Biologicals and biosimilars: a review of the science and its implications
Published on 2012/02/21
Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(1):13-6.
| Abstract: Biopharmaceuticals are medicines whose active drug substance is made by living cells. Copies of these drugs, called biosimilars, are not identical to their reference drug and therefore specific regulatory requirements for registration apply. Whereas pharmaceutical quality evaluation requires a full dossier and a detailed comparative analysis to the reference drug, non-clinical and clinical requirements are much less extensive compared to the requirements for an innovator. Limited clinical experience and their complex nature exclude biosimilars from being considered interchangeable with the reference drug.. |
Submitted: 20 April 2011; Revised manuscript received: 22 September 2011; Accepted: 14 October 2011
Introduction
Biopharmaceuticals, also called ‘biological medicinal products’ or ‘biological medicines’, are medicines whose active drug substance is made by a living organism or derived from a living organism by means of recombinant DNA and/or controlled gene expression methods. These products are polypeptides, (glyco-)proteins, and/or nucleic acids and their molecular characteristics are much more complex than traditional chemical drugs.The final biopharmaceutical product is influenced by many variables, such as the type of expression system, e.g. bacteria, yeast, and mammalian cells; the growth conditions, the purification process, the actual formulation and the conditions during storage and transport. Post-translational modifications occur during cellular synthesis, such as glycosylation, phosphorylation, sulphation, methylation, acetylation and hydroxylation which may affect biological activity and which results in an intrinsic molecular heterogeneity. It can be calculated, theoretically, that these modifications may result in more than one million product-related variants. Since this structural variability is substantial and can be very subtle, the currently available analytical techniques are insufficient to fully characterise the end product. In contrast, ‘traditional low molecular weight chemical drugs’ are produced by well-controlled and highly reproducible chemical reactions and are molecules with a small, well-defined and stable chemical structure, which can be completely characterised by analytical methods.The heterogeneity of biopharmaceuticals is further increased by the fact that these products are quite sensitive to ‘external’ conditions. The latter can affect product integrity and stability, leading, for example, to varying degrees of peptide denaturation, aggregation, oxidation, and degradation. Such modifications are less likely to occur in traditional non-biopharmaceutical drugs because they are much smaller and can be better controlled and are more predictable by nature [1-3].Importantly, and in contrast to traditional chemical drugs, biopharmaceuticals are potentially immunogenic. In this respect it is important to note that subtle structural differences, for example, consequent to small differences in the number and type of product variants, may significantly affect the immunogenic potential of the drug product [4-6]. Additionally, product- or process-related impurities can provoke an immune response [2, 7].The main differences between low molecular weight (chemical) drugs and biological drugs are summarised in Table 1.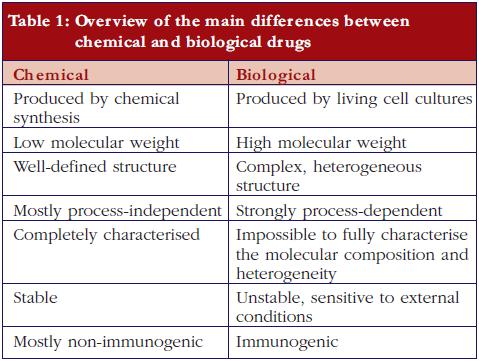
Biosimilars
After the expiration of patent(s) for the first approved biopharmaceuticals, ‘copying’ and marketing of these biological substances can be offered by other biotech companies and might possibly, as with generics, reduce cost to patients and social security systems. However, biopharmaceuticals are made by living cells. Because of their intrinsic complexity and because no two cell lines, developed independently, can be considered identical, biopharmaceuticals cannot be fully copied. This is recognised by the European regulatory authorities and has resulted in the establishment of the term ‘biosimilar’ in recognition of the fact that, whilst biosimilar products are similar to the original product, they are not exactly the same [8, 9].European legislation has included specific guidelines for the approval of biosimilars since 2005. The Australian Therapeutic Goods Administration has adopted the European guidelines. Canadian health authorities have recently published a guidance document for approval of biosimilars, mainly based on the European guidelines [10]. Adaptation of US legislation concerning biosimilar approval processes is still under development. In March 2010, the Public Health Service Act was amended to create an abbreviated approval pathway for biosimilars [11]. Also WHO has issued guidelines for the evaluation of biosimilars [12].Thus a biosimilar, sometimes called ‘similar biological medicinal product’ or ‘follow-on biologic’ (Japan and USA) or ‘subsequent entry biologic’ (Canada), is a medicine that is similar to a biopharmaceutical that has already been authorised (the ‘reference product’). Since the active substance of a biosimilar is similar but not identical to the active substance of the reference product the regulatory requirements for approval of generics are inadequate to demonstrate the quality, efficacy, and safety of biosimilars.For a generic low molecular weight chemical drug, it is sufficient to demonstrate comparable quality, for example, content and purity, and a comparable clinical pharmacokinetic (PK) profile, i.e. relative bioavailability/bioequivalence, with a reference (innovator) product to obtain regulatory approval [13]. For biosimilars, EMA not only requires comparative quality and clinical PK studies, but also non-clinical studies, clinical pharmacodynamic (PD) studies, and limited toxicology studies, as well as comparative clinical efficacy and tolerability studies [8, 14, 15]. However, non-clinical PKs, safety profile, reproduction toxicology, mutagenicity and carcinogenicity studies are not mandatory for approval of a biosimilar, in contrast to what is required for reference biopharmaceuticals. A comparison of the European regulatory requirements for a marketing authorisation application of a biosimilar versus a reference is shown in Table 2.The guidelines for quality requirements for a biosimilar product claiming to be similar to an approved and marketed biopharmaceutical product published by the Committee for Medicinal Products for Human Use (CHMP) [14] states that the active substance in the biosimilar should be similar to the one in the reference product. Demonstration of similarity requires comparability exercises versus the chosen reference product. This implies the use of appropriately selected state-of-the-art analytical methods that are able to detect ‘slight differences relevant for quality evaluation.’ These comparability exercises also include the comparative evaluation of physicochemical parameters, biological activity using relevant bioassays and a qualitative and quantitative comparative assessment of purity and impurity profiles. The guideline also indicates that ‘… it is not expected that the quality attributes in the similar biological and reference medicinal products will be identical …’. Indeed, the quality attributes of the final biological product inherently vary with the type of host cell, the growth conditions, the purification process, the formulation, and the storage conditions. However, the CHMP requires that any difference in the quality attributes between the biosimilar and its reference product, such as variability in post-translational modifications or differences in impurity profiles should be justified in relation to its potential impact on efficacy and tolerability. The existence of differences in quality attributes between a biosimilar and the reference product is reported in the public assessment reports (EPAR) (visit www.ema.europa.eu) made available upon approval of the biosimilar. For example, for a biosimilar of epoetin alfa, differences are reported with respect to glycosylation (higher levels of phosphorylated high-mannose-type structures, lower levels of N-glycolylneuraminic acid and diacetylated neuramic acids) and oxidation (lower levels of the oxidised variant). For a biosimilar of somatropin, differences in impurities are reported as well as a higher level of deamidated variants.The general non-clinical and clinical requirements for a biological medicinal product claiming to be similar to an approved biopharmaceutical were also published by the CHMP in 2006 [15]. These are much less comprehensive compared to the requirements for an innovator, see Table 2, and evaluation is mainly based upon data obtained by comparative studies—biosimilar versus reference. In addition to the general non-clinical and clinical guidelines, product class-specific annexes to these guidelines have also been adopted for biosimilars containing recombinant interferon alfa, recombinant granulocyte-colony stimulating factor, recombinant somatropin, recombinant insulin, low molecular weight heparins, and recombinant erythropoeitins as the active substance. Draft guidelines for biosimilars containing monoclonal antibodies [16], as well as concept papers for biosimilars containing recombinant interferon beta [17] and recombinant follitropin [18] have been released for consultation.In general, non-clinical comparative tests should comprise in vitro studies, e.g. receptor-binding studies or cell-based assays, as well asin vivo PD studies. In addition, given the immunogenic potential of biopharmaceuticals, at least one repeat dose toxicity study should be performed including toxicokinetic measurements such as determination of antibody titres, cross-reactivity and neutralising capacity[6]. Such tests are particularly useful to detect the presence of hostcell proteins and/or impurities in the product. However, in contrast to the reference products, the approval process of biosimilars does not require safety pharmacology, reproduction toxicology, mutagenicity and carcinogenicity studies, see Table 2. Clinical studies required for regulatory approval of a biosimilar should include comparative PK and PD studies in healthy volunteers, followed by comparative efficacy and tolerability trials. The guideline states specifically, ‘… the clinical comparability exercise is a stepwise procedure that should begin with PK and PD studies followed by clinical efficacy and safety trial(s) or, in certain cases, PK/PD studies for demonstrating clinical comparability.’ The latter studies are usually performed only in the most sensitive and most relevant target patient population(s). In some cases, PK/PD studies alone might be considered sufficient [15] and in other cases, for example, where it is assumed that the mechanism of action of the drug is only dependent on its interaction with one single binding partner as the target, therapeutic similarity demonstrated in one indication may be extrapolated to other indications of the reference product. Extrapolation, however, remains a matter of debate especially when different indications imply the use of significant different doses [19], or different patient populations, for example, children versus adults, or when extrapolation to use in healthy individuals is concerned, for example, use of filgrastim for stem cell mobilisation and collection in healthy donors.On the other hand, the guidelines put special emphasis on assessment of the clinical tolerability of a biosimilar because of its potential immunogenicity. Indeed, differences in quality attributes between the biosimilar and its reference product cannot always be detected during the quality control process, and their clinical consequences cannot always be predicted from non-clinical animal studies [6]. Hence, clinical trials that extensively evaluate the tolerability and immunogenicity of the biosimilar are indispensable. These assessments require optimal antibody testing, characterisation of the observed immune response, and evaluation of the correlation between antibodies and their effects on PKs, PDs, efficacy and tolerability. It is also important to realise that for one product the risk of immunogenicity may differ depending on the therapeutic indication. In most cases, pre-approval data over at least a six-month period are requested and a post-approval commitment to provide data up to 12 months. Since immunogenicity is a long-term event, gathering of immunogenicity data after marketing authorisation remains an important prerequisite. Consequently, within the authorisation procedure, the company applying for regulatory approval of a biosimilar, as for any newly approved biopharmaceutical, should also provide plans for post-marketing surveillance including a risk management programme.
Clinical studies required for regulatory approval of a biosimilar should include comparative PK and PD studies in healthy volunteers, followed by comparative efficacy and tolerability trials. The guideline states specifically, ‘… the clinical comparability exercise is a stepwise procedure that should begin with PK and PD studies followed by clinical efficacy and safety trial(s) or, in certain cases, PK/PD studies for demonstrating clinical comparability.’ The latter studies are usually performed only in the most sensitive and most relevant target patient population(s). In some cases, PK/PD studies alone might be considered sufficient [15] and in other cases, for example, where it is assumed that the mechanism of action of the drug is only dependent on its interaction with one single binding partner as the target, therapeutic similarity demonstrated in one indication may be extrapolated to other indications of the reference product. Extrapolation, however, remains a matter of debate especially when different indications imply the use of significant different doses [19], or different patient populations, for example, children versus adults, or when extrapolation to use in healthy individuals is concerned, for example, use of filgrastim for stem cell mobilisation and collection in healthy donors.On the other hand, the guidelines put special emphasis on assessment of the clinical tolerability of a biosimilar because of its potential immunogenicity. Indeed, differences in quality attributes between the biosimilar and its reference product cannot always be detected during the quality control process, and their clinical consequences cannot always be predicted from non-clinical animal studies [6]. Hence, clinical trials that extensively evaluate the tolerability and immunogenicity of the biosimilar are indispensable. These assessments require optimal antibody testing, characterisation of the observed immune response, and evaluation of the correlation between antibodies and their effects on PKs, PDs, efficacy and tolerability. It is also important to realise that for one product the risk of immunogenicity may differ depending on the therapeutic indication. In most cases, pre-approval data over at least a six-month period are requested and a post-approval commitment to provide data up to 12 months. Since immunogenicity is a long-term event, gathering of immunogenicity data after marketing authorisation remains an important prerequisite. Consequently, within the authorisation procedure, the company applying for regulatory approval of a biosimilar, as for any newly approved biopharmaceutical, should also provide plans for post-marketing surveillance including a risk management programme.
Being biosimilar is not equal to being interchangeable
In general, when copies of chemical drugs (generics) have been approved, approval has been based on demonstrated bioequivalence compared to the reference product. Having an identical structure and a proven bio-equivalence implies that the generics and reference product, as well as any two generics, are interchangeable. For biopharmaceuticals, however, the situation is completely different since two independently developed biopharmaceuticals demonstrated to be bio-equivalent will not have identical pharmaceutical quality attributes and therefore cannot be considered interchangeable in the absence of evidence gathered from adequately designed clinical studies. Indeed, potential differences in immunogenicity can only be observed in large study populations and switching between biological preparations from different origins may increase the risk of antibody development. On the other hand, it should also be realised that, in contrast to various generics of the same reference product which can be considered identical, two biosimilars, independently developed and compared to the same reference product cannot be considered biosimilar to each other. It is obvious that demonstration of similarity between biosimilar A and reference product on the one hand and between biosimilar B and reference product on the other hand does not allow any conclusions with respect to a possible similarity between the two biosimilars, i.e. the degree of similarity between A and B. Thus, from a scientific point of view as well as for the sake of patient safety, biopharmaceuticals, irrespective of their regulatory status as biosimilar or reference, should not be considered interchangeable in the absence of solid clinical data. This is also enforced in the new US Health Care Reform Bill, which clearly states that more data are required for a product to be labelled interchangeable rather than the mere fact of being biosimilar [20]. It must be stressed that if interchangeability has been proven between two biopharmaceuticals, e.g. between two biosimilars or between a biosimilar and its reference, this remains strictly valid only for the two specific products that were evaluated.It may also be of interest to note that, in this context, it is often claimed that approved manufacturing changes form the proof for a generalisation of interchangeability for biosimilars. For a variety of reasons this argument contains some major flaws. Firstly, all independently developed biosimilars have so far been proven to be different from their reference product with respect to particular quality aspects. In contrast, in most cases, manufacturing changes are accompanied by the demonstration that the majority, if not all, quality attributes remain within preset specification limits. In cases where manufacturing changes would have resulted in a significant qualitatively different composition, demonstration of clinical safety will be required [21]. Secondly, any company producing a biopharmaceutical must handle hundreds of various (process-)specific quantitative and qualitative criteria that need to be taken into account in the comparison of the products obtained before and after the manufacturing change. Therefore, a comparison (‘before’ versus ‘after’) can be made at various stages in the process. This is in contrast to the comparability exercises for biosimilars which only involve the end product. Thirdly, introduction of a manufacturing change will, at most result in only one switch for the patient in only one direction, i.e. a drug produced by the ‘old process’ to a drug produced by the ‘new process’, not vice versa. Fourthly, approval of a manufacturing change should not be interpreted as a proof that both versions can be safely switched back and forward. Taken together, even though it needs to be realised that some manufacturing changes could result in safety concerns, the putative safety risk associated with a manufacturing change can, in general, be considered a few orders of magnitude smaller compared to that associated with the differences between two independently developed biopharmaceuticals [22]. It should be stressed that a risk assessment for any biopharmaceutical always needs to be considered on a case-by-case basis.
Conclusion
The production of biopharmaceuticals involves complex processes and includes the development of an engineered cell line, the production of the active substance through large scale culturing of cells, the purification of the protein including a wide variety of downstream processing steps, and its formulation. Consequently, any two independently developed biopharmaceuticals starting from the same DNA sequence will be characterised by particular differences in composition.Approval of biosimilars is contingent upon a full and detailed demonstration of pharmaceutical quality, a comparative analysis with a reference product, limited non-clinical and clinical evaluations, and a post-approval follow-up. In the absence of specific data concerning interchangeability, any measures taken, e.g. by health insurance companies and/or reimbursement authorities, to control budgets by stimulating the use of less expensive biopharmaceuticals should contain a mechanism that prevents switching between products in a patient.For patientsBiopharmaceuticals are complex medicines produced by living cells. Copies of approved biopharmaceuticals have been introduced recently. Because of their intrinsic complexity such copies are similar but not identical to the reference medicine and are therefore called ‘biosimilars’. Approval of biosimilars requires a full quality analysis including a detailed comparison to the reference whereas non-clinical and clinical evaluations are less extensive. It is important to keep in mind that inherently related to the complex nature of biopharmaceuticals, similarity is not equal to interchangeability. Therefore, switching between similar biopharmaceuticals in a patient should be prevented.
References
1. Crommelin DJ, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm. 2003;266:3-16.
2. Crommelin D, Bermejo T, Bissig M, et al. Pharmaceutical evaluation of biosimilars: important differences from generic low-molecular-weight pharmaceuticals. Eur J Hosp Pharm Sci. 2005;11(1):11-7.
3. Sekhon BS, Saluja V. Biosimilars: an overview. Biosimilars. 2011;1:1-11.
4. Kessler M, Goldsmith D, Schellekens H. Immunogenicity of biopharmaceuticals. Nephrol Dial Transplant. 2006;21(5):9-12.
5. Sauerborn M, Brinks V, Jiskoot W, Schellekens H. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol Sci. 2010;31:53-9.
6. European Medicines Agency [homepage on the Internet]. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. 2007 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf
7. Schellekens H, Casadevall N. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol. 2004;251 Suppl 2:II4-9.
8. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products. 2006 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
9. Declerck PJ. Biotherapeutics in the era of biosimilars: what really matters is patient safety. Drug Saf. 2007;30:1087-92.
10. Health Canada [homepage on the Internet]. Guidance for sponsors: Information and submission requirements for subsequent entry biologics. 2010 [cited 2011 Dec 11]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/seb-pbu/notice-avis_seb-pbu_2010-eng.php
11. US legislation H.R. 1548: Pathway for Biosimilars Act. [cited 2011 Dec 11]. Available from: http://www.govtrack.us/congress/bill.xpd?bill=h111-1548
12. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 2010 [cited 2011 Dec 11]. Available from: http://www.who.int/ biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
13. Meredith PA. Potential concerns about generic substitution: bioequivalence versus therapeutic equivalence of different amlodipine salt forms. Curr Med Res Opin. 2009;25:2179-89.
14. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues. 2006 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003953.pdf
15. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2006 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
16. European Medicines Agency [homepage on the Internet]. Guideline on similar biological medicinal products containing monoclonal antibodies Draft. 2010 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/11/WC500099361.pdf
17. European Medicines Agency [homepage on the Internet]. Concept paper on similar biological product containing recombinant interferon beta. 2010 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089208.pdf
18. European Medicines Agency [homepage on the Internet]. Concept paper on similar biological medicinal products containing recombinant follicle stimulation hormone. 2010 [cited 2011 Dec 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089208.pdf
19. Declerck P, Darendeliler F, Goth M, Kolouskova S, Micle I, Noordam C, et al. Biosimilars: controversies as illustrated by rhGH. Curr Med Res Opin. 2010;26:1219-29.
20. Sensabaugh SM. Requirements for biosimilars and interchangeable biological drugs in the United States. Drug Inf J. 2011;45(2):155-62.
21. The international conference on harmonisation of technical requirements for registration of pharmaceuticals for human use 2004, ICH Harmonised tripartite guideline comparability of biotechnological/biological products subject to changes in their manufacturing process Q5E. [cited 2011 Dec 11]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf
22. Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2006;22:1383-91.
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
| Author: Professor Paul J Declerck, PhD, Laboratory for Pharmaceutical Biology, Faculty of Pharmaceutical Sciences, Katholieke Universiteit Leuven, Campus Gasthuisberg, O&N2, PO Box 824, 49 Herestraat, BE-3000 Leuven, Belgium |



venkata yeturu
I would like to get the PDF access
May 1, 2020 at 1:57 pm
would you please, send me pdf copy
thanks
May 11, 2020 at 1:33 am
its good article
May 11, 2020 at 8:14 am
Dear Dr Venkata Yeturu,
We very much appreciate your kind feedback. Article PDF is available on the website for free download.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office
March 10, 2021 at 1:58 pm
Dear Dr Hamdi,
We very much appreciate your kind feedback. Article PDF is available on the website for free download.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office
March 10, 2021 at 2:05 pm
Dear Raju,
We very much appreciate your kind feedback.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office