval. In this context, there is no reason to assume that biosimilars would behave differently from the reference product in clinical practice if the respective regulatory and scientific principles have been properly applied [40]. In addition, according to a review of an extensive set of clinical trials and post-authorization safety data, no specific safety concerns have yet been identified for switching from one authorized biological to another for the same indication [43, 44]. However, since many safety-related issues of biological drugs only occur in very rare situations, which often relate to long-term use, more experience is needed with distinct product classes.
To help manufacturers provide reliable efficacy and safety data for their biosimilar products in the most cost-effective way, and in order to give physicians and patients’ confidence in globally comparable safety profiles, harmonization regarding these topics is highly desirable.
Competing interests: The authors are employees of PharmaLex GmbH. The authors declare that there are no conflicts of interest that are directly relevant to the content of this manuscript. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Provenance and peer review: Not commissioned, externally peer reviewed.
Tobias Blank, PhD
Tilo Netzer, PhD
Wolfram Hildebrandt, MSc, MA
Angela Vogt-Eisele, PhD
Professor Marietta Kaszkin-Bettag, PhD
PharmaLex GmbH,13-15 Joseph-Meyer-Straße, DE-68167 Mannheim, Germany
References
1. GaBI Online – Generics and Biosimilars Initiative. US$67 billion worth of biosimilar patents expiring before 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Jul 16]. Available from: http://www.gabionline.net/Biosimilars/General/US-67-billion-worth-of-biosimilar-patents-expiring-before-2020
2. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04. 2005 [homepage on the Internet]. 2005 [cited 2013 Jul 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
3. World Health Organization. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs); Geneva, Switzerland [homepage on the Internet]. 2009 [cited 2013 Jul 16]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
4. Declerck PJ. Biologicals and biosimilars: a review of the science and its implications. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;
1(1):13-6. doi:10.5639/gabij.2012.0101.005
5. Kaszkin-Bettag M. Regulatory Requirements and Challenges for Biosimilars in Europe. In: Bingenheimer N, editor. Life Science in the Capital Market, Biosimilars: Deutsche Vereinigung für Finanzanalyse und Asset Management (DVFA e.V.); 2010. p. 13-7.
6. Wang J, Chow S-C. On the regulatory approval pathway of biosimilar products. Pharmaceuticals. 2012;5(4):353-68.
7. Giezen TJ, Mantel-Teeuwisse AK, Straus SM, Schellekens H, Leufkens HG, Egberts AC. Safety-related regulatory actions for biologicals approved in the United States and the European Union. Jama. 2008;300(16):1887-96.
8. Schellekens H. Biosimilar therapeutics-what do we need to consider? NDT Plus. 2009;2(Suppl_1):i27-i36.
9. Hincal F. An introduction to safety issues in biosimilars/follow-on biopharmaceuticals. J Med CBR Def. 2009;7.
10. Lekkerkerker F. Are there safety concerns for biosimilars? Int J Risk Saf Med. 2009;21(1-2):47-52.
11. Locatelli F, Roger S. Comparative testing and pharmacovigilance of biosimilars. Nephrol Dial Transplant. 2006;21 Suppl 5:v13-6.
12. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 [homepage on the Internet]. 2006 [cited 2013 Jul 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
13. Schellekens H. Immunologic mechanisms of EPO-associated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18(3):473-80.
14. Gascon P. Presently available biosimilars in hematology-oncology: G-CSF. Target Oncol. 2012;7 Suppl 1:S29-S34.
15. Zuniga L, Calvo B. Biosimilars: pharmacovigilance and risk management. Pharmacoepidemiol Drug Saf. 2010;19(7):661-9.
16. Jimenez A, Brake B. Biosimilars in the European Union – regulatory perspectives; ICH GCG ASEAN Training Workshop on ICH Q5C; 30-31 May 2011; Kuala Lumpur: European Medicines Agency – Bfarm (German Federal Institute for Drugs and Medical Devices); 2011.
17. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. EMA/CHMP/BMWP/403543/2010 [homepage on the Internet]. 2012 [cited 2013 Jul 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
18. European Medicines Agency. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. EMA/CHMP/BMWP/86289/2010 [homepage on the Internet]. 2012 [cited 2013 Jul 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf
19. European Medicines Agency. Similar-biological-medicine applications: questions and answers [homepage on the Internet]. 2013 [cited 2013 Jul 16]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl pages/regulation/general/general_content_000529.jsp&mid WC0b01ac0580533e0b
20. Gal A, Sonnenfeld A, Stephano P. Biosimilars: review of FDA guidance documents; generally supportive of biosimilar industry development. Bernstein Research. 2012.
21. US Food and Drug Administration. Guidance for industry. Biosimilars: questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009 [homepage on the Internet]. 2012 [cited 2013 Jul 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM273001.pdf
22. US Food and Drug Administration. Biologics Price Competition and Innovation Act (BPCI Act) [homepage on the Internet]. 2010 [cited 2013 Jul 16]. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/Therapeutic
BiologicApplications/Biosimilars
23. Chow SC, Ju C. Assessing biosimilarity and interchangeability of biosimilar products under the Biologics Price Competition and Innovation Act. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1):20-5. doi:10.5639/gabij.2013.0201.004
24. GaBI Online – Generics and Biosimilars Initiative. US FDA defends biosimilar substitution [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Jul 16]. Available from: www://gabionline.net/Biosimilars/General/US-FDA-defends-biosimilar-substitution
25. GaBI Online – Generics and Biosimilars Initiative. Greece says no to automatic substitution of biological [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Jul 16]. Available from: www.gabionline.net/Biosimilars/News/Greece-says-no-to-automatic-substitution-of-biologicals
26. US Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product [homepage on the Internet]. 2012 [cited 2013 Jul 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
27. US Food and Drug Administration. Guidance for industry on biosimilars: Q & As regarding implementation of the BPCI Act of 2009 [homepage on the Internet]. Questions and Answers Part II. 2012 [cited 2013 Jul 16]. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm259797.htm
28. McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3(2):209-17.
29. Ofosu FA. A review of the two major regulatory pathways for non-proprietary low-molecular-weight heparins. Thromb Haemost. 2012;107(2):201-14.
30. US Food and Drug Administration. Abbreviated New Drug Application (ANDA): Generics [homepage on the Internet]. 2012 [cited 2013 Jul 16]. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNew
DrugApplicationANDAGenerics
31. Gray E, Mulloy B. Biosimilar low molecular weight heparin products. J Thromb Haemost. 2009;7(7):1218-21.
32. Kalodiki E, Fareed J. New and generic anticoagulants and biosimilars: safety considerations. Clin Appl Thromb Hemost. 2011;17(2):136-9.
33. Walenga JM, Jackson CM, Kessler CM. Low molecular weight heparins differ substantially: impact on developing biosimilar drugs. Semin Thromb Hemost. 2011;37(3):322-7.
34. European Biopharmaceutical Enterprises (EBE), European Federation of Pharmaceutical Industries and Associations (EFPIA). EBE–EFPIA Position paper: labelling of biosimilar medicinal products 2007 [homepage on the Internet]. 2008 [cited 2013 Jul 16]. Available from: http://www.biofarmacos.org/files/contenido/Ebe-Efpia_Biosimilars_labelling_position_paper_28_mar_07.pdf
35. European Commission. Directive 2012/52/EU laying down measures to facilitate the recognition of medical prescriptions issued in another Member State [homepage on the Internet]. 20 December 2012 [cited 2013 Jul 16]. Available from: http://ec.europa.eu/health/cross_border_care/docs/impl_directive_presciptions_2012_en.pdf
36. Rader RA. Nomenclature of new biosimilars will be highly controversial. Bioprocess International. 2011;9(6):26-33.
37. US Food and Drug Administration. Guidance for industry: quality considerations in demonstrating biosimilarity to a reference protein product [homepage on the Internet]. 2012 [cited 2013 Jul 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
38. Kozlowski S. Biosimilars – An update focused on quality considerations. 2012 Aug 8 [cited 2013 Jul 16]. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM315764.pdf
39. European Medicines Agency. Concept paper. Guideline on the revision of the guideline on similar biological medicinal products containing biotechnology derived proteins as active substance: non-clinical and clinical issues. (EMA/CHMP/BMWP/572828/2011) [homepage on the Internet]. 2011 [cited 2013 Jul 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2011/09/WC500112835.pdf
40. Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, et al. Biosimilars: what clinicians should know. Blood. 2012:120(26):5111-7.
41. Greer F. Biosimilar development: from science to market. SGS, Life science, Technical bulletin. 2011;(47).
42. GaBI Online – Generics and Biosimilars Initiative. Biotech firms try to limit biosimilar substitution in US [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Jul 16]. Available from: www.gabionline.net/Biosimilars/News/Biotech-firms-try-to-limit-biosimilar-substitution-in-US
43. Ebbers HC, Mantel-Teeuwisse AK, Sayed-Tabatabaei FA, Moors EH, Schellekens H, Leufkens HG. The role of Periodic Safety Update Reports in the safety management of biopharmaceuticals. Eur J Clin Pharmacol. 2013;69(2):217-26.
44. Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12(11):1473-85.
|
Author for correspondence: Professor Marietta Kaszkin-Bettag, PhD, Head of Preclinical and Medical Aff airs, PharmaLex GmbH, 13-15 Joseph-Meyer-Straße, DE-68167 Mannheim, Germany |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/safety-and-toxicity-of-biosimilars-eu-versus-us-regulation.html
Copyright ©2025 GaBI Journal unless otherwise noted.