Transforming healthcare: CinnaGen’s leadership in follow-on biologicals/biosimilars development and market expansion
|
Abstract:
CinnaGen, the largest biopharmaceutical company in the MENA region, is a leader in developing follow-on biologicals/biosimilars. Dr Haleh Hamedifar, Chairperson of CinnaGen, spoke to GaBI (Generics and Biosimilars Initiative) about the company’s strategic focus, which includes expanding its product portfolio, entering highly regulated global markets, and advancing affordable treatments for conditions such as multiple sclerosis and immunological diseases—transforming health care in underserved regions..
|
Submitted: 27 August 2024; Revised: 23 October 2024; Accepted: 23 October 2024; Published online first: 7 November 2024
Introduction
Biosimilars have emerged as an important sector in medicine, improving global healthcare access in areas ranging from diabetes to cancer. They present one of the greatest opportunities to control drug costs, enhance patient care, and expand access to essential treatments.
CinnaGen, established in 1994, is the largest Iranian biopharmaceutical company and the biggest in the Middle East and North Africa (MENA) region. CinnaGen Group currently consists of eight companies that are active in different pharmaceutical areas including recombinant proteins, monoclonal antibodies, high-tech chemical drugs and niche dietary supplements, covering stages from early R & D to production, clinical studies, sales and marketing. CinnaGen Group entered the biosimilars field in 2006 when it manufactured its first follow-on biological (FOB)/biosimilar product in collaboration with the Fraunhofer Institute in Germany.
The company focuses its efforts on development of FOBs in the areas of immunological diseases, infertility, hormone disorders (endocrinology), haematology, neurology, oncology, ophthalmology and rare disease, in addition to providing contract research and development services.
In an interview on 27 August 2024, Dr Haleh Hamedifar, Chairperson of CinnaGen, discussed her views on FOBs and the company’s plans regarding them with GaBI (Generics and Biosimilars Initiative).
Clinical development programme for follow-on biologicals/biosimilars
A clinical development programme for biosimilars involves rigorous testing to demonstrate that a new biosimilar product is highly similar to an existing, approved biological in terms of safety, efficacy, and quality.
The company is actively engaged in clinical development for FOBs/biosimilars in the MENA region, India, Commonwealth of Independent States (CIS) region, and Europe. These efforts encompass several key drugs in the treatment areas of autoimmune diseases, blood disorder, and cancer, including efmoroctocog alfa, adalimumab, ocrelizumab, pembrolizumab, eptacog alfa, laronidase, see Table 1.
These clinical studies aim to demonstrate the safety and efficacy of the FOBs/biosimilars. Once approved, these products will offer patients more affordable treatment options. The focus of this clinical development programme is also on increasing the availability of FOBs/biosimilars in these regions, thereby improving patient access to essential therapies amidst economic challenges and the rising demand for biological treatments.
CinnaGen ensures the safety and efficacy of its FOBs/biosimilars by adhering to the European Medicines Agency (EMA) guidelines. The stepwise approach in developing the FOB/biosimilars includes analytical characterization to confirm similarity with the reference product, non-clinical studies to assess potential risks and side effects, clinical pharmacology studies to evaluate pharmacokinetics and pharmacodynamics, clinical trials to compare safety and efficacy, and post-marketing surveillance in real-world settings. Additionally, CinnaGen employs advanced technology in molecular biology, protein engineering, and process development to develop high-quality biosimilars.
Biopharmaceutical drug development and commercialization
Building on its clinical development success, CinnaGen has made significant progress in drug development, particularly for multiple sclerosis (MS) treatment. One of the company’s key achievements was the successful launch Xacrel, an FOB/biosimilar to Ocrevus (ocrelizumab), on 17 March 2021 in Iran. Xacrel is approved for various forms of MS, including relapsing-remitting disease, active secondary progressive disease, and primary progressive MS in adults.
The launch in Iran has been well received, with many MS patients switching to Xacrel, leading to an estimated exposure of 5,356 patients. To date, 43,320 vials of Xacrel have been sold in the MENA region, providing treatment for an estimated 7,220 patients. This success represents a more accessible treatment option for MS patients.
CinnaGen is actively seeking approval for Xacrel in other countries and expects to secure it within the next few years.
In a similar vein, CinnaGen achieved another milestone with the launch of its FOB for Aldurazyme (laronidase) on 13 February 2024, in Iran. This marks the first enzyme replacement therapy biosimilar developed in the MENA region. Laronidase is used to treat Mucopolysaccharidosis I (MPS I), targeting patients with Hurler and Hurler-Scheie forms of the disease, as well as those with the Scheie form who exhibit moderate to severe symptoms.
Long-term vision and a 10-year plan in the biopharma space
CinnaGen’s long-term vision focuses on expanding its global footprint and enhancing its product portfolio over the next five to 10 years. Central to this vision is entering highly regulated markets such as Australia, the European Union, and Japan, which will broaden the company’s patient base and increase its market presence. These markets offer substantial opportunities for growth, but navigating complex regulatory frameworks will require strategic partnerships and compliance with stringent standards.
Achieving this vision will involve substantial regulatory and strategic planning. Success in these markets demands a deep understanding of regional market dynamics, building relationships with regulatory authorities, and adhering to strict quality standards. CinnaGen plans to develop local capabilities, forge partnerships, and leverage its expertise in biopharmaceutical development and manufacturing to ensure compliance and maximize opportunities in these competitive regions.
In addition to geographical expansion, CinnaGen is committed to developing the next generation of biological products. By investing in cutting-edge research and advanced technology, the company aims to meet evolving patient needs and improve treatment outcomes. This commitment, combined with its strategic partnerships [1], will ensure CinnaGen’s sustained growth and leadership in the global biopharmaceutical landscape.
Follow-on biologicals/biosimilars in the MENA region: the case of adalimumab
In the MENA region, Iran has emerged as a leader in adopting FOB/biosimilar products, notably with the launch of CinnoRA, an adalimumab FOB, in September 2016. This launch has driven significant growth in the adalimumab market, with adoption rates exceeding initial expectations. It underscores the potential of biosimilars to enhance patient care and expand access to essential treatments in the region.
CinnoRA has had a considerable impact on the market since its launch, with sales figures demonstrating that the product has exceeded initial expectations, with an estimated 79,711 patients to have used CinnoRA since its launch.
Competitive advantage
At CinnaGen, the distinct combination of strengths distinguishes the company within the biosimilar industry. Key competitive advantages include:
- Large-Scale Manufacturing Capacity: With over 50,000 liters of bioreactor capacity in commercial operation, the company’s production scale ranks among one of the largest globally. This enables the company to deliver products more competitively.
- Intellectual Property Flexibility: Since Iran is not part of the World Trade Organization, the company can develop and market its products faster than competitors. For international partnerships or joint ventures, potential partners benefit from access to real-world data and de-risked, commercialized assets, enhancing investment confidence.
- Sophisticated Product Portfolio: With over 19 FOB/biosimilars on the market, see Table 1, and more than 25 in development, CinnaGen boasts one of the most comprehensive FOB/biosimilar portfolios in the MENA and CIS regions [2], covering therapeutic areas like endocrinology, haematology, immunology, neurology, ophthalmology, oncology, and rare diseases.
Current FOB/biosimilar products development of CinnaGen
The company has launched over 19 FOB/biosimilar products in the MENA region [2], see Table 1, and secure good manufacturing practices (GMP) certification from stringent regulatory authorities such as the EMA in 2018 [3].
Furthermore, AryoGen, a member company of CinnaGen, produced a range of FOBs notably bevacizumab (Stivant), rituximab (Zytux), trastuzumab (AryoTrust), etanercept (Altebrel), see Table 2, which are available in the MENA region.
Conclusion
CinnaGen’s strategic focus on innovation, expansion of its FOB/biosimilar portfolio, and entry into highly regulated global markets positions the company as a key player in the biopharmaceutical landscape. Its commitment to affordable, accessible health care through the development of high-quality biosimilars is transforming patient care, particularly in underserved regions like MENA. Coupled with its strong corporate responsibility initiatives, the company’s long-term vision ensures that CinnaGen is not only shaping the future of biopharmaceutical manufacturing in the MENA region but also contributing to sustainable healthcare solutions worldwide. As CinnaGen continues to grow, its leadership in biosimilar development is set to create lasting, positive impacts on global health.
Competing interests: This article was funded by CinnaGen.
Provenance and peer review: Article prepared based on the interview conducted on 27 August 2024; internally peer reviewed.
References
1. GaBI Online – Generics and Biosimilars Initiative. CinnaGen to invest in Turkish pharmaceuticals sector [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2024 Oct 23]. Available from: www.gabionline.net/pharma-news/CinnaGen-to-invest-in-Turkish-pharmaceuticals-sector
2. CinnaGen [homepage on the Internet]. [cited 2024 Oct 23]. Available from: https://www.cinnagen.com/
3. GaBI Online – Generics and Biosimilars Initiative. MENA region biologicals maker CinnaGen receives Eu GMP certification [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2024 Oct 23]. Available from: www.gabionline.net/pharma-news/MENA-region-biologicals-maker-CinnaGen-receives-EU-GMP-certification
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2024 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/transforming-healthcare-cinnagens-leadership-in-follow-on-biologicals-biosimilars-development-and-market-expansion.html
Repurposing non-oncology drugs for cancer treatment
Submitted: 15 March 2021; Revised: 17 March 2021; Accepted: 18 March 2021; Published online first: 24 March 2021
Cancer is one of the leading causes of mortality in the world today. The development of new drugs can reduce death rates, but these products are extremely expensive in terms of development time and money. This has led to the strategy of drug repurposing; whereby drug products already approved for noncancer indications are identified as potential cancer therapies. A review, published in Signal Transduction and Targeted Therapy [1], presents various promising repurposed non-oncology drugs for clinical cancer management.
The study also summarizes approaches used for drug repurposing and discusses the main barriers to uptake [2].
Drug repurposing and cancer treatment: an overview
When an already approved drug product is repurposed, pharmacokinetic, pharmacodynamic, and toxicity profiles of drugs have been already established in preclinical and phase I clinical studies. These drugs can thus rapidly progress to phase II and phase III clinical studies and the associated development time and cost of the cancer treatment can be significantly reduced.
The study notes that our understanding of cancer biology and the associated characteristics of cancer is increasing. If this is successfully coupled with repurposing studies that apply systematic screening of the entire pharmacopoeia together with advanced bioinformatics, new drugs for cancer treatment can be identified faster and at reduced cost.
The study provides a summary of non-oncology compounds that are already used for cancer therapy which are outlined below. It highlights the specific cancer characteristics targeted and distinguishes between those agents suitable for monotherapy or as drug combinations. For all the treatment options outlined, the study also provides a detailed discussion of how they effectively regulate at least one characteristic of cancer; or exert comprehensive anticancer effects by regulating multiple targets mediated by various alternative signalling routes.
Non-oncology drugs suitable for cancer monotherapy
Proliferative signal inhibitors
Cancer cells have an ability to maintain chronic proliferation. Non-oncology drugs which work by inhibiting proliferative signalling include rapamycin, prazosin, indomethacin. These had original applications as an immunosuppressant and anti-restenosis agent; in hypertension; and in rheumatic disease, respectively.
Cancer cell death inducers
Apoptosis causes cell death and prevents tumourigenesis once the cell is damaged or is placed under various physiologic stresses. Non-oncology drugs which work by inducing cell death include artemisinin, chloroquine and their related derivatives. Both of these medications are used to treat malaria and chloroquine is also a treatment for rheumatoid arthritis.
Cellular metabolism regulators
Cancer cells often have reprogrammed energy metabolism which supports malignancy by sustaining key characteristics of cancer, including uncontrolled cell proliferation, evading growth suppressors, and resisting cell death. Non-oncology drugs which work by regulating of cellular metabolism include etformin and disulfiram. The former is used to treat obese type-2 diabetes and the latter is an alcohol aversion drug.
Antitumour immunity activators
Some cancers, especially virus-induced cancers, can avoid immune surveillance or limit being eliminated by the immune system by somehow regulating both the innate and adaptive immune systems. Non-oncology drugs which work by activation of antitumour immunity include infectious disease vaccines:
Non-oncology drugs suitable for drug combination therapy
The study notes that, in some cases, compounds may not be considered for drug repurposing screening due to their low anticancer activity at known tolerated plasma drug doses described in previous indications. However, drugs that may be effective at higher dosage can be utilized through drug combination therapy. Here, a synergistic effect is produced by targeting alternative signalling pathways associated with certain cancer characteristics.
Tumour suppressor reactivators
There is now a bank of evidence that indicates a lack of crucial tumour suppressors can stimulate tumour growth. Many non-oncology drugs are being repurposed to target cancer cells that evade growth suppressors. Non-oncology drugs which work by reactivating growth suppressors include quinacrine and ritonavir. The former has previous indications for malaria, giardiasis, rheumatoid arthritis, and the latter for human immunodeficiency virus (HIV) treatment.
Cancer cell division interrupters
In cancer cells, the specialized DNA polymerase, telomerase, is expressed at high levels to counteract the normal cell growth and the division cycle. Non-oncology drugs which work by targeting telomerase and interfering with replication include curcumin, used to treat dermatological diseases, and genistein, used in the treatment of the menopause, osteoporosis and obesity.
Angiogenesis reducers
Tumour cells also stimulate angiogenesis to generate neovasculature, an important mechanism by which tumours obtain nutrients and evacuate waste products. Non-oncology drugs which work by decreasing angiogenesis include the infamous thalidomide, originally used to treat the symptoms of morning sickness, which is now used as a sedative and antiemetic drug; and itraconazole, an antifungal agent.
Cell invasion and metastasis suppressors
Tumour invasion is the mechanism by which tumour cells spread to the surrounding environment, while tumour metastasis is where cancer cells leave the primary tumour and migrate to a new location where they generate new (secondary) tumours. Some non-oncology drugs work by suppression of invasion and metastasis. These include Barberine, used to treat bacterial diarrhoea and niclosamide, an antihelminthic drug.
Genome instability disruptors
Genome instability is a major characteristic of malignancy. This allows some favourable mutant tumour genotypes to survive under stress conditions, particularly those induced by traditional cancer treatment methods, such as chemo- and radiotherapy. As such, to enhance the therapeutic index of traditional cancer therapies, drugs could be repurposed as sensitizers of genotoxic therapy to inhibit DNA damage response. Non-oncology drugs which work by disrupting the DNA damage response include spironolactone and mebendazole, which are respectively diuretic and antihelminthic drugs.
Tumour-promoting inflammation reducers
Inflammation is typically associated with tumourigenesis as it supports and accelerates tumour growth. Some non-oncology drugs work by targeting tumour-promoting inflammation. These include aspirin, a common pain and fever relief medication, and thiocolchicoside, used to treat rheumatologic and orthopaedic disorders.
Conclusions
The study stresses the urgent need to develop effective, safe, cheaper, and readily available anticancer agents. The repurposed drugs outlined that act on specific characteristics of cancer, show that there is therapeutic potential in this strategy.
Editor’s note
Oncology drugs
Proliferative signal inhibitors, cancer cell death inducers, cellular metabolism regulators, antitumour immunity activators,
Non-oncology drugs suitable for drug combination therapy
Tumour suppressor reactivators, cancer cell division interrupters, angiogenesis reducers, cell invasion and metastasis suppressors, genome instability disruptors, and tumour-promoting inflammation reducers
Competing interests: The work of the paper [1] was supported by grants from the Chinese NSFC (nos. 81821002, 81790251, and 81773143), and Guangdong Basic and Applied Basic Research Foundation (2019B030302012).
Provenance and peer review: Article abstracted based on published scientific or research papers recommended by members of the Editorial Board; internally peer reviewed.
Alice Rolandini Jensen, MSci, GaBI Journal Editor
References
1. Zhang Z, Zhou L, Xie N, Nice EC, Zhang T, Cui , et al. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct Target Ther. 2020;5(1):113.
2. GaBI Online – Generics and Biosimilars Initiative. Technological approaches to drug repurposing for cancer treatment [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2021 Mar 17]. Available from: www.gabionline.net/Generics/Research/Technological-approaches-to-drug-repurposing-for-cancer-treatment
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2021 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/repurposing-non-oncology-drugs-for-cancer-treatment.html
Biosimilars markets: US and EU compared
|
Abstract:
Differences between the US and European regulatory processes can lead to differences in the time taken for biosimilar approval. The European Medicines Agency is in some cases quicker to approve biosimilar drugs and Europe has overall more approved biosimilars, whilst the US approved the first biosimilar in 2015. Whatever the market, there are a number of strategies that can be employed to help address patient and healthcare provider concerns, such as educational programmes. This paper provides some insights on these aspects.
|
Submitted:29 February 2020; Revised: 29 April 2020; Accepted: 4 May 2020; Published online first: 18 May 2020
The US and Europe have a number of key differences in the way in which they approve biosimilar drugs. These were discussed at a recent conference [1], in addition to some strategies that can be used to address healthcare provider and patient concerns regarding biosimilars.
US and EU biosimilars markets compared
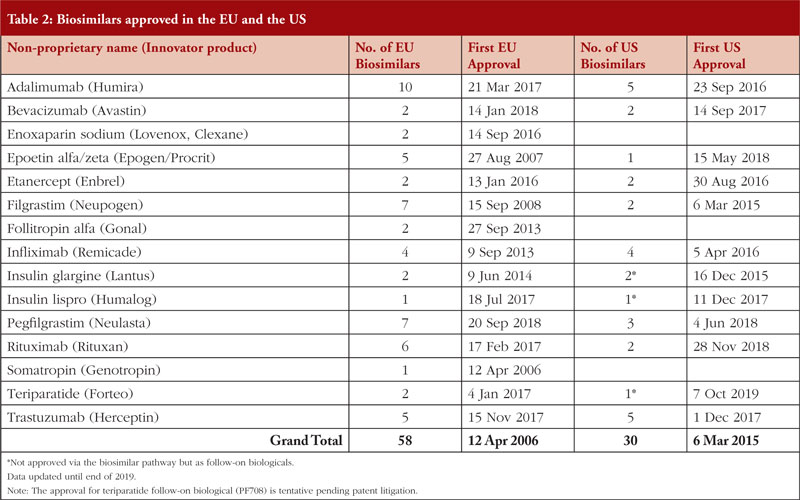
Comparisons of the time taken to authorize (or recommend approval of) a selection of nine biosimilars by the European Medicines Agency (EMA) and the US Food and Drugs Administration (FDA) are shown in Table 1.

EMA approved their first biosimilar, somatropin, in April 2006. The following year, an epoetin alfa biosimilar was approved by EMA in August, followed by FDA approval in May 2018; 11 years later. Similarly, a filgrastim biosimilar was approved first in the European Union (EU) in September 2008, followed by US approval in March 2015. Filgrastim was the first biosimilar approved in the US, nine years after EMA’s first approved biosimilar, somatropin.
However, more recently, approval times in the US have caught up. For example, a bevacizumab biosimilar was first approved in the US in September 2017, while EMA approved a bevacizumab biosimilar a few months later in January 2018.
Table 1, however, does not include five biologicals shown in Table 2, i.e. enoxaparin sodium, follitropin alfa, insulin glargine, insulin lispro, somatropin, which were not approved via the biosimilar pathway in the US [2] at the time of EU approval. By end of 2019, there are 26 biosimilars approved in the US and 58 approved in the EU [3].
In Europe, biosimilar competition has significantly reduced prices for biologicals, as demonstrated by the change in price (per treatment day) in the EU in the year of the first biosimilar launch, see Figure 1.

Total market prices for erythropoietin, for example, decreased by around 30%, which was similar to the reductions in prices seen for granulocyte colony-stimulating factor (G-CSF) after the launch of a biosimilar version.
In the US, the opportunities for biosimilars can be particularly lucrative. For an overview of biosimilar opportunities in the US until 2023, see Figure 2.

There are 71 patents on biologicals due to expire in the US by 2023, with a total of US$55 billion in sales open to competition from biosimilars manufacturers [1].
Together, expiry of the patents on Humira (adalimumab), Kadcyla (trastuzumab), Lumizyme (alglucosidase alfa) and Stelara (ustekinumab) are worth an estimated US$20 billion to biosimilars manufacturers.
Patents on Amevive (alfacept), Lemtrada (alemtuzumab) and Rituxan (rituximab), meanwhile, have already expired in the US. Biosimilars for these products have an estimated value of US$4.5 billion. Of these, so far only rituximab (Pfizer and Celltrion/Teva) and trastuzumab (Celltrion/Teva; Amgen; Biocon/Mylan; Merck/Samsung Bioepis; Pfizer) have licensed biosimilars in the US [2]. Amgen/Allergan have additionally submitted an application to FDA for their rituximab biosimilar [4].
Addressing concerns about biosimilars
Although the biosimilars market is advancing well in both EU and the US, concerns about the use of biosimilars remain widespread. A number of strategies can be employed to address the concerns of both healthcare providers and patients.
Utilizing the EU’s experience in biosimilars can be helpful, especially in cases where there are limited data. Almost two times as many biosimilars have been approved in the EU compared to the US, see Table 2.
The EU monitoring system for adverse events, in particular, can help to provide evidence for the safety of biosimilars. Furthermore, EMA states that evidence it has acquired over 10 years shows that biosimilars can be used ‘as safely and effectively in their approved indications as their reference biological medicines’ [5]. The European experience is testament to the value of multi-stakeholder participation and engagement at every stage of the process, from the regulatory framework through education all the way to procurement policy decisions and treatment guidelines [6].
Gaining support from physician leaders within an organization (or other key opinion leaders) can also help to alleviate concerns about biosimilars, while educational initiatives aimed at healthcare providers and patients can help to minimize the nocebo effect (when negative expectations of a treatment, in this case a biosimilar, lead the patient to believe it is less effective).
Training healthcare providers to be well informed and confident in the use of biosimilars can lead to the transfer of confidence to the patient. FDA materials on biosimilars can be useful for such educational purposes.
A further recommendation is to transition patients to biosimilars in a conservative manner in order to gain experience and support. Monitoring the outcomes of conversion can also improve confidence in the use of biosimilars. Finally, it is important to advocate for comparable patient assistance programmes with biosimilar manufacturers.
Competing interests: None.
Provenance and peer review: Commissioned; internally peer reviewed.
Eleanor Bird, MSc; GaBI Journal Editor; Michelle Derbyshire, PhD, GaBI Online Editor
References
1. Gutierrez A. Payor strategies to drive biosimilars access and savings. GRx+Biosims Conference 2019; 4 6 November 2019; North Bethesda, Maryland, USA.
2. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in the US. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 29]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-the-US
3. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 May 9]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
4. GaBI Online – Generics and Biosimilars Initiative. Rituximab biosimilar ABP 798 submitted to FDA [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 29]. Available from: www.gabionline.net/Biosimilars/News/Rituximab-biosimilar-ABP-798-submitted-to-FDA
5. GaBI Online – Generics and Biosimilars Initiative. Improving understanding of biotherapeutics and biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 29]. Available from: www.gabionline.net/Biosimilars/General/Improving-understanding-of-biotherapeutics-and-biosimilars
6. Reilly MS, Schneider PJ. Policy recommendations for a sustainable biosimilars market: lessons from Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):76-83. doi:10.5639/gabij.2020.013
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/biosimilars-markets-us-and-eu-compared.html
Clinical trials for trastuzumab biosimilars
|
Abstract:
Clinical trials must demonstrate biosimilarity for regulatory marketing approval to be granted. The GaBI Journal has conducted a search for clinical trials that have been, or are being, carried out for trastuzumab biosimilars.
|
Submitted: 26 March 2020; Revised: 2 April 2020; Accepted: 2 April 2020; Published online first: 15 April 2020
Roche’s originator trastuzumab biological (Herceptin) was approved by the US Food and Drug Administration (FDA) in September 1998 and by the European Medicines Agency (EMA) in August 2000 [1]. The patents on Herceptin expired in Europe in July 2014 and in the US in June 2019 [1].
Trastuzumab is a monoclonal antibody that binds to and inactivates the human epidermal growth factor receptor 2 (HER2)/neu receptor. In some cancers, notably certain types of breast cancer, HER2 is overexpressed and causes cancer cells to reproduce. Trastuzumab is therefore used to treat HER2 positive (HER2+) breast cancers.
EMA developed the first guidelines for the approval of biosimilars via an abbreviated registration process during 2005 to 2006, and since then EMA has developed many general and specific guidelines for biosimilars [2]. In the US, a legal framework for approving biosimilars was established in 2009, via the Biologics Price Competition and Innovation Act of 2009 (BPCI Act). Since then, FDA has issued several guidance documents on biosimilars [3].
In order to gain regulatory approval for biosimilars a package of data that includes analytical, non-clinical and clinical data is required. A comprehensive comparison of the quality attributes of the biosimilar with the reference product using state-of-the-art methods must support the conclusion that the products are biosimilar on a structural and functional level [2, 3]. However, even with robust analytical data, the biosimilar can be approved only with additional non-clinical and clinical data. Hence the need for clinical trials.
Clinical trials aim to resolve any uncertainties that may remain following non-clinical development regarding the similarity of the proposed biosimilar with the reference product. Pharmacokinetic and pharmacodynamic studies form the backbone of early clinical development and serve to inform phase III clinical development. Factors to be considered in clinical development include study population, design, endpoint, sample size, duration and analytical methods.
To demonstrate biosimilarity for regulatory approval, clinical trials of biosimilars should therefore use sensitive endpoints with the aim of ensuring that there are no clinically meaningful differences in efficacy, safety or immunogenicity.
Many companies have developed trastuzumab biosimilars [4]. These include major players such as South Korean biotechnology company Celltrion, which gained European Commission (EC) approval for its trastuzumab biosimilar Herzuma (CT-P6) in February 2018 [5] and FDA approval in December 2018 [6].
Other major players who have completed or are carrying out clinical trials for candidate biosimilars include US-based biotech giant Amgen, Korea-based Samsung Bioepis (Samsung and Biogen’s joint venture), as well as collaborations such as that between Japan-based Daiichi Sankyo and UK-based AstraZeneca.
Clinical trials being carried out by these companies include those listed in Table 1. As of 30 March 2020, 19 clinical trials relevant to trastuzumab biosimilar development covering 13 different candidate biosimilars and non-originator biologicals were identified by a search of ClinicalTrials.gov and the EU Clinical Trials Register using the search terms ‘trastuzumab’ AND ‘biosimilar’. The searches were carried out on 26 March 2020 and included all clinical trials recorded in the databases until that date.





Editor’s comment
European Medicines Agency regulatory requirements ensure the same high standards of quality, safety and efficacy for biosimilars as for originator biologicals, and also include a rigorous comparability exercise with the reference product but they are not universally accepted by regulatory bodies outside of the European Union (EU). It should be noted that non-originator biologicals approved only outside of Europe might not have been authorized if they had been subjected to the strict regulatory processes required for approval of biosimilars in the EU.
Competing interests: None.
Provenance and peer review: Commissioned; internally peer reviewed.
Michelle Derbyshire, PhD, GaBI Online Editor
References
1. Derbyshire M. Patent expiry dates for biologicals: 2018 update. Generics and Biosimilars Initiative Journal (GaBI Journal). 2019;8(1):24-31. doi:10.5639/gabij.2019.0801.003
2. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 2]. Available from: www.gabionline.net/Guidelines/EU-guidelines-for-biosimilars
3. GaBI Online – Generics and Biosimilars Initiative. US guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 2]. Available from: www.gabionline.net/Guidelines/US-guidelines-for-biosimilars
4. GaBI Online – Generics and Biosimilars Initiative. Biosimilars of trastuzumab [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 2]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-of-trastuzumab
5. GaBI Online – Generics and Biosimilars Initiative. EC approval for trastuzumab biosimilar Herzuma [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 2]. Available from: www.gabionline.net/Biosimilars/News/EC-approval-for-trastuzumab-biosimilar-Herzuma
6. GaBI Online – Generics and Biosimilars Initiative. FDA approves trastuzumab biosimilar Herzuma [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 2]. Available from: www.gabionline.net/Biosimilars/News/FDA-approves-trastuzumab-biosimilar-Herzuma
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/clinical-trials-for-trastuzumab-biosimilars.html
Top developments in biosimilars during 2019
Submitted: 3 February 2020; Revised: 12 February 2020; Accepted: 18 February 2020; Published online first: 25 February 2020
2019 has been a busy year for biosimilars; there have been a record number of approvals and the start of many clinical trials. It has been a particularly busy year for the oncology and anti-inflammatory therapeutic areas, with numerous approvals and launches for key molecules including adalimumab, infliximab, bevacizumab and trastuzumab. There have also been a number of promising new investments and collaborations, some of which will bear fruit in 2020. Yet challenges remain for the global biosimilars market. Focus areas include improving education, streamlining the regulatory process and ensuring the long-term sustainability of the market, among others.
Biosimilar approvals in 2019
By end of 2019, there were 26 biosimilars and four follow-on biologicals (with one tentative approval) approved in the US and 58 biosimilars approved in Europe [1, 2], not to mention the growing numbers of approvals elsewhere around the globe, see Table 1.
 Adalimumab
Adalimumab
Adalimumab, sold under the brand name Humira, is a tumour-necrosis factor-alpha (TNF-α) inhibitor used to treat inflammatory conditions including rheumatoid arthritis, psoriasis and inflammatory bowel disease (IBD). In Europe, Fresenius Kabi, the generics unit of German healthcare giant Fresenius, received approval for their adalimumab biosimilar Idacio in April 2019. The product was launched in Germany soon after, at the beginning of May 2019. Mylan’s biosimilar Hulio, which gained European Medicines Agency’s (EMA) recommendation for approval in September 2018 was launched in major European markets in October 2018 and in Spain in July 2019. This was Mylan’s first biosimilar to launch in Spain. In the US, the US Food and Drug Administration (FDA) has approved two adalimumab biosimilars during 2019; Hadlima from Samsung Bioepis, followed by Pfizer’s Abrilada. Finally, in China, the National Medical Products Administration (NMPA) approved Qletli in November 2019. Qletli is produced by Bio-Thera Solutions and is the company’s first biological to receive approval.
Bevacizumab
EMA was the first regulatory agency to approve a bevacizumab biosimilar in 2019. The originator drug, marketed as Avastin, targets vascular endothelial growth factor A (VEGF-A) and is used to treat a number of cancer types. Patents on Avastin expired in the US in July 2019 and will expire in Europe in January 2022. Pfizer’s biosimilar version, sold as Zirabev, was approved for sale in the European Union (EU) in February 2019. EMA accepted an application from Samsung Bioepis in July 2019, but the drug has not yet been approved. FDA approved Zirabev for sale in the US soon after, in June 2019. Zirabev was approved in Japan in June 2019 and a further bevacizumab biosimilar was approved in September 2019. The second biosimilar is developed by Amgen but will be distributed and commercialized in Japan by Tokyo-based Daiichi-Sankyo. Zirabev was also approved in Australia in November 2019. A bevacizumab similar biologic, named Versavo (Dr Reddy’s Laboratories), was launched in India in August 2019.
Darbepoetin alfa/Epoetin alfa
Darbepoetin alfa and epoetin alfa are both recombinant forms of human erythropoietin, also known as EPO, which stimulates red blood cell production. Both treatments are used in the treatment of anaemia, typically in patients with chronic kidney failure. South Korean company PanGen gained approval from the Malaysian National Pharmaceutical Regulatory Agency for its epoetin alfa biosimilar Erisa in February 2019. In Japan, three darbepoetin alfa biosimilars were approved in 2019. JCR Pharmaceuticals developed a long-acting injectable version of the drug in collaboration with Kissei Pharmaceuticals. The other darbepoetin alfa biosimilars that were approved were produced by South Korean companies Chong Kun Dang (which will be distributed via Mylan’s Japanese arm) and Dong-A ST (with Japanese partner Sanwa Kagaku Kenkyusho).
Dornase alfa
Russian biotech company Generium and Swiss cell-line producer Selexis received approval for their dornase alfa non-originator biological Tigerase in September 2019. The drug is a non-originator biological of Roche’s Pulmozyme and is used to treat cystic fibrosis by cleaving the DNA in the mucus of patients’ lungs.
Eculizumab
In April 2019, the Russian Ministry of Health approved an eculizumab non-originator biological called Elizaria. The product, which is used to treat rare blood and immune disorders, is produced by Generium. They claim to be the first company in the world to develop and market a non-originator biological of eculizumab.
Etanercept
There were four approvals for etanercept biosimilars in 2019. First, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) approved Lupin’s biosimilar of the TNF inhibitor in March 2019. The drug (YLB113) is distributed by YL Biologics, a joint venture between Lupin and Japanese generics maker Yoshindo. Lupin is awaiting approval for its etanercept biosimilar in Europe. In the US, FDA approved Samsung Bioepis etanercept biosimilar Eticovo in April 2019. Finally, China approved an etanercept copy biological in a pre-filled syringe dosage form. The drug, Yisaipu, was developed by Sunshine Guojian Pharmaceutical and had previously been approved as a powder.
Filgrastim/Pegfilgrastim
In 2019, the European Commission (EC) approved three pegfilgrastim biosimilars. In January they approved Fulphila from Biocon and Mylan; in April they approved Grasustek made by Indian manufacturer USV; and in October they approved Pegfilgrastim Mundipharma (now called Cegfila), produced by Mundipharma Biologics. They are all biosimilars of Amgen’s Neulasta (pegfilgrastim), a PEGylated form of the recombinant human granulocyte colony-stimulating factor (G-CSF) analogue filgrastim. The treatment is used to stimulate bone marrow to produce more neutrophils, which help to fight infection in immunocompromized patients, such as those undergoing chemotherapy. In the US, FDA approved Sandoz’s pegfilgrastim biosimilar Ziextenzo in November 2019, following a previous rejection of the application in 2016. FDA also rejected an application for a filgrastim biosimilar from Taiwan-based biosimilars maker Tanvex in 2019. The company plans to work closely with FDA in order to resubmit the application as soon as possible. In Australia, three pegfilgrastim biosimilars were approved in 2019: Lapelga and Neutropeg from Apotex and Ziextenzo from Sandoz.
Heparin
There was one biosimilar of the anticoagulant heparin approved in 2019 in Canada. Canadian pharmaceuticals company Valeo Pharma won the rights to market a biosimilar of low molecular weight heparin in August 2019. Health Canada stated that heparins should be categorized as biosimilars (rather than generics) in 2013 [3].
Infliximab
Infliximab is a monoclonal antibody treatment for autoimmune conditions including IBD and rheumatoid arthritis. The EC approved an infliximab biosimilar from South Korean company Celltrion in September 2019, following a positive EMA opinion based on phase I/III studies. The product is sold as Remsima. In the US, FDA approved Amgen’s infliximab biosimilar Avsola in December 2019. The approval was based on evidence that included pharmacokinetic (PK) similarity and a clinical study in patients with rheumatoid arthritis.
Insulin
In October 2019, Biocon/Mylan were the first to launch an insulin glargine biosimilar in Australia. The product is approved in over 40 countries and is available in Australia on the Pharmaceutical Benefits Scheme.
Rituximab
There were three approvals for rituximab biosimilars in 2019. Rituximab is a monoclonal antibody treatment for autoimmune diseases such as arthritis and some types of cancer. It has been sold under the brand name Rituxan by Genentech (Roche). Teva Canada Innovation’s biosimilar version Truxima was approved by Health Canada in April 2019, making it the first rituximab biosimilar approved in Canada. In the US, a biosimilar created by Pfizer called Ruxience was approved in July 2019, based on data from patients with CD20-positive, low tumour burden follicular lymphoma. A biosimilar approved in China was developed by Shanghai Fuhong Hanlin Bio-Pharmaceutical (Hanlin) and sold under the name Hanlikon – the first rituximab biosimilar approved in the country. Two further biosimilars are also in development in China.
Teriparatide
The first teriparatide biosimilar launched in 2019 was in Europe. Hungarian company Gedeon Richter launched their biosimilar Terrosa in August 2019. The product is used to activate osteoblasts, stimulating bone formation. Not long after, in the US, FDA approved a follow-on biological from US-based company Pfenex. Finally, in South Korea Daewon Pharmaceutical received regulatory approval to market Gedeon Richter’s product in late 2019. It is the first teriparatide biosimilar to receive regulatory approval in South Korea and the first approval for Daewon.
Trastuzumab
There were a number of new trastuzumab biosimilars approved in 2019. The monoclonal antibody is primarily a treatment for breast cancer and has been marketed by Roche as Herceptin. In the US, FDA approved four new trastuzumab biosimilars in 2019: Herzuma (Celltrion/Teva), Kanjinti (Amgen/Allergan), Ontruzant (Samsung Bioepis) and Trazimera (Pfizer). Likewise, Canada approved Mylan’s biosimilar version Ogivri in June 2019, followed shortly after by Herzuma, the third of Celltrion’s biosimilars approved in the country. In Australia, Mylan was the first to launch a trastuzumab biosimilar Ogivri in August 2019 for the treatment of human epidermal growth factor receptor 2-positive (HER2+) breast and gastric cancers, and Pfizer’s trastuzumab biosimilar Trazimera was approved in August 2019. A similar biologic Eleftha was launched in India by Intas Pharmaceuticals in April 2019, at an impressive 65% discount. In Europe, a biosimilar from Singapore-based Prestige has been accepted for review by EMA, but not yet approved. In May 2019, Brazil’s Health Surveillance Agency approved the trastuzumab biosimilar Ontruzant which is produced by Samsung Bioepis, and Celltrion’s trastuzumab biosimilar Herzuma.
Clinical studies for biosimilars
In 2019, results were reported from a number of clinical studies investigating the efficacy and safety profile of biosimilars including adalimumab, aflibercept, bevacizumab, denosumab, eculizumab, infliximab, insulin, natalizumab, rituximab, trastuzumab and ustekinumab. Some of these clinical studies are summarized below.
Adalimumab
Positive phase III data for adalimumab biosimilars was presented at the 2019 European Congress of Rheumatology, which was held in Madrid, Spain in June. China-based Hisun revealed the results of a multicentre trial of patients with ankylosing spondylitis, which showed their copy biological (HS016) to have comparable efficacy, safety and immunogenicity to the originator. Another China-based company Innovent in September 2019 published positive results from a phase III equivalence trial. The results showed that 75% of the copy biological-treated group met the primary outcome compared to 72% in the originator-treated group. In March 2019, Amgen published data on the long-term safety of their biosimilar ABP 501, showing persistent benefit with the biosimilar and no loss of efficacy or safety.
2019 also saw the start of two new clinical trials for adalimumab biosimilars. Both Alvotech (Iceland) and Celltrion (South Korea) initiated phase III trials for adalimumab biosimilars in 2019. In March 2019, Alvotech enrolled their first patient while Celltrion finished registering patients. Alvotech aims to enrol 400 participants in the trial at 30 sites and expects the trial to be completed in August 2020. Celltrion is aiming to enrol 564 participants in their trial, which is being carried out in Bulgaria and should be completed in June 2020.
Aflibercept
South Korean biologicals company Alteogen began a phase I trial for an aflibercept biosimilar (ALT-L9) towards the end of 2019. Aflibercept is a recombinant protein used to treat age-related macular generation (sold as Eylea). The trial, which is being conducted in South Korea, will compare the safety, efficacy and PK of ALT-L9 versus Eylea in 30 patients.
Bevacizumab
There were multiple trials for bevacizumab in 2019. China’s Bio-Thera Solutions published positive phase I results for its copy biological in June. The study involved 128 healthy male volunteers and showed similarity to both US- and EU-licensed originator products. Singapore-based Prestige started a phase III trial for its candidate, HD204, at the end of 2019. The randomized controlled trial will compare HD204 to the reference product, EU-licensed Avastin, in 500 patients with non-squamous non-small cell lung cancer (NSCLC). An already completed study of Amgen’s biosimilar Mvasi showed no statistically significant differences in clinical efficacy, safety, immunogenicity and PK with Avastin. Innovent presented results from a phase III trial at the 2019 Annual Meeting of the American Society of Clinical Oncology (ASCO). They showed no significant difference in safety profile or immunogenicity between their copy biological IBI305 and the originator. Finally, Samsung Bioepis’ biosimilar (SB8) showed promising results from a phase III trial, presented at the 2019 European Society for Medical Oncology (ESMO) Congress. Their study assessed response in patients with metastatic or recurrent NSCLC and showed no statistically significant differences in efficacy to the reference product, Avastin.
Denosumab
Denosumab is a monoclonal antibody treatment for osteoporosis, as well as certain tumours of the bone. The originator drug Prolia was developed by Amgen. China-based Qilu has started a phase III clinical trial for their copy biological, which will compare efficacy and safety to the originator product. It is expected to be completed in July 2021. Sandoz is to start a phase III trial for their denosumab biosimilar soon, with an expected completion date of July 2022.
Eculizumab
Samsung Bioepis started a phase III clinical trial for an eculizumab biosimilar following approval from the Korean Ministry of Food and Drug Safety in June 2019. The study will compare the efficacy, safety, PK and immunogenicity of SB12 versus the originator drug, Soliris. It will include 50 patients with the rare blood disorder paroxysmal nocturnal haemoglobinuria (PNH).
Infliximab
Given its efficacy and wide range of treatment indications, infliximab is a popular biosimilar candidate for pharmaceutical companies and a number of clinical studies are ongoing. In June 2019, Celltrion presented real-world safety data on their infliximab biosimilar CT-P13. In one study, 329 patients were recruited at 38 sites across Europe and Canada. Results showed no statistically significant difference in adverse events compared to the originator. A second study collected additional safety data from 1,579 patients treated with at least one dose of CT-P13. Based on these results, the authors concluded that treatment with CT-P13 is well tolerated in the real-world setting. A switching study conducted in IBD patients in Spain showed that most patients switching from originator to CT-P13 had a good profile of efficacy and safety, after a 2-year follow-up period. A final study in Brazil, which included 40 psoriasis patients who required or were already treated with infliximab, showed that the introduction or switch to CT-P13 appears to be safe and non-inferior to the originator.
Insulin
In June 2019, US generics company Lannett started a phase I trial of its insulin glargine biosimilar in South Africa. The single-dose study aimed to compare the PK and pharmacodynamics (PD) of the candidate with the US originator Lantus (Sanofi) in 27 healthy male adult volunteers. The trial was the first clinical study in humans to directly compare Lannett’s insulin glargine biosimilar to Lantus and results showed that the biosimilar was well tolerated and showed no significant differences to the reference product in terms of PK, PD and safety.
Natalizumab
Polish company Polpharma began a phase III trial for its natalizumab biosimilar, PB006, in 2019, with plans to complete in August 2021. The product can be used to treat multiple sclerosis (MS) and Crohn’s disease. The trial includes approximately 260 patients with relapse-remitting MS and will compare safety and efficacy to the reference product Tysabri.
Rituximab
Amgen, together with Allergan, announced positive results from a phase III trial of its rituximab biosimilar in January 2019. Their randomized, double-blind trial of 311 patients found no statistically significant differences to PK, safety or efficacy compared to the originator. In a second study, they showed that the biosimilar is safe and effective in the treatment of non-Hodgkin’s lymphoma. In May 2019, Eli Lilly (and its partner Innovent) announced positive date from two studies of their rituximab copy biological, IBI301. Phase I and III studies both met their primary endpoints (PK profile and objective response rate, respectively). Finally, data presented at the 2019 European Congress of Rheumatology revealed positive results for rituximab non-originator biological AcellBia (Biocad) and copy biological Hanlikon (Hanlin). Biocad published further results on AcellBia in November, showing equivalent efficacy to the originator in lymphoma. In Europe, mAbxience Research reported on a clinical trial of their rituximab biosimilar (RTXM83) in July 2019. They showed comparable biological activity to the EU originator MabThera, and no significant differences in adverse events or immunogenicity.
Trastuzumab
Data published in the British Journal of Cancer in January 2019 showed that Pfizer’s trastuzumab biosimilar Trazimera was non-inferior to the originator product, Herceptin (Roche). The randomized controlled trial was international and included over 700 subjects. The results so far show that, when given as first-line treatment for HER2+ metastatic breast cancer, the biosimilar is equivalent to the originator in terms of objective response rate (ORR). The drug has been approved in the EU and Japan but was rejected by FDA in 2018. Results from Biocon and Mylan’s trastuzumab follow-on biological Zedora approved in Brazil in December 2017 were presented at the 2019 ASCO Annual Meeting showing that the safety of Zedora is consistent with that of the originator. Finally, Prestige announced positive phase II data from their trastuzumab biosimilar (HD201) in June 2019. The trial included over 500 patients and confirmed that ‘HD201 is the most equivalent biosimilar of Herceptin’. The drug was submitted to EMA in May 2019.
Ustekinumab
Ustekinumab, sold under the brand name Stelara (Janssen Immunology), is a monoclonal antibody treatment for psoriasis, which has also been approved to treat IBD in some countries. Australian biosimilars manufacturer NeuClone began a phase I trial of their proposed ustekinumab biosimilar in October 2019, recruiting over 200 healthy volunteers. German company Formycon announced the start of a phase I clinical trial for a rival biosimilar FYB202 at the same time. Dosing of subjects has begun in the trial, which compares PK, safety and tolerability to the reference product Stelara. A trial for an ustekinumab copy biological was approved in December 2019, this time produced by China-based Bio-Thera Solutions. The trial will involve administration of a single dose of the copy biological and is expected to enrol 270 healthy volunteers.
Reports and non-clinical research
There have been a number of non-clinical studies in 2019 investigating several biosimilar issues including:
European biosimilars market
Several reports from 2019 have assessed the European market for biosimilars. One analysis showed an increase in the share of the EU biologicals market that is subject to competition from biosimilars, as well as an increase in patient use of biologicals overall and reductions in prices for biologicals.
However, maintaining long-term sustainability of the market in Europe will be challenging. IQVIA explored some of these challenges, including the fact that fewer new blockbuster biologicals will become available for biosimilars competition over the next five years compared to the last five years. In line with this, the EC held a workshop focussing on sustainability in October 2019, attended by patients, healthcare professionals, authorities and the pharmaceutical industry.
In terms of regulation, one assessment of the European regulatory framework found that it could be difficult for physicians, pharmacists and patients to understand the biosimilar pathway and regulators should therefore improve their communication strategy. Analysis of biosimilars licensed in Europe published in January 2019 also found that the packaging of many did not meet EU guidelines on readability. This is an important issue as unnecessarily complex drug information leaflets could lead to some patients not reading them.
At the 12th Pharmacovigilance Conference in January 2019, EMA reviewed their pharmacovigilance process, outlining strengths and weaknesses of the system and detailing future plans such as increasing the use of real-world data, including from electronic health records and even wearable technology.
A series of roundtable discussions held with clinicians, industry and government in 2018 identified measures to help EU policymakers to access the full potential of biosimilars [4, 5]. Recommendations were made for supply-side incentives, e.g. design more flexible procurement and reimbursement mechanisms, demand-side incentives, e.g. use of physician quotas, and gainsharing, e.g. develop more experience in the design of gainsharing arrangements.
Prescribing
A study reported in February 2019 assessed the barriers and facilitators to biosimilar prescribing in the UK, identifying concerns about safety and efficacy to be a major barrier and departmental cost savings as an important facilitator. A review of pharmacy practice published in August 2019 suggested that pharmacists working in all settings should take a key role in advocating for biosimilars. An important part of this role is to educate patients, providers and healthcare practitioners about biosimilars.
Switching
Switching from originator to biosimilar therapy is one of the most controversial aspects of biosimilar use. Encouragingly, a number of reports in 2019 support safe switching to biosimilar treatments. Data on switching to etanercept biosimilars presented at the 2019 European Congress of Rheumatology, including real-world data, support the safety, efficacy and economic advantage of switching. A review also concluded that switching to biosimilar infliximab (CT-P13) is safe and acceptable, complemented by an 18-month study of switching in ankylosing spondylitis patients.
However, concerns remain about such a strategy. In Denmark for example, government mandated non-medical switching of all patients treated with the originator etanercept Enbrel to biosimilar etanercept Benepali. However, a significant percentage of patients were still being treated with the originator drug one year later. This led some to conclude that mandatory switching is likely not the most efficient strategy for increasing biosimilar use, at least in the context of chronic rheumatic disease. Additional commentary, however, suggested the data may not be useful to assess non-medical switching, due to methodological defects causing ‘misleading biases’. More studies are needed in this area.
Education
Education is a commonly cited factor when it comes to increasing biosimilar use. A survey carried out by ESMO identified important knowledge gaps among oncologists. Gaps identified included knowledge on biosimilar development, clinical trial design and requirements for extrapolation, which should be points of focus for educational initiatives.
A literature review published in January 2019 also concluded that clinician education is critical to increase knowledge surrounding biosimilars and ultimately increase their use. Indicative of this, an additional systematic review found that clinicians in Europe and the US do not primarily support the use of biosimilars as safe and effective therapies in patients already receiving originator biological treatment. At the 17th Biosimilar Medicines Conference 2019, the World Health Organization (WHO) representatives further emphasized the importance of education to increase the use of biosimilars.
US biosimilars market
A number of critical assessments of the US biosimilars market were made in 2019. A January 2019 review discussed key considerations for payers in the US, including factors promoting biosimilar uptake as well as key issues such as interchangeability and naming. Particularly important considerations were lack of understanding of the regulatory process, limited education, concerns over the documentation of adverse events and insurance coverage barriers.
President of the Biosimilars Forum Juliana Reed gave a presentation on ‘The State of the Biosimilars Market’ at the 2019 Drug Information Association’s (DIA) Biosimilars Conference. She described various anti-competitive behaviours and market dynamics that discourage uptake of biosimilars in the US, including exclusionary contracting and the lengthy time from FDA approval to market launch. She also described measures that could be used to achieve a more sustainable and competitive biosimilars market in the country, including enabling fair and easy access to the market and increasing the breadth of insurance coverage.
Regulation and guidance on biosimilars
National
It has been a busy year for regulatory changes in the US. Particularly prominent were changes to the regulation of insulin products. In May 2019, FDA held a public meeting to discuss access to affordable insulin. One of the outcomes of this meeting was plans to transition biologicals currently approved under the Food, Drug, and Cosmetic Act as drugs to instead approve them as biologicals, in order to open the pathway to market new products that are biosimilar to, or interchangeable with, these transitioned products. This change includes all insulin products (insulin, insulin mix and insulin analogue products) and will come into effect in March 2020. In November 2019, FDA released additional guidance on immunogenicity considerations for insulin, to help facilitate the development of biosimilar insulin products. 2019 also saw the release of the Insulin Price Reduction Act, a new piece of legislation which would hold pharmaceutical companies, pharmacy benefit managers and insurers accountable for rising insulin prices. These changes look to be effective, as both Eli Lilly and Novo Nordisk launched lower price insulin products in 2019.
In terms of substitution, despite the fact that FDA has not yet approved a biosimilar as interchangeable with its reference product, 45 US states have passed their own substitution laws. The states have passed laws that permit or require pharmacists to dispense an interchangeable biological product in certain situations, but only if the prescriber has not designated on the prescription that substitution is prohibited.
Other changes related to biosimilars include the release of final guidance on demonstrating interchangeability with a reference product, guidance on quality-related considerations which outlines essential factors for consideration when performing comparative analytical assessments (and allows for sponsors to use non-US licensed comparators in some cases). FDA also released new patient-facing guidance to help explain to the public that biosimilars are just as safe and effective as their reference counterparts.
In March 2019, FDA issued new guidelines on the naming of biologicals. The agency had previously said that it would assign a non-proprietary name to all biologicals and biosimilars that includes an ‘FDA-designated suffix’. The March 2019 guidelines importantly stated that already approved biologicals will not need to change their names. For biosimilars, the guidelines state that FDA will designate a proper name, which is a combination of the core name and a distinguishing suffix composed of four lowercase letters. However, the guidance was not unanimously well received, with some stakeholders suggesting the changes would be confusing. A literature review, however, suggested the differential naming of originator and biosimilar products supports pharmacovigilance and promotes biosimilar uptake.
Similar discussions were held in Canada in 2019 where an online consultation on naming, proposing three different options for the naming of biologicals. As a result of this consultation, Health Canada announced that all biologicals (including biosimilars) will be identified by their unique name and non-proprietary name, without the addition of a product-specific suffix. They will also continue to have a unique Drug Identification Number (DIN) which identifies key characteristics such as manufacturer, ingredients, strength and dosage form.
Canada has also been leading the way in switching. In 2019, British Columbia stopped coverage of certain originator biologicals and switched patients to biosimilars. The province’s healthcare system, PharmaCare, stopped providing coverage for originator biologicals for ankylosing spondylitis, diabetes, plaque psoriasis, psoriatic arthritis or rheumatoid arthritis on 25 November 2019. The move was publicly commended by the Canadian Biosimilars Forum and Green Shield Canada, prompting the province of Alberta to consider stopping coverage of originator biologicals for IBD. For those interested, a paper published in GaBI Journal provides a comprehensive update on the regulation and reimbursement of biosimilars in Canada [6].
Australia’s Therapeutic Goods Administration (TGA) launched a similar consultation on naming in 2017, offering four options for the naming of biologicals. As a result, the agency decided in 2019 to continue using the same system: the Australian biological name (without a specific suffix). However, they also require the product’s trade name and non-proprietary name to be stated when reporting an adverse event in order to improve pharmacovigilance. During the Generic and Biosimilar Medicines Association’s Biosimilar Week in Australia, the 2019 Biosimilar Education Program for healthcare professionals was launched, alongside a dedicated Biosimilar Hub, which provides guidance for patients, prescribers and pharmacists.
In England, the National Health Service (NHS) updated its ‘What is a Biosimilar Medicine?’ document in June 2019, for both clinical and non-clinical stakeholders. The document aims to support the safe, effective and consistent use of biosimilars. In Ireland, an initiative has been introduced which offers hospitals Euros 500 for each patient they switch to a biosimilar. The incentive scheme relates to adalimumab and etanercept biosimilars, which were approved in the EU in 2016/7. In the scheme, money will be paid directly to hospitals, which it is hoped will help save over eight million Euros. Germany meanwhile introduced a bill which provides a legal framework for the automatic substitution of biosimilars by pharmacists.
Elsewhere, representatives of the Gulf countries (which include Saudi Arabia, United Arab Emirates (UAE), Yemen, Kuwait and Qatar) met with experts from Europe and the US to discuss the global harmonization of biosimilars, discussing issues related to production, regulation and nomenclature across the globe [7]. The UAE’s regulatory strategy for biosimilars was also discussed at the Generics and Biosimilars Initiative’s 2nd MENA Stakeholder Meeting on Regulatory Approval, Clinical Settings, Interchangeability and Pharmacovigilance of Biosimilars [8].
International
WHO began 2019 by releasing a new Q&A on biosimilars evaluation. The document deals with issues including licensing, evaluation and pharmacovigilance and is based on questions frequently asked by regulators.
Shortly after, the organization released an update to their Global Benchmarking Tool (which is used to evaluate regulatory systems around the world). The revised tool included the move towards ‘WHO-listed authorities’ (WLAs), replacing the previous ‘Stringent Regulatory Authorities’ (SRAs). The changes mean all existing SRAs will now be regarded as ‘WHO-listed’ and additional authorities will be designated, based on the Global Benchmarking Tool and a confidence-building process. The decision to become a WLA will be a voluntary process, undertaken at the request of the country.
In March 2019, WHO provided the results for their pilot project on the prequalification of two anticancer biosimilars: rituximab and trastuzumab, among the first monoclonal antibody treatments to be listed in the WHO Model List of Essential Medicines. WHO’s prequalification programme offers guidance on the acceptability of medical products ahead of their procurement to UN agencies and WHO Member States. All prequalified products must meet WHO requirements on quality, safety and efficacy.
Near the end of 2019, WHO announced a groundbreaking prequalification scheme for insulin. The scheme aims to increase the flow of quality-assured insulin products, which is especially important for low-income countries. WHO invited drug manufacturers who wish to have their products evaluated by the programme to submit their information to the prequalification team.
Collaborations, agreements and investments in biosimilars
Kicking off 2019, Samsung Bioepis announced in January that it was expanding into China through a licensing agreement with 3SBio. The agreement covers multiple candidates, including a bevacizumab copy biological. In the same month, Coherus announced an agreement with AbbVie giving Coherus global rights to commercialize their adalimumab biosimilar, CHS-1420, given that they pay royalties to AbbVie.
In March 2019, Biogen entered into an agreement with Fujifilm. Under the agreement, Fujifilm acquired Biogen’s manufacturing facility in Hillerød, Denmark. It will become Fujifilm’s fourth biopharmaceutical contract development and manufacturing site and will retain the existing workforce of around 800 employees.
April 2019 saw Iceland’s Alvotech and Japan’s Fuji Pharma make a binding agreement for the commercialization of Alvotech’s ustekinumab biosimilar, Stelara, in Japan. Fuji Pharma invested in Alvotech in 2018, giving them a 4.2% stake in the company. Two Belgian companies, SYnAbs and Univercells, announced on 20 April 2019 the signing of an agreement for an undisclosed biosimilar. Under the agreement, contract research organization SYnAbs will develop an assay to measure features of a biosimilar monoclonal antibody developed by Univercells. On 30 April 2019, Sandoz signed an agreement with EirGenix of Taiwan to market a trastuzumab biosimilar. The agreement gives Sandoz the exclusive rights to globally commercialize EirGenix’s proposed trastuzumab biosimilar, EG12014.
South Korea also announced a massive investment in April 2019. The government announced that it would be investing almost Won 3 trillion (US$2.6 billion) into biotech-related research and commercialization, 2.9% on-year increase from 2018.
In May 2019, Boehringer Ingelheim signed a long-awaited licensing deal for its adalimumab biosimilar Cyltezo. The deal, signed with originator company AbbVie, will allow Boehringer to launch Cyltezo on the US market in July 2023. At the end of May 2019, German generics manufacturer Stada Arzneimittel and Swedish biotech company Xbrane Bioscience announced an expansion to their biosimilar development partnership, to focus on products with patent expiration dates between 2025−2030.
Pharmapark announced in July 2019 a deal to exclusively supply Prestige’s trastuzumab biosimilar (Tuzune) in Russia, followed by the news that Celltrion and the Nan Fung Group had formed a joint venture (named Vcell Healthcare) to develop and sell copy biologicals in China. On 29 July 2019, Alvotech and Cipla Gulf announced an exclusive partnership for the commercialization of an adalimumab biosimilar (AVT02) in a range of emerging markets. The product is in phase III development and will be filed with EMA and FDA in 2020.
In September 2019, Sandoz and Polpharma announced a worldwide agreement giving Sandoz the commercialization rights to a natalizumab biosimilar. Following regulatory approval, Sandoz will commercialize and distribute the biosimilar in all markets via an exclusive global licence. Similarly, Shanghai Pharma and Biocad agreed to a joint venture to develop, make and sell cancer and autoimmune copy biologicals in China. Finally, Biocon announced in September 2019 that its subsidiary, Biocon Biologics, had acquired a Pfizer research facility in India. The facility houses facilities for cell-line development and drug substance process development and is expected to employ around 250 scientists.
Alvogen in October 2019 announced exclusive commercialization agreements to distribute and market a teriparatide biosimilar in Canada, Israel and South Korea, while its subsidiary Alvotech formed an agreement with Stada for the commercialization of seven biosimilar candidates.
In December 2019, Japanese companies Gene Techno Science and Kishi Kasei announced an agreement for the joint development of an Eylea (aflibercept) biosimilar. The agreement also includes Fuso Pharmaceuticals, who have developed an aflibercept-producing cell line. Finally, Samsung Bioepis and Biogen expanded their agreement to include two ophthalmology biosimilars and to enable Biogen to commercialize Samsung Bioepis’ anti-inflammatories in Europe for an additional five years.
Summary and recommendations
2019 has been a successful year for many in the biosimilars market, not only those in industry but also for patients. Instances of success were highlighted by a presentation at the DIA’s Biosimilars Conference in September 2019; trastuzumab for example, has halved recurrences of HER2+ breast cancer, while TNF-α inhibitors have revolutionized care in IBD, and β-interferons have reduced relapses in MS. Increasingly available biosimilars of these drugs are increasing access and reducing costs to patients around the world.
However, many challenges remain for the biosimilars market, not least the fact that the number of blockbuster biologicals with patents due to expire is decreasing. Going forward, it is important to take action to shape a sustainable and competitive biosimilars market in the long term.
Key recommendations in this regard include changes to market dynamics, such as considering removing mandatory price cuts for originators, regulatory changes such as reducing the time it takes to get a biosimilar to market, implementing provider incentives or prescription quotas to promote biosimilar use, as well as awareness raising initiatives to improve education for both patients and physicians. Perceptions of biosimilars are improving – a recent survey of gastroenterologists showed that only 19.5% felt little or no confidence in the use of biosimilars, compared to 63% in 2013 – but more work needs to be done in this area.
Amid all of this, however, it is essential that it is the decision of the patient – with the input of their healthcare provider – whether to use a biosimilar or an originator biological.
Finally, efforts to achieve regulatory harmonization will be essential in enabling the consistent use of biosimilars worldwide. Initiatives are paving the way for worldwide standards, which will ultimately build consumer confidence in biosimilars.
Editor’s comment
European Medicines Agency regulatory requirements ensure the same high standards of quality, safety and efficacy for biosimilars as for originator biologicals, and also include a rigorous comparability exercise with the reference product but they are not universally accepted by regulatory bodies outside of the European Union (EU). It should be noted that copy biologicals approved in China, similar biologics approved in India, or non-originator biologicals approved in Russia might not have been authorized if they had been subjected to the strict regulatory processes required for approval of biosimilars in the EU.
Competing interests: None.
Provenance and peer review: Article prepared with data from GaBI Online and GaBI Journal; internally peer reviewed.
Eleanor Bird, MSc, GaBI Journal Editor
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in the US [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Feb 12]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-the-US
2. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Feb 12]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
3. Government of Canada. Policy statement: clarifying the appropriate regulatory pathway for subsequent entry low molecular weight heparins [homepage on the Internet]. [cited 2020 Feb 12]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/lmwh-pol-hfmm-eng.php
4. Simeons S, Le Pen C, Boone N, et al. How to realize the potential of off-patent biologicals and biosimilars in Europe? Guidance to policymakers. Generics and Biosimilars Initiative Journal. 2018;7(2):70-4. doi:10.5639/gabij.2018.0702.014
5. GaBI Online – Generics and Biosimilars Initiative. How can EU policymakers access the potential of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Feb 12]. Available from: www.gabionline.net/Biosimilars/Research/How-can-EU-policymakers-access-the-potential-of-biosimilars
6. Siu ECK, Tomalin A, West K, et al. An ever-evolving landscape: an update on the rapidly changing regulatory and reimbursement of biosimilars in Canada. Generics and Biosimilars Initiative Journal. 2019;8(3):107-18. doi:10.5639/gabij.2019.0803.014
7. Trifirò G, Al-Foheidi M, Alhomaidan AM, Aljedai AH, Alkholief MA, Alsenaidy MA, et al. First GCC stakeholder meeting on approval process, interchangeability/substitution and safety of biosimilars 2017 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(4):158-63. doi:10.5639/gabij.2018.0704.032
8. Laslop A, Wang J, Thorpe R. 2nd MENA stakeholder meeting on biosimilars 2018 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2019;8(2):76-87. doi:10.5639/gabij.2019.0802.009
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/top-developments-in-biosimilars-during-2019.html
Top developments in biosimilars during 2018
Submitted: 5 February 2019; Revised: 14 March 2019; Accepted: 20 March 2019; Published online first: 2 April 2019
The global market for biosimilars is valued at approximately US$4 billion, and it is growing rapidly as patents expire on an increasing number of biological drugs.
A number of important milestones were achieved during 2018 regarding approval of biosimilars.
Biosimilar approvals in 2018
Argentina
Argentina’s Administración Nacional de Medicamentos, Alimentos y Tecnología Médica approved Laboratorio Elea’s ophthalmic medicamento biológico similar Lumiere (bevacizumab biosimilar) in August 2018.
Asia
In Asia, a number of biosimilars were approved and launched in 2018. Japan’s medicines regulatory agency, the Pharmaceuticals and Medical Devices Agency (PMDA), approved its first darbepoetin alfa biosimilar as well as Pfizer’s infliximab biosimilar (marking Pfizer Japan’s first biosimilar approval in the country). Two South Korean firms have since announced that they have submitted applications to PMDA for approval of their proposed darbepoetin alfa biosimilars. The first etanercept biosimilar was launched in Japan in 2018 (Etanercept BS), as well as trastuzumab biosimilar 2 and agalsidase beta BS. The launch of CKD Pharma’s darbepoetin alfa biosimilar is also underway in Japan. In South Korea, the insulin glargine biosimilar Glarzia and etanercept biosimilar Eucept were approved by the Korean Ministry of Food and Drug Safety, and the trastuzumab biosimilar Samfenet and insulin glargine biosimilar Glarzia were launched. The China Food and Drug Administration (CFDA) accepted applications for approval of adalimumab copy biologicals from Hisun, Bio-Thera and Innovent. In India, a ‘similar biologic’ of adalimumab, Mabura, was launched in January 2018, and the ‘similar biologic’ of trastuzumab Hervycta was launched in July.
Australia
Australia’s drug regulator, the Therapeutic Goods Administration (TGA) approved Mylan’s insulin biosimilar Semglee, and trastuzumab biosimilars Truxima and Herzuma in 2018.
Canada
Health Canada approved Apotex’s pegfilgrastim biosimilar Lapelga in 2018. Sandoz’s etanercept biosimilar Erelzi was added to Quebec’s public drug plan in early 2018 after gaining approval from Health Canada in 2017.
Europe
The European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) recommended marketing authorization for two pegfilgrastim biosimilars, Mylan/Biocon’s Fulphila and Accord Healthcare’s Pelgraz, as well as Amgen/Allergan’s trastuzumab biosimilar Kanjinti, and Pfizer’s bevacizumab biosimilar Zirabev. EMA is reviewing Mabion’s rituximab biosimilar Mabion CD20.
The European Commission (EC) approved a number of biosimilars in 2018, including adalimumab biosimilars Halimatoz, Hefiya and Hyrimoz from Sandoz and Cyltezo from Boehringer Ingelheim. Trastuzumab biosimilars Herzuma from Celltrion, Trazimera from Pfizer and Ogrivi from Mylan/Biocon, and pegfilgrastim biosimilars Udenyca from Coherus BioSciences, Pelmeg from Cinfa Biotech and Ziextenzo from Sandoz were also approved, as well as Amgen’s bevacizumab biosimilar Mvasi and Sandoz’s infliximab biosimilar Zessly.
Biosimilars launched in Europe in 2018 include adalimumab biosimilars Amgevita from Amgen, Imraldi from Samsung Bioepis, and Hulio from Mylan/Fujifilm. Samsung Bioepis’s trastuzumab biosimilar Ontruzant and Mylan/Biocon’s insulin glargine biosimilar Semglee launched in the UK.
USA
In 2018, the US Food and Drug Administration (FDA) approved a number of biosimilars including its first rituximab biosimilar Truxima from Celltrion, the follow-on insulin lispro Admelog from Sanofi, trastuzumab biosimilar Herzuma, and adalimumab biosimilar Hyrimoz. FDA also approved the pegfilgrastim biosimilars Fulphila and Udenyca, and two biosimilars from Pfizer – its epoetin alfa biosimilar Retacrit and its filgrastim biosimilar Nivestym.
FDA rejected a number of biosimilar applications in 2018. These include initial rejections of rituximab biosimilar Truxima and trastuzumab biosimilar Herzuma. FDA also rejected applications for trastuzumab biosimilars from Pfizer and Amgen, Sandoz’s rituximab biosimilar and Evolus’s botulinum toxin biosimilar.
FDA is reviewing Samsung Bioepis’s submission for its adalimumab biosimilar Imraldi. Taiwan-based Tanvex BioPharma submitted its first biosimilar application to FDA for its filgrastim biosimilar TX01 in October 2018.
Clinical studies for biosimilars
In 2018, results were reported from a number of clinical studies investigating the efficacy and safety profile of biosimilars including adalimumab, bevacizumab, filgrastim, infliximab, pegfilgrastim, rituximab and trastuzumab. Some of these clinical studies are summarized below.
Adalimumab
Adalimumab is the most commonly prescribed biological. It is approved for the treatment of rheumatoid arthritis (RA). as well as psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis. Results from the VOLTAIRE-RA equivalence phase III trial, which compared outcomes in patients with active RA randomized to Boehringer’s adalimumab biosimilar Cyltezo or AbbVie’s reference product Humira, and patients switched from Humira to Cyltezo, were published in 2018. The results indicate that efficacy outcomes and the safety and immunogenicity profiles were similar between the groups. In September, Boehringer announced positive phase III data for Cyltezo as a treatment for moderate-to-severe chronic plaque psoriasis. The results, according to Boehringer ‘confirm that Cyltezo is equivalent to Humira, with no clinically meaningful differences in efficacy, safety and immunogenicity in people with moderate-to-severe chronic plaque psoriasis’.
A phase III trial compared the efficacy and safety profile of Sandoz’s adalimumab biosimilar, Hyrimoz (GP2017) with reference adalimumab in patients with moderate-to-severe RA with inadequate response to disease modifying anti-rheumatic drugs. The results from the study demonstrate, according to the authors, that Hyrimoz is of equivalent efficacy to the reference adalimumab in this patient group and that the ‘safety and immunogenicity of GP2017 and reference adalimumab were similar and consistent with clinical experience with reference adalimumab’.
Samsung Bioepis and Biogen conducted an analysis combining data from three phase III trials that compared the efficacy and safety of anti-tumour necrosis factor (TNF) biosimilars to their reference biologicals. The pooled analysis included data from Benepali (etanercept biosimilar), Flixabi (infliximab biosimilar) and Imraldi (adalimumab biosimilar) and assessed the impact of anti-drug antibodies (ADAs) on efficacy and tolerability, as well as radiographic progression by disease activity state. Immunogenicity data from 1,710 patients with moderate-to-severe RA revealed that the incidence of ADAs was comparable between the biosimilars and their reference products, and that the development of ADAs is associated with reduced clinical efficacy and increased incidence of injection site reactions/infusion-related reactions in patients with RA.
Bevacizumab
Researchers from mAbxience presented data from a phase I trial supporting the pharmacokinetic (PK) bioequivalence of their bevacizumab biosimilar BEVZ92 with originator bevacizumab Avastin as a first-line treatment in patients with metastatic colorectal cancer.
A phase III clinical trial comparing Pfizer’s candidate bevacizumab biosimilar PF 06439535 with originator bevacizumab in patients with advanced non-squamous non-small cell lung cancer found that PF 06439535 and reference bevacizumab had similar objective response rates, progression-free survival, overall survival, and safety and immunogenicity profiles.
Epoetin alfa
In January 2018, Japan-based collaborators Kissei and JCR announced positive results from the phase III trial of their candidate darbepoetin alfa biosimilar, JR 131 in renal anaemia patients with chronic kidney disease. The results showed that ‘JR 131 demonstrated equivalence in efficacy and safety compared with darbepoetin’, according to the companies.
Researchers in Italy carried out a prospective cohort study to compare the safety profiles of biosimilars with respect to their reference products in a nephrology setting. The results confirm the comparable safety profiles of originator and biosimilar epoetin alfa drugs when used in patients receiving dialysis.
Etanercept
Results from YL Biologics’s global phase III trial showed that its etanercept biosimilar YLB113 had similar efficacy and a similar safety profile to the reference Enbrel in Japanese patients with RA.
Filgrastim
Researchers from Germany compared results from the phase III PIONEER trial of Sandoz’s biosimilar filgrastim (Zarxio) with post-approval data from the MONITOR-GCSF study, concluding that Zarxio ‘prevented febrile neutropenia in a randomized controlled trial and in real-world practice in breast cancer patients receiving (neo-)adjuvant chemotherapy’.
Infliximab
The efficacy and safety profile of Amgen’s infliximab biosimilar ABP 710 was compared with the originator biological infliximab, Johnson & Johnson/Merck’s Remicade, in patients with moderate-to-severe RA in a phase III clinical trial. According to Amgen, the results confirm that ABP 710 was non-inferior to Remicade but could not rule out superiority based on the primary endpoint which measured response by a 20% or greater improvement defined by the American College of Rheumatology Criteria. Amgen believes that the totality of the evidence generated ‘supports ABP 710 as highly similar to the reference product’. Overall, the safety and immunogenicity profiles of ABP 710 and Remicade were comparable.
In June 2018, Sandoz announced positive results from its phase III REFLECTIONS B537 02 trial for its infliximab biosimilar Zessly in patients with moderate-to-severe active RA. The results showed, according to Sandoz, that Zessly was comparable to the reference infliximab Remicade in terms of safety profile, efficacy and quality.
Celltrion is developing a subcutaneous version of its infliximab biosimilar Remsima (CT-P13). In June 2018, Celltrion announced that data from its phase I study showed that the subcutaneous administration of Remsima was comparable with intravenous administration of Remsima/Inflectra in terms of efficacy outcomes and safety profile in patients with active Crohn’s disease. In August, Celltrion announced the completion of a phase III trial with subcutaneous Remsima.
Insulin
Authors of a systematic review assessing the similarity of biosimilar insulins to their reference products concluded that the studies identified ‘suggest similar clinical efficacy and safety’ compared with their reference products. They added that ‘these biosimilars may be considered as alternative options for non-basal and basal insulin therapy in patients with type 1 and type 2 diabetes’.
Omalizumab
Indian generics maker Glenmark announced in July 2018 that results from a phase I study suggest similarity in PK, PD and safety and immunogenicity profiles between its proposed omalizumab biosimilar GBR 310 and the originator product Xolair.
Pegfilgrastim
Canada-based Apobiologix reported results of an international phase III trial investigating biosimilarity between its proposed pegfilgrastim biosimilar Lapelga and Amgen’s US-licensed and EU-approved reference product Neulasta in early stage breast cancer patients. The results demonstrated that the biosimilar was highly similar to Neulasta with regard to the efficacy and safety endpoints investigated in the study.
Rituximab
In a phase III clinical trial in patients with low tumour burden follicular lymphoma (FL), Celltrion’s rituximab biosimilar CT-P10 and the originator rituximab, Roche’s MabThera/Rituxan demonstrated equivalent efficacy and PKs. Celltrion also recently presented positive phase III results for CT-P10 in patients with advanced FL, reporting that the efficacy outcomes and safety profile of CT-P10 was comparable to originator rituximab.
The efficacy and safety profile of Shanghai Henlius Biotech’s rituximab copy biological HLX01 in previously untreated patients with CD20+ diffuse large B-cell lymphoma was investigated in a phase III trial. The authors concluded that the results ‘successfully demonstrate equivalence in PK and pharmacodynamics (PDs), efficacy and safety between HLX01 and originator rituximab sourced from China’.
Pfizer’s proposed rituximab biosimilar PF 05280586 was compared with originator rituximab in a phase III trial in patients with CD20+, low tumour burden FL. The results of the study demonstrated, according to the authors, that the ‘efficacy, safety and immunogenicity, PK and PD of PF 05280586 and rituximab-EU were similar’.
A multinational phase III trial comparing PK and PD of mAbxience’s proposed biosimilar RTXM83 with originator rituximab in patients with diffuse large B-cell lymphoma supported the claim of biosimilarity.
Studies assessing rituximab biosimilars tend to be large and expensive clinical trials assessing PK and PD. Researchers from Austria and Canada piloted a different approach using the half-maximal effective dose (ED50) in healthy volunteers to investigate the effects of a proposed biosimilar rituximab with the originator product. The authors concluded that their trial demonstrates an alternative, cost-effective, more sensitive approach to comparing PD of biosimilars of rituximab using pharmacological principals.
Trastuzumab
Celltrion presented phase III clinical trial data supporting the PK similarity of its trastuzumab biosimilar CT-P6 to the originator trastuzumab (Roche’s Herceptin) in patients with human epidermal growth factor receptor positive (HER2+) early-stage breast cancer.
In early 2017, Mylan and Biocon reported that the results from their phase III HERiTAge study ‘confirm the efficacy, safety and immunogenicity of Ogivri, in comparison to branded trastuzumab’. In 2018, the authors reported the results of the secondary endpoints of safety and immunogenicity, concluding that ‘maintenance monotherapy with FDA-approved trastuzumab-dkst [Ogivri] after combination with taxane was well tolerated, with safety and efficacy profiles similar to originator trastuzumab’.
Results from a clinical trial assessing Samsung Bioepis’s trastuzumab biosimilar Ontruzant in patients with HER2+ early-stage breast cancer ‘demonstrate the long-term safety profile’ of Ontruzant, according to the authors.
A multinational phase III trial investigated the efficacy, safety and immunogenicity profile of Shanghai Henlius Biotech’s trastuzumab copy biological HLX02 and originator trastuzumab in HER2+, locally recurrent or previously untreated metastatic breast cancer patients. The results showed, according to the authors, that the ‘three-way PK and safety equivalence of HLX02 and reference trastuzumab were demonstrated’.
Regulation and guidance on biosimilars
Regulation
The EC announced a proposal to refine the intellectual property rules in Europe in May 2018. This included an ‘export manufacturing waiver’ to Supplementary Protection Certificates (SPCs; which extend the protection of a product protected by an underlying patent also held by the applicant). The waiver allows EU-based companies to manufacture a generic or biosimilar version of an SPC-protected medicine during the term of the certificate, if done exclusively for the purpose of exporting to a non-EU market where protection has expired or never existed. The EC claims the waiver ‘will support Europe’s pioneering role in pharmaceutical research and development,’ however, some industry groups expressed concerns. The European Federation of the Pharmaceutical Industries and Associations (EFPIA) stated that the waiver ‘jeopardises patient access to innovative treatments’ while the European Biopharmaceutical Enterprises said that it ‘reduces intellectual property rights and de-values the incentives framework’.
In July 2018, the Australian TGA made an amendment to its biological’s regulation. Under the new regulation, a substance will be regulated as a biological rather than a blood component if it is subject to processing involving ‘more than minimal manipulation’. This broader definition incorporates conditioned serum and adipose-derived cell extracts, which had previously been excluded from regulation. This regulation is similar to EMA’s approach for stem cell products and also mimics similar regulations in the US. As of September 2018, manufacturers are no longer able to request parallel processing when submitting a biosimilar application for approval until after the TGA has made a determination of biosimilarity.
In August 2018, the US announced a new ‘understanding’ on trade with Mexico as part of efforts to re-negotiate the North American Free Trade Agreement between Canada, Mexico and the US. This includes providing 10 years of exclusivity for biologicals which could potentially hinder competition and access to biosimilars.
Guidance
As part of plans to provide additional clarification on its 2009 guidelines on the evaluation of biosimilars, the World Health Organization (WHO) drafted a document in 2018 providing answers to questions that regulators have posed over the past eight years.
In June 2018, FDA published guidance providing recommendations to industry on formal meetings between FDA and sponsors or applicants relating to the development and review of biosimilars or interchangeable biological products. It also issued final guidance on the Biosimilar User Fee Act (BsUFA) II fee structure, finalizing the changes introduced since BsUFA I. Also, in June, FDA announced that it had withdrawn its draft guidance on analytical studies of biosimilars following public comments. In July, FDA issued two new guidance documents providing recommendations to industry on labelling requirements for prescription drugs and biologicals. In December 2018, the agency released four guidance documents and a proposed rule on the definition of a ‘biological product’. This guidance provides greater clarity on scientific and regulatory considerations for the development of biosimilar and interchangeable products, and describes how it plans to transition certain biological products approved as drugs to be licensed as biologicals. FDA also clarified that products reclassified as biologicals will not receive the 12-year exclusivity period afforded to newly licensed biologicals.
A proposed amendment to the Hatch-Waxman Act was filed in June 2018 by Senator Hatch, aiming to restore the careful balance the Hatch-Waxman Act struck to incentivize generics development. The amendment requires that a generic drug applicant must not use an inter partes review or post-grant review for a patent covering the innovator drug if it plans to use the abbreviated Hatch-Waxman pathway to approval.
South Korea’s financial authority released new ‘relaxed’ guidelines for how drug companies should list research and development ‘R & D’ spending as assets in September 2018. The guidelines say that drug companies can capitalize their research spending after regulatory approval of phase III trials, and biosimilar makers can capitalize their research spending from the phase I stage.
Position statements
In March 2018, the Spanish Society of Gastroenterology (Sociedad Española de Patología Digestiva) updated its position statement in support of the development and use of biosimilars for the treatment of inflammatory bowel disease (IBD). This reflects the growing body of evidence supporting the biosimilarity of Remsima/Inflectra (CT-P13) with originator infliximab as a treatment for IBD. It follows the 2017 update to the European Crohn’s and Colitis Organisation position statement which supports switching from originator Remicade to biosimilar infliximab in IBD patients.
In March 2018, the American Society of Clinical Oncology (ASCO) issued a position statement on biosimilars in oncology. The statement reflects the society’s views on issues such as naming, labelling, safety and efficacy, interchangeability, switching and substitution, and prescriber and patient education. In May, the Brazilian Society of Clinical Oncology issued a position statement on ‘follow-on biological products’ in oncology addressing similar issues to those ASCO addressed, as well as the potential impact that ‘follow-on biological products’ may have on the financial burden in healthcare.
The Association of British Clinical Diabetologists issued a position statement on the use of biosimilar insulin in December 2018. The statement summarises information on the advantages and disadvantages of using biosimilar insulins and gives the association’s position on when biosimilar insulins should be used.
Biologicals naming
Naming of biologicals remained a contentious issue during 2018. WHO first proposed the concept of a biological qualifier (a four-letter alphabetic code for all biologicals) in 2014. In December 2018, authors from the US urged WHO to finalise its guidance, stating that ‘precise naming will improve patient safety by reducing confusion and mishaps in prescribing and dispensing’ [1].
The FDA’s finalized guidance on naming biologicals assigns a non-proprietary or ‘proper’ name to all originator biologicals, related biologicals and biosimilars. The proper name applies prospectively and retrospectively and consists of a combination of the ‘core name’ and a meaningless suffix composed of four lowercase letters. Concerns have been raised that this naming convention creates the impression that the clinical effects of a biosimilar may differ meaningfully from the reference product. Groups such as the International Generic and Biosimilar Medicines Association, have called for biosimilars to share the proper name with the reference biological.
Following an open consultation on how to name biologicals, Australia’s TGA concluded in February 2018 that ‘implementation will involve making the product’s trade name, as well as the non-proprietary name, a mandatory field when reporting an adverse event to the TGA in order to provide product specificity’.
Interchangeability and substitution
According to FDA, ‘an interchangeable biological product, in addition to meeting the biosimilarity standard, is expected to produce the same clinical result as the reference product in any given patient, and for a product that is given to a patient more than once, the risk in terms of safety and effectiveness of alternating or switching between the interchangeable and the reference product is not greater than the risk of using the reference product without alternating or switching’. Interchangeability is important because without it, payers are unlikely to encourage switching from an originator product to a biosimilar, even if they pursue formulary policies which favour biosimilars for new start patients.
A review published in early 2018 reported that the slow development of clear US guidance on interchangeability is limiting price competition. At the FDA biosimilars meeting in September 2018, there was a call to include interchangeability information in the ‘Purple Book’.
The debate over substitution practices continues. Pfizer conducted an internal global survey of 82 countries in which it examined biosimilar pharmacy-mediated substitution in 2017 and found that no universal position is held worldwide on substitution of biosimilars [2, 3].
According to the US Biologics Price Competition and Innovation Act of 2009 (BPCI Act), only a biological that has been approved as interchangeable may be substituted for the reference product without the intervention of the healthcare provider who prescribed the reference product. Many US states are considering, or have introduced, laws related to the substitution of biosimilars at the pharmacy level. In February and March 2018, bills in Michigan, South Dakota and West Virginia were passed to authorize pharmacists to substitute a biosimilar for a prescribed biological product if the biosimilar has been approved by FDA as ‘therapeutically equivalent’ and the prescriber has not indicated ‘d.a.w.’ (dispense as written) on the prescription.
Switching
Switching is defined as a decision by the treating physician to exchange one medicine for another medicine with the same therapeutic intent in patients who are undergoing treatment. This can involve switching between a reference product and a biosimilar, or between biosimilars. Attitudes to switching continue to change.
The European Specialist Nurses Organisations launched a guide for switching from reference biologicals to biosimilars in June 2018. The guide is specifically directed towards specialized nurses and aims to provide information to facilitate interactions with patients switching from a reference biological to a biosimilar (or vice versa), or between biosimilars of the same reference product.
Some of the trials investigating switching to biosimilars which were published in 2018 are summarized below.
Adalimumab
Results from the VOLTAIRE-RA equivalence phase III trial in patients with active RA showed that a switch from reference adalimumab Humira to biosimilar Cyltezo had no significant impact on efficacy outcomes. Safety and tolerability profiles were similar in the group continuing treatment with Humira and the group switched to Cyltezo.
Etanercept
Results from a UK study suggest that switching from originator etanercept Enbrel to biosimilar Benepali is safe and effective in a hospital setting for patients with RA, ankylosing spondylitis and psoriatic arthritis.
Results from the phase III EQUIRA study suggest that there is no impact on efficacy, safety or immunogenicity outcomes when patients with moderate-to-severe RA are switched from reference etanercept to Sandoz’s etanercept biosimilar, Erelzi.
Infliximab
Following the landmark NOR-SWITCH clinical trial which failed to find inferiority when switching from originator infliximab Remicade to biosimilar Remsima/Inflectra, a number of studies have investigated the switch from Remicade to other biosimilars.
The phase III REFLECTIONS B537–02 trial evaluated the effects of switching from Remicade to the biosimilar Zessly in patients with moderate-to-severe active RA. The research indicated that patients who switched from Remicade to Zessly had similar efficacy outcomes and a similar safety and immunogenicity profile to patients who continued to use Remicade.
French researchers assessed the long-term retention rate of Remsima/Inflectra after switching from Remicade. They compared this with the retention rate observed in a historic cohort of patients treated with Remicade from a previous study carried out by their group. According to the authors, the results suggest that a nocebo effect occurred in the first weeks after the switch which initially lowered the biosimilar retention rate. After this initial nocebo effect, originator and biosimilar retention rates appeared to be identical, supporting the safety, efficacy and acceptability of the switch in the long term. The authors conclude that ‘initial information provided before the switch appears to be crucial for biosimilar acceptance and therefore reducing the nocebo effect’.
Results from a retrospective cohort study conducted by Italian and Spanish researchers on the immunogenicity of Remsima/Inflectra suggest that there is full interchangeability between infliximab biosimilars with respect to immunogenicity profiles.
Insulin
Researchers carried out a retrospective chart review using the electronic medical records of patients hospitalized at a Chinese hospital from 2005 to 2016. They found that switching from copy insulin glargine biological Basalin to originator Lantus is effective in patients with diabetes. However, in light of another recently published study that had contrasting findings, the authors recommend that further studies be carried out.
Cost savings associated with biosimilar use
Use of biosimilars can result in significant savings to health systems.
US-based biosimilars specialist Coherus announced in November 2018 that it would price its pegfilgrastim biosimilar at a 33% discount to the originator product, Amgen’s Neulasta. Also, in November, it was reported that Pfizer launched its epoetin alfa biosimilar, Retacrit, in the US at a significant discount. According to reports, Pfizer began shipping Retacrit to wholesalers at a 57% discount compared with Amgen’s Procrit and a 33.5% discount compared with Johnson & Johnson’s Epogen.
A study assessing the use and costs of targeted therapies for cancer treatment in Southern Italy from 2010 to 2014 estimated that, assuming an uptake of biosimilar trastuzumab and rituximab of 50%, almost Euros 1 million could be saved annually.
Researchers from Canada compared the cost-effectiveness of infliximab biosimilar Inflectra to originator Remicade for the management of Crohn’s disease. Using a Markov model with a 5-year time horizon and a willingness-to-pay threshold of CA$50,000 per quality-adjusted life year, they found that Inflectra has an 83% probability of being cost-effective while Remicade has a 17% probability of being cost-effective.
In February 2018, Canadian not-for-profit healthcare benefits specialist, Green Shield Canada, announced that it was ‘the first major benefits carrier to preferentially list biosimilars’. In September 2018, New Zealand’s Pharmac announced that it will stop funding Janssen’s brand-name epoetin alfa biological Eprex and will only fund the biosimilar epoetin alfa Binocrit supplied by Novartis.
Barriers to uptake of biosimilars
The outlook for biosimilars looks promising. There are an increasing number of global biosimilar approvals based on positive efficacy and safety data, and cost analyses are generally finding that biosimilars have the potential to provide significant savings for healthcare systems globally. A number of countries have implemented strategies to stimulate the biosimilars market. In July 2018, FDA released its Biosimilars Action Plan, which aims to improve the efficiency of the biosimilar and interchangeable product development and approval process. France is strongly in favour of biosimilars and aims to reach 80% biosimilar penetration by 2022 as part of its 2018–2022 National Health Strategy, representing an increase from the previous year’s 70% target. This follows a revamp of the substitution policy in 2017 and the creation of a biosimilar registry. Meanwhile, Ireland has updated its National Biosimilars Medicine Policy in a bid to increase biosimilar use in the country.
However, barriers to the uptake of biosimilars still exist.
One of the main barriers is the perception that biosimilars may not have been studied thoroughly enough and therefore that they may not be safe. This perception, according to a recent review, comes from a lack of knowledge about the scientific principles underlying the development and licensing of biosimilars, and the inappropriate labelling of non-originator and copy-version products as biosimilars. The authors recommend that regulatory authorities develop a specific regulatory framework for approving biosimilars that is distinct from the regulatory procedures previously applied to copy-version products. Communication and education of patients is also important, and the review recommends the publication of public assessment reports on biosimilars as a way to increase transparency and public trust in biosimilars. The Biosimilar Working Group of the International Pharmaceutical Regulators Forum published a template for biosimilar assessment reports that could be used by regulators worldwide, entitled Public Assessment Summary Information for Biosimilars.
In a move to increase confidence in biosimilars, EMA and the EC published new material in September 2018 including an animated video for patients that explains key facts on biosimilars and how EMA works to ensure that biosimilars are as safe and effective as their reference biologicals. EMA also published translations of its biosimilar guide for healthcare professionals in Dutch, French, German, Italian, Polish, Portuguese and Spanish.
A recent study found that only 3% of US spending on biologicals is subject to competition from biosimilars. There are therefore huge potential savings to be made with increased use of biosimilars. The study reported that current obstacles to the development and uptake of biosimilars in the US include uncertainty over naming, the slow development of clear guidance on interchangeability, and secrecy about manufacturing processes. In addition, the author believes that consolidation of a reference product and all its biosimilars under a single billing code ‘would probably generate strong price competition’.
Prescribing incentives can be used to increase the uptake of biosimilars. In September 2018, the Australian Pharmaceutical Benefits Scheme announced the implementation of two specific drivers to encourage the use of biosimilars, which it says will complement the Biosimilar Awareness Initiative. These drivers are: 1) encouraging prescribing of a biosimilar brand rather than the reference biological brand for treatment naïve patients; and 2) providing a simpler and faster approval process for prescribing biosimilar brands (streamlined authority), while maintaining an existing higher-level authority requirement for the reference biological brand (written authority). Further, the TGA announced that it will require mandatory reporting of drug shortages from sponsors of medicines in Australia, starting from 1 January 2019.
Knowledge sharing is also an important factor for promoting use of biosimilars. The Spanish Association of Biosimilar Medicines and the Federation of Spanish Medical Scientific Associations signed a collaboration agreement in March 2018 that includes the creation of a joint working group to share up-to-date information on biosimilars. The agreement aims to generate and disseminate knowledge about biosimilars for medical professionals.
Collaborations, agreements, and investment in biosimilars
Many collaborations were formed in 2018, and a number of important investments in biosimilars were made.
In January 2018, Biocon and Sandoz announced a global partnership to develop, manufacture and commercialize new next-generation biosimilars.
Fujifilm Kyowa Kirin Biologics announced in April 2018 that it will partner with Mylan to commercialize its adalimumab biosimilar Hulio. Mylan will be granted exclusive commercialization rights for Hulio in Europe.
US-based biotech Pfenex and China’s NT Pharma announced an agreement in April 2018 concerning PF708, a candidate biosimilar of Eli Lilly’s osteoporosis treatment Forteo (teriparatide). Pfenex granted NT Pharma non-exclusive development and exclusive commercialization rights to PF708 in mainland China, Hong Kong, Singapore, Malaysia and Thailand.
US generics maker Amneal and biosimilar specialist mAbxience announced in May 2018 that they have signed an exclusive licensing and supply agreement in the US for mAbxience’s candidate bevacizumab biosimilar.
India-based Lupin announced in June 2018 that Mylan will commercialize its proposed etanercept biosimilar in a number of markets.
In July 2018, German generics giant Stada and Swedish biotech company Xbrane announced that they had entered into a co-development agreement for Xlucane, a proposed ranibizumab biosimilar.
In September 2018, Iceland-based biopharmaceutical company Alvotech announced that it was entering into a joint venture with China-based Changchun High & New Technology Industries Group, which will enable Alvotech to develop, manufacture and commercialize its biosimilar portfolio in China.
In November 2018, Alvotech and Japan-based Fuji Pharma announced they had entered into a partnership whereby Alvotech will develop and supply biosimilars from its current pipeline and Fuji Pharma will then be responsible for registering and selling the biosimilars in Japan.
AbbVie signed yet more deals regarding biosimilar versions of Humira (adalimumab) in 2018. In July, AbbVie announced patent license agreements with Mylan which will allow Mylan to launch its biosimilar adalimumab in the US in 2023. In October, AbbVie announced global resolution of patent disputes with Sandoz and Fresenius Kabi and in November, it announced agreements with Momenta and Pfizer. These agreements will allow Fresenius Kabi and Sandoz’s adalimumab biosimilars to be launched in the US in September 2023, and Momenta and Pfizer’s biosimilars to be launched in the US in November 2023. Samsung Bioepis reached a settlement with AbbVie in April 2018 under which it could launch its biosimilar Imraldi in the EU in October 2018, and in the US in June 2023. Imraldi has been making waves in Europe, gaining more than 60% of the adalimumab market in Germany as of early 2019. The only company still resisting a deal with AbbVie is Boehringer.
The Orion Group announced in October 2018 that it will distribute Amgen’s adalimumab biosimilar Amgevita in Finland.
According to a study into how the landscape of biosimilars development is changing, manufacturing is moving to Asia, with biosimilars developed in Korea contributing 43% of the global biosimilars value, up from 0% in 2012. Indian generics maker Cadila and Indonesia-based Kalbe are both making advances into the biosimilars field. In February 2018, Cadila announced that it was carrying out a phase I clinical trial for its pegfilgrastim similar biologic, and Kalbe inaugurated its factory in Cikarang, West Java, which will produce biotechnology-based medicines. In March 2018, Lupin announced that it had entered into an agreement with the Council of Scientific and Industrial Research-National Chemical Laboratory and the Department of Science and Technology to conduct research on a continuous purification process development of a ‘similar biologic’ monoclonal antibody. In April 2018, China-based WuXi Biologics, announced that it is expanding its biologicals manufacturing capacity with new facilities planned in China, Ireland, Singapore and the US. Biocon has stated that its biosimilars plant in Bangalore has received a clean bill of health, indicating that it is now compliant with FDA manufacturing requirements. Samsung BioLogics announced in July 2018 that it had been licensed by FDA to manufacture a monoclonal antibody drug product at its first plant.
Discontinuation of biosimilars
The development of some biosimilars was abandoned in 2018.
In October 2018, Merck pulled out of a deal with Samsung Bioepis, terminating the development and commercialization agreement between the two companies for insulin glargine biosimilar Lusduna. Merck has faced significant challenges in bringing Lusduna to the market in the US, including patent litigation from originator manufacturer Sanofi.
German generics giant Stada Arzneimittel announced in March 2018 that it would halt development of its adalimumab biosimilar as part of a move to focus its speciality pharmaceuticals on oncology, central nervous system, diabetes and ophthalmology. US-based biotechnology firm Momenta announced in October 2018 that it would be focusing on two key biosimilars and halt the development of five others. In November 2018, Sandoz announced that it would no longer pursue approval for its rituximab biosimilar (GP2013) in the US ‘at this time’. Boehringer has said that it will not commercialize its approved adalimumab biosimilar Cyltezo in Europe and is discontinuing all biosimilar development activities outside the US.
Summary and recommendation
Key considerations for increasing biosimilar market penetration include increasing harmonization around the world with regard to regulatory approval guidance, and approach to interchangeability and switching practices. Reaching consensus on naming of biologicals and biosimilars is a key factor in ensuring safe dispensing and tracking.
Government policies need to be introduced to support increased uptake of biosimilars, for example, prescribing incentives for healthcare professionals. Patient confidence in biosimilars is key to acceptance, therefore interventions aimed at improving patient education on the efficacy and safety of biosimilars are likely to increase market adoption.
A 2018 review has suggested a number of recommendations to FDA to encourage the faster development and adoption of biosimilars in the US, which were the subject of a citizen petition filed to FDA. These include waiver bridging studies (currently required where there is a non-US product as a reference product), encouraging substitution for naïve patients, allowing in vivo immunogenicity study waivers, making PK profiling clinically relevant, modifying tier testing criteria for analytical similarity, clarifying analytical testing validation, encouraging development of novel testing methods, accepting smaller batch sizes and minimizing clinical studies [4].
Editor’s comment
It should be noted that ‘copy biologicals’ approved in China, ‘similar biologics’ approved in India, and ‘similar biotherapeutic products’ approved in Latin America might not have been authorized following as strict a regulatory process as is required for approval of biosimilars in the European Union. The EMA (European Medicines Agency) regulatory requirements ensure the same high standards of quality, safety and efficacy for biosimilars as for originator biologicals, and also include a rigorous comparability exercise with the reference product.
Competing interests: None.
Provenance and peer review: Article prepared with data from GaBI Online and GaBI Journal; internally peer reviewed.
Sophie Shina, MSc, GaBI Journal Editor
References
1. Pitts PJ. Reilly MS. Medicines regulation in the MENA region and the importance of the World Health Organization’s INN proposal of Biological Qualifier. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):97-100. doi:10.5639/gabij.2018.0703.021
2. Giezen TJ. Global policies on pharmacy-mediated substitution of biosimilars: a summary. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4): 155-6. doi:10.5639/gabij.2017.0604.033
3. Larkin H, Macdonald J, Lumsden R. Pharmacy-mediated substitution of biosimilars – a global survey benchmarking country substitution policies. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):157-64. doi:10.5639/gabij.2017.0604.034
4. Niazi SK. Rationalizing FDA guidance on biosimilars—expediting approvals and acceptance. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):84-91. doi:10.5639/gabij.2018.0702.008
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2019 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/top-developments-in-biosimilars-during-2018.html
US generic prescription drug markets 2004‒2016
Submitted: 6 December 2018; Revised: 12 December 2018; Accepted: 14 December 2018; Published online first: 19 December 2018
Generic drugs are currently central to disease treatment in the US. However, recent policy concerns have focussed on generics supply inadequacies and the perceived increase in prices of some generic drug products. To further explore the changing landscape of US generic prescription drug markets, a study published by the National Bureau of Economic Research (NBER) in the US examines manufacturer entry and exit, the extent of competition and the relationship between supply structure and inflation adjusted prices among generic drugs, between 2004–2016 [1].
In recent years, policy developments in the US have resulted in a shift towards generic drug use. Their share of all retail and mail order dispensed drugs increased from 36% in 1994 to 87% in 2015. In their early years following inception, generic drug products largely provided increased access to safe and effective treatments at prices that continued to decrease. However, after 2009, the prices of some generics started to increase for a number of reasons. It is posited that these include low generics profit margins, concentrated buying power, and pharmaceutical plants failing to meet standards set out by the US Food and Drug Administration (FDA). Since 2012, massive price increases of generics have been observed.
With this in mind, the authors of the study who are based at NBER, sought to answer the following questions: ‘How competitive are markets for generic drugs?’, and ‘How has the competitive market structure varied over time and across drug formulations and therapeutic classes?’. To do this and better understand the landscape of generic prescription drugs in the US, they conducted an empirical evaluation using national quarterly data from QuintilesIMS’s National Sales Perspectives™ (NSP) database between 2004 Q4 and 2016 Q3, on US prescription drug sales. They undertook several descriptive and statistical analyses.
Overall, the team found that between 2004–2016, approximately 500–650 manufacturers supplied prescription drugs (with a steady increase over this period).
Total annual sales revenue derived by both brand and generics manufacturers increased substantially over time, from approximately US$300 billion in 2004 to US$450 billion in 2016. The study found that there was a statistically significant increase in the price of generics in the US over time. This became particularly evident following the implementation of the 2010 Affordable Care Act (ACA) and the 2012 Generic Drug User Fee Amendments (GDUFA I). There was a positive correlation between price increase and the reduction in generics manufacturers over time. Their analyses also isolated four sets of important novel findings relevant to the US generics markets which are summarized in the following sections.
Small generics markets
Through defining the product market by molecule dosage form (that may be manufactured or marketed by multiple suppliers), the team found that the size of generics markets are surprisingly small. Here, the median sizes of drug markets have inflation adjusted sales revenues starting under US$10 million per annum but increasing over time.
Generic entry and exit rates have differing time trends
The number and rate of generic drug products entering the market increased up to 2013, after which they decreased. In contrast, the number and rate of generic drug products exiting has continually increased over time.
Decreasing number of generics manufacturers
The evaluation showed that the median number of generics manufacturers in each market is approximately two and the mean approximately four. Authors of the study consider these numbers to be relatively low. Their evaluation also provides evidence suggesting that there have been decreasing numbers of generic drug suppliers/manufacturers between 2004–2016, particularly following implementation of the Affordable Care Act (ACA) and Generic Drug User Fee Amendments (GDUFA I), both of which have contributed to more drug exits and less entries over time. In addition, evidence suggests that approximately 40 per cent of markets are supplied by just one generics manufacturer. In regard to this, it was also found that there are likely to be two or fewer suppliers of non-oral formulations of generic drugs, whereas for oral formulations are likely to have more suppliers.
Low levels of competition in the generics market
The level of competition in the generics market between 2004–2016 was determined by calculating the Herfindahl-Hirschman Index (HHI), a measure of market concentration. In this case, market concentration is the extent to which sales in a drugs market are dominated by one or more manufacturers. During the study period, the team’s analysis showed a decline in concentration for most therapeutic classes of generic drugs. As such, the HHI values indicate distinctly low levels of competition between manufacturers of generic drug products.
Monopolies reign over US generics
As a result of these findings, the authors conclude that the US generic drugs market is in a relatively steady state with low levels of competition between generics manufactures. This contrasts with previous assumptions and published works that assume a high level of competition involving four or more competitors following patent expiry of originator molecules. The generic drugs market is made up of relatively small revenue products that have a monopoly, or at most a duopoly. The authors note that, considering the lack of competition, it is perhaps surprising that the price of generics has not risen further.
The study concludes with a summary of a number of testable hypotheses based on economic theory that may explain the US generics landscape observed. The authors encourage further investigation of these hypotheses in order to better understand generics markets and the impact of policy on keeping products affordable.
Competing interests: None.
Provenance and peer review: Article abstracted based on published scientific or research papers recommended by members of the Editorial Board; internally peer reviewed.
Alice Rolandini Jensen, MSci, GaBI Journal Editor
Reference
1. Berndt ER, Conti RM, Murphy SJ. The landscape of US generic prescription drug markets, 2004–2016. NBER Working Paper No. 23640.
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/us-generic-prescription-drug-markets-2004%e2%80%922016.html
How does generic drug policy affect market prices?
Submitted: 21 November 2018; Revised: 22 November 2018; Accepted: 28 November 2018; Published online first: 4 December 2018
A study published by the World Health Organization (WHO) compares the effect of different pricing policies for generic medicines in four countries [1]. Using atorvastatin as a case study, their results show that price cuts combined with price-disclosure policies lead to lasting price reductions, while price cuts alone are less effective over the long term.
There are a range of policies available to facilitate the use of generic drugs, such as mandatory generics substitution, where pharmacists must, where possible, substitute a generic drug product for a branded one; reference pricing, where the price of a generic drug is set based on similar medicines; and percentage price reductions, where the price of the generic drug is reduced by a set amount over time. However, little is known about how these policies actually influence the price of generic drugs over time.
This study investigated this issue in the context of atorvastatin, a cholesterol lowering drug marketed by Pfizer as ‘Lipitor’ and the world’s best-selling medication between 1996 and 2012.
Using Australia, New Zealand, the Republic of Korea (South Korea) and Singapore as case studies, the researched evaluated the annual price of atorvastatin, per defined daily dose (DDD, the assumed average maintenance dose per day) supplied, across each country.
They assessed data between 2006 (two years before the generic drug was introduced in all countries) and 2015 (four years after the generic drug was introduced).
Each country uses a slightly different pricing policy. In New Zealand for example, the government applies a tendering system, whereby the market is typically limited to one brand, alongside the use of preferred medicines, which means a subsidy is offered for just one or two medicines within a class.
Before generic atorvastatin reached the market, New Zealand had the lowest price for the drug (just US$0.10/DDD), while the Republic of Korea had a price almost 30 times that (US$2.89/DDD).
The Republic of Korea mandates a 30% reduction in the price of an originator drug the year after patent expiry. The first generic drug product to enter the market is priced 15% below the originator, with a further 10% reduction in both the originator and generic medicine one year after generics entry.
As expected, prices fell in all countries when generic atorvastatin was introduced – except in New Zealand, where prices were already remarkably low. The biggest drop in price after generic entry was in Singapore, where prices decreased by almost 50% in the first year after the generic entered the market. Singapore operates free pricing for manufacturers in the private market, while there is competitive tendering in the public sector. Although there is no mandatory generic substitution in Singapore, public sector institutions are encouraged to use generic drugs.
Four years after generic atorvastatin was introduced, the price had fallen in all countries, however, there were still significant price differences between countries. Prices in New Zealand remained the lowest (US$0.03/DDD) and highest in Korea (US$1.43/DDD). In the middle of the range were Australia (US$0.14/DDD) and Singapore (US$0.47/DDD).
The authors relate these price differences to policy. They suggest that the competitive tendering system utilized by New Zealand is most effective at reducing prices and led to low prices for atorvastatin even before Pfizer’s patent had expired.
However, because prices were already low in New Zealand, they did not decrease after generic versions entered the market.
Pricing policies in Australia, Korea and Singapore were more impactful after the generic entered the market and prices reduced in all countries within four years of generics entry. Prices for atorvastatin decreased by as much as 80% in Australia and Singapore, compared to just 46% in Korea.
Singapore, like New Zealand, employs a tendering system, while Australia applies mandatory price cuts on generic entry combined with price-disclosure policies. Korea meanwhile employs mandatory price cuts alone. This led to immediate price reductions for atorvastatin (25% in the first year) but the impact of this policy was not enduring, and was associated with the smallest price reduction after four years. Overall then, this study suggests that a mix of strategies for pricing generic medicines are most effective and can maximise value for money.
Competing interests: None.
Provenance and peer review: Article abstracted based on published scientific or research papers recommended by members of the Editorial Board; internally peer reviewed.
Eleanor Bird, MSc, GaBI Journal Editor
Reference
1. Roughead EE, Kim DS, Ong B, Kemp-Casey A. Pricing policies for generic medicines in Australia, New Zealand, the Republic of Korea and Singapore: patent expiry and influence on atorvastatin price. WHO South East Asia J Public Health. 2018;7(2):99-106.
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/how-does-generic-drug-policy-affect-market-prices.html
EU risk-sharing agreements between 2000−2015
Submitted: 20 June 2018; Revised: 14 August 2018; Accepted: 18 August 2018; Published online first: 31 August 2018
The ageing population, longer life expectancies, and the increasing cost of drugs are expected to significantly increase the burden on healthcare systems in the coming years. To reduce this burden and increase access to new, innovative and effective drugs in the European Union (EU), risk-sharing agreements (RSAs) between national healthcare payers, health technology assessment (HTA) agencies and the pharmaceutical industry, have been put in place. A recent study published in PharmacoEconomics – Open [1] explores RSAs in the EU from 2000–2015. It shows that during this period there was increased interest in, and uptake of, RSAs and discusses the reasons for this increase.
Risk sharing agreements to facilitate universal access to medicines
In the EU, near universal healthcare coverage is the norm and budgets are limited. To ensure universal access to new, innovative and effective drugs at an acceptable and manageable cost, innovative approaches to pricing and reimbursement of drugs are needed. Pharmaceutical manufacturers are increasingly expected to demonstrate the real-world value for money of their products. Their attempts to demonstrate value are often assessed by HTA agencies, but generally, there are limited data available on products at the time of launch. This means that HTA decisions can be slow, meaning manufacturers can lose out financially and/or payers are left only able to access more expensive and less effective medicines. To address this problem, at the time of launch when the value of a new or innovative medicine is not fully understood, national healthcare payers, HTA agencies and the pharmaceutical industry, have formal agreements by which they share the financial risk associated with the medicine’s launch and introduction. These are called managed entry agreements (MEAs); which are most commonly referred to as RSAs.
An extensive literature review the history of RSAs in the EU
Over the last 15 years (2000 to 2015), an increase in RSAs has been reported. By carrying out a systematic literature review, researchers at the University of Copenhagen in Denmark and Ghent University in Belgium have identified the underlying trends in RSAs that took place in the EU during this time. To do this review, Trevor Piatkiewicz of the University of Copenhagen and first author of the paper, looked at ‘grey literature’ from Google, Google Scholar, and the official websites of international organizations such as the World Health Organization (WHO), the International Society for Pharmacoeconomics and Outcomes Research (ISPOR), and the Organization for Economic Cooperation and Development (OECD) to create a list of keywords that could be used to identify relevant peer-reviewed articles. Once the keywords had been defined, Piatkiewicz used them to identify peer-reviewed articles about RSAs published between 2000–2015 by searching some commonly used databases (PubMed, Scopus, Web of Science and Embase). All articles that were included in this review were published in English and were conducted or published in and/or about EU Member States. In addition, only agreements or schemes relating to pharmaceutical products and/or involving the sale of pharmaceutical products were included, and only if the title of the article included or alluded to at least one of the keywords selected to indicate the content of the article was relevant to the objective of the review. There were also a number of exclusion criteria that are laid out in detail in the paper.
Potentially relevant articles found in the literature search were then divided into two groups, ‘quantitative articles’ which were used to explore changes in the level of interest in RSAs, and ‘qualitative articles’ which were used to explore the underlying reasons for the changes.
The literature screening enabled data from 238 scientific articles published between 2000 and 2015 to be extracted. Of these, 100 were said to contain quantitative data of interest and 138 qualitative data of interest. Overall, the literature search and evaluation showed that there has been a rise in the number of publications related to RSAs during this period. Piatkiewicz and co-authors Marie Traulsen and Tove Holm-Larsen, found that the increase in the number of publications related to RSAs was particularly significant between 2014–2015, which they attribute to an overall increased interest in, and knowledge of, RSAs.
Four key factors which influenced RSAs over time
A qualitative analysis of the articles was used to identify information about possible underlying reasons for the fluctuations in the yearly number of articles published between 2000–2015. Four overall themes emerged that appeared to influence interest in RSAs over time. These are summarized as follows:
1. 2000–2007: A push for value-based pricing (VBP)
During the early 2000s, across Europe there were increasing demands for VBP made by national healthcare payers. Publications on the subject during this time generally reflected VBP policies discussed by the UK and many articles questioned the necessity of such schemes.
2. 2007–2009: The economic crisis and a greater push to contain costs
Following the economic crisis of 2007–2008, healthcare resources came under increased stress and across Europe budgets were cut. Healthcare providers and HTAs became more concerned with cost-effectiveness and the budget impact of product approvals. Pharmaceutical companies experienced increase pressure to ensure the value of new drug products. Many countries implemented cost-containment strategies to reduce the price of new pharmaceuticals. The economic crisis during this time period pushed VBP implementation and ended the era of free pharmaceutical pricing in Europe. In addition, HTAs started to play an ever more important role as intermediaries between healthcare providers and pharmaceutical companies. These actions prompted an increase in reporting and discussion of VBPs, as has been reflected in an increase in publications during this period.
3. 2009–2011: General discussion about RSAs
From 2009–2010, there was a spike in the number of qualitative articles discussing the pros and cons of European RSAs. Schemes that had been introduced in preceding years were analysed, discussed and criticized. With the increased level of interest in RSAs, came valid arguments and questions surrounding their use. This resulted in a steady increase in the overall number of publications on performance-based schemes.
4. 2011–2015: RSAs diversified to be fit for purpose
Between 2011–2015 a steady increased interest in, and discussion of, RSAs was maintained. This is reflected by the increasing number of publications discussing RSAs during this time. Many existing schemes were adapted and developed across Europe and were made fit for purpose. Many studies noted there is no ‘one size fits all’ approach to RSAs that can be adopted, and each situation should be addressed individually, from both a country and product perspective. The total number of schemes reported plateaued between 2011–2013. Many existing schemes were adapted during this period, rather than new ones being created.
Overall, the review identified several countries that have had a major influence on RSA development and uptake between 2000–2015. These include France, Germany, Italy and the UK, although Eastern European nations began to play an increasing role in the latter years. Each country within the EU has adopted different strategies and developed different relationships with HTAs, which have led to noticeable differences in drug benefit evaluations, recommendations, and overall access to EU markets.
The uncertain future of RSAs
The review showed that increased reporting on pricing and reimbursement practices across the EU has led to an improved understanding of RSAs and that RSAs have evolved dramatically since their first conception and implementation. The report concludes by stating that RSAs may play an increasingly important role in the future of drug availability, particularly when it comes to high-priced personalized medicine options. However, the authors point out that pharmaceutical companies could negate the necessity for RSAs if they become better at creating and gathering the data necessary to allow them to build stronger approval arguments for their new drug products. Such data are needed to lower the risks of being rejected by HTA agencies. If successful, RSA conception, uptake and evolution will have played an important role in helping to reduce the financial burden on EU healthcare systems and allowing patients increased access to new, innovative and effective drugs.
Competing interests: None.
Provenance and peer review: Article abstracted based on published scientific or research papers recommended by members of the Editorial Board; internally peer reviewed.
Alice Rolandini Jensen, MSci, GaBI Journal Editor
Reference
1. Piatkiewicz TJ, Traulsen JM, Holm-Larsen T. Risk-sharing agreements in the EU: a systematic review of major trends. Pharmacoecon Open. 2018;2(2):109-23.
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/eu-risk-sharing-agreements-between-2000-2015.html
A call for coherence in EU legislation to promote generics
Submitted: 6 May 2018; Revised: 7 May 2018; Accepted: 8 May 2018; Published online first: 18 May 2018
Data and market exclusivity for originator medicines in the European Union (EU) create a barrier to market entry for generic medicines. Legislation means that generics cannot reach the market for at least 10 years following the originator marketing authorization. There are no exceptions to this rule which is a cause for concern for many EU Member States. Even high-income countries struggle to afford high-cost medicines under patent to the detriment of patients and overall health care. A recent study published in the Journal of Pharmaceutical Policy and Practice, looks at the current state of EU pharmaceuticals legislation and creates a proposal for greater coherence in this legislation [1].
Regulation concerning the marketing authorization of a generic medicine in the EU currently prohibits the use of the originator’s preclinical and clinical test data for an eight-year period. This is data exclusivity. Following this, market exclusivity means that a generic medicine in the EU cannot then reach the market until 10 years after the originator marketing authorization has passed. In some cases, an additional one year of market exclusivity can also be granted. There are no exceptions made to this rule, which is also known as the 8+2+1 rule.
Current EU medicines legislation goes beyond the requirements of the World Trade Organization (WTO) Trade-Related Aspects of Intellectual Property Rights (TRIPS) Agreement Art. 39.3. The TRIPS Agreement was set up to protect undisclosed test data in registration submissions of new chemical entities against unfair commercial use. However, it does not require countries to grant originator companies exclusive rights over data related to marketing approval. The legislation in place in the EU does grant such exclusive rights, creating an obstacle to the effective use of compulsory licensing.
As part of the WTO TRIPS Agreement, governments can allow third parties or themselves, the right to use a patent without the patent holder’s permission. Such TRIPS flexibilities were clarified in the 2001 WTO Doha Declaration on the TRIPS Agreement and Public Health. This has since resulted in low- and middle-income countries making use of these flexibilities to enable the supply of low-cost generic medicines for the treatment of HIV (human immunodeficiency virus). These flexibilities have a lot of potential which is being taken note of by many WTO countries. Since 2001, the UN High Level Panel on Access to Medicines has recommended their use and that legislation be implemented to enable compulsory licences to be issued that are ‘designed to effectuate quick, fair, predictable and implementable compulsory licenses for legitimate public health needs’.
The EU is yet to make use of TRIPS flexibilities. However, many European health services are currently being restricted by the high price of patented medicines. This results in reduced or denied access to certain medicines in many Member States. There have been requests for governments to address patent barriers made by many higher-income EU Member States.
In the EU, it is possible for individual nations to issue a compulsory licence to overcome patents. However, the regulatory requirements for marketing authorization within the EU, that include data exclusivity, are a matter of European pharmaceutical legislation. These systems together, lack coherence. This has led to cases such as that which occurred in Romania in 2016. Here, the Romanian Government considered issuing a compulsory licence for sofosbuvir, which is used to treat hepatitis C. However, in accordance with EU legislation, it is not possible to register a generic version of this medicine until 2022 due to data exclusivity, meaning that a compulsory licence could not be issued.
Some voluntary licences include a data exclusivity waiver to ensure effective availability of generic medicines. Such waivers are also provided in some middle- and high-income countries that are members of the WTO. These hope to aid generic medicines registration and sales when required to protect public health. They are explicit medicines regulations data exclusivity waivers or relate to the use of compulsory licences in the patent laws of different countries. Data exclusivity waivers are in existence in some non-EU countries including Chile, Colombia, Guatemala and Malaysia and should be considered in the EU.
In the US, data and marketing exclusivity rules are similar to those in the EU. Small molecule generics have a marketing exclusivity period of five years and this is 12 years for biologicals, with a data exclusivity period for the first four. In 2007, the US New Trade Policy authorized a public health exception to data/market exclusivity in the event of a compulsory licence or other requirements of public health. This allows countries that have signed US developing-country free-trade agreements to disregard data/marketing exclusivity as long as they take measures to protect public health. This can occur even in circumstances where a compulsory licence should be issued, however, its implementation does depend on the status of the patent of the medicine being considered.
In the EU, data and market exclusivity waivers do exist in accordance with the ‘EU regulation on compulsory licensing of patents for the manufacture of pharmaceutical products for export to countries with public health problems outside the EU’. Some EU trade agreements include provisions that can enable exceptions to exclusivity for reasons of public interest or under situations of national emergency or extreme urgency. Under these circumstances, third parties can be allowed access to certain data. Such an agreement exists between the EU and Peru.
Overall, despite the right that EU governments have to grant compulsory licences, there is a lack of legal coherence within the EU which prevents generic medicines being able to enter the market early. This is primarily due to data exclusivity and a lack of data exclusivity waivers. EU governments should attach conditions to licensing that enable the use of waivers within the EU and EU legislation needs to be amended to facilitate this. Such waivers do exist for pharmaceutical products exported from the EU. The burden on health budgets across the EU is considerable and EU law needs to act to ensure that there is easy access to new essential medicines to protect and promote public health.
Competing interests: None.
Provenance and peer review: Article abstracted based on published scientific or research papers recommended by members of the Editorial Board; internally peer reviewed.
Alice Rolandini Jensen, MSci, GaBI Journal Editor
References
1. ‘t Hoen EFM, Boulet P, Baker BK. Data exclusivity exceptions and compulsory licensing to promote generic medicines in the European Union: A proposal for greater coherence in European pharmaceutical legislation. J Pharm Policy Pract. 2017;10:19.
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/a-call-for-coherence-in-eu-legislation-to-promote-generics.html
Copyright ©2024 GaBI Journal unless otherwise noted.
Generics and Biosimilars Initiative (GaBI)
Tel: +32 474989572 | Fax: +32 14 583 048