Assessment of the interchangeability between generics
Author byline as per print journal: Luther Gwaza1,2, BPharm, MPhil; John Gordon3, PhD; Henrike Potthast4, PhD; Marc Maliepaard5, PhD; Jan Welink5, Hubert Leufkens1,5, PhD; Matthias Stahl6, MD; Alfredo García-Arieta7, PhD
|
Abstract:
Generic medicines are approved by regulatory authorities based on demonstration of bioequivalence with the innovator, however, direct comparison between all available generics of the same innovator to ensure interchangeability between them is not feasible. With this in mind, the recent use of indirect comparison in investigating the differences in bioavailability between generics was reviewed. Among the available methods for performing indirect comparisons, the adjusted indirect comparison is the simplest and most suitable method for bioequivalence studies, because it uses publicly available data, and partly preserves the power of randomized controlled trials. The homoscedastic method is the most conservative approach, thus recommended for calculating the width of the confidence intervals for adjusted indirect comparisons.
In the present review, the majority of adjusted indirect comparisons of the generic antimalarial artemether/lumefantrine, first-line antituberculosis, and the first-line antiretroviral medicines prequalified by World Health Organization (WHO), and generics approved in the European Union were within the typical acceptance limits of ±20%, and none exceeded the ±30% range, despite the reduced precision of indirect estimates. To ensure interchangeability between generics, the original studies should be sufficiently powered, i.e. > 80%, and the point estimate ratios should not exceed the 7% difference. Thus, a point estimate constraint in the original studies is recommended where it is important to ensure generic drug interchangeability, e.g. narrow therapeutic index drugs.
In conclusion, adjusted indirect comparison is a useful tool to compare relative bioavailabilities between generics that have been compared with a common reference in direct comparison to ensure interchangeability between the generics.
|
Submitted: 21 March 2016; Revised: 11 April 2016; Accepted: 11 April 2016; Published online first: 25 April 2016
Introduction
Direct comparison within well-designed and well-conducted randomized controlled trials (RCT) is the gold standard for comparing health interventions. The usual practice to obtain marketing authorization for new health interventions is to compare them to placebo, or standard of care, but not with all available health interventions. Some interventions are developed simultaneously, thus it is not feasible to perform comparison between them during this development phase to support marketing authorizations. Moreover, in some cases, there is a relatively large number of available interventions making direct comparison through RCTs between them impractical. Therefore, in these instances where multiple interventions exist, there is often lack of, or insufficient, evidence of direct comparison (head-to-head comparison) to evaluate relative effectiveness of all the available health interventions; indirect comparisons are then employed [1–3].
Indirect treatment comparison is defined as an evaluation of different health interventions using information from independent studies. This is useful when there are no data on direct comparison, or to provide supplementary evidence when the data from direct comparison are insufficient [4]. Indirect comparison can be categorized into naïve (unadjusted), informal indirect comparison, and adjusted indirect comparison [3]. Naïve indirect comparison evaluates the data from the independent studies as if the data are from the same study ignoring the between-study variance. For this reason, evidence from naïve indirect comparison is equivalent to observational studies, prone to bias and it may over- or underestimate the treatment effect; thus this approach is not recommended for analysing data from RCTs [2–4]. In informal indirect comparison, the results from the independent studies are compared directly, and relative effects or statistical significance are not formally calculated [3]. Adjusted indirect treatment comparison evaluates different treatments tested in independent studies modified based on the results of their direct comparison with a common control, partly preserving the power of RCTs [3] without the added cost of actual direct RCT comparison. However, adjusted indirect comparisons are less precise as reflected in wider confidence intervals [4], thus wherever possible direct comparisons should be performed.
Figure 1 illustrates the direct and indirect comparison of health interventions. Suppose there are four different treatments, A, B, C and D compared in three different trials. If treatment A was compared in an RCT with treatment B, treatment C in another RCT with treatment B, and treatment C with D in another RCT, adjusted indirect treatment comparison can be used to compare treatment A and C since both were tested in two independent trials with the common treatment B. Likewise, treatment B and D can be compared using adjusted indirect comparison since both were compared in direct comparison with common treatment C.
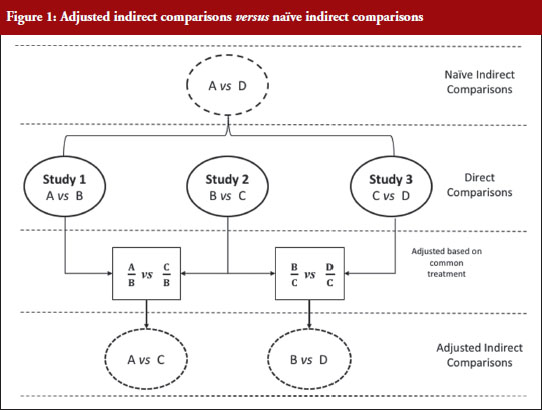
Generic medicines are approved nationally following the expiry of patents or market exclusivity period for the innovator products based on demonstration of bioequivalence with the innovator product. A bioequivalence study comparing a generic and an innovator drug is a form of direct comparison. With time, the number of approved generic medicines per active substance generally increases. Thus, requirement for direct comparison through bioequivalence studies between generics is impracticable since several generics of the same drug are licensed at various times, often without knowledge of the other generics under development. For this reason, adjusted indirect treatment comparison is a useful approach to identify those generic drug products whose interchangeability can be assured, by comparing the different generic drug products that have been demonstrated to be bioequivalent with the same reference product, to support or ensure switchability in clinical practice without concerns on efficacy or safety due to the switching.
Methods used in indirect comparisons
Several methods are available for performing indirect comparisons: (1) naïve or unadjusted indirect comparisons; (2) statistical methods using aggregate data such as simple weighted combination of separate estimates as suggested by Bucher et al. [1] (adjusted indirect comparison); (3) modelling approaches based on the individual patient data (meta-regression); and (4) mixed treatment comparisons based on Bayesian statistics (logistic regression) [2, 5].
In meta-regression analysis, the estimated difference between the groups (treatment effect) is modelled as a function of one or more study characteristics as the predictor variable. Estimated treatment effect in each study is weighted according to the inverse of its variance. A simple approach for meta-regression is weighted linear regression, and the residual heterogeneity is estimated using random effects. Meta-analysis can be performed using the fixed-effects to describe the residual heterogeneity. The key assumption for fixed effect meta-analysis is that the different trials estimated the same effect, for example, the effect of treatment A relative that of treatment B. Regression methods such as logistic regression can be used to perform indirect comparison using the generalized linear models and individual patient data. With regression modelling, one can adjust for other variables available for each study. While full individual patient data, which are required for logistic regression for indirect comparison, or the estimated treatment effect, its variance and covariates for each trial, which are necessary to perform meta-regression, are rarely publicly available, adjusted indirect comparison is performed using the summarized data available in published articles and approved product labelling. It is the simplest and most appropriate methodological approach when only two interventions are to be compared indirectly as it is the case for bioequivalence studies [6].
When using summarized data extracted from published data, first the data are extracted or calculated using appropriate summary statistics, e.g. confidence intervals, mean ratios, for each set of studies. For bioequivalence studies, the extracted data are study products, sample sizes preferably in each sequence, confidence intervals and study design (fasting or fed study, parallel or crossover, single or multiple dose studies). The confidence intervals of the bioequivalence studies are converted to log scale and used to estimate the point estimate and standard error of the treatment effects. Lastly, the data are combined to provide an overall comparison. The standard statistical result, i.e. the variance of the difference between the two independent estimates, is sum of the two variances (variance is square of the standard error), which is similar to a 2-sample t-test. Thus, using the illustration in Figure 1, if you have the two estimated effects for A υs B and B υs C as θAB and θBC, respectively, the effect of the comparison A υs C is estimated as θAC = θAB − θBC and var (θAC) = var (θAB) + var (θBC) [1]. The scale of the effect θ relates to the scale on which the data would be analysed, such as risk difference, log risk ratio, log odds ratio for binary data, the means, mean difference, mean change for continuous data and log hazard ratio for time-to-event data. In bioequivalence studies, the pharmacokinetic outcome measure is continuous data, and the effect θ is the ratio of log-transformed geometric means of the treatments, e.g. A and B (point estimate). The 90% confidence interval (CI) of the ratio of the log-transformed geometric means is the standard statistical result. Therefore, the 90% CI for the indirect comparison 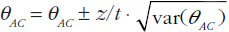 , where z/t in this equation is the z value of standardized normal distribution or the t value of the Student’s t distribution that corresponds to the desired level of confidence (90% in case of bioequivalence studies) and the degrees of freedom in the case of the Student’s t distribution.
, where z/t in this equation is the z value of standardized normal distribution or the t value of the Student’s t distribution that corresponds to the desired level of confidence (90% in case of bioequivalence studies) and the degrees of freedom in the case of the Student’s t distribution.
We explored the different approaches for calculating adjusted indirect comparisons [7]. In this study, we compared six methods that can be used to calculate the width of the confidence intervals based on z distribution (z0.9) or Student’s t distribution (t0.9, d.f.). Four methods that assumed small sample sizes with Student’s t distribution are: (a) Chow and Liu meta-analysis method [8], which assumes all studies had 2 × 2 crossover design and homogeneity of the distribution of reference product data in all studies; (b) homoscedastic method which assumes homogenous variances; (c) heteroscedastic method which assumes heterogeneous variances; and (d) the pragmatic approach which does not require the assumption of homogeneity of variances between studies with small sample sizes. The two methods which assumes large sample sizes with a standardized normal distribution (z0.9) are: (a) Chow and Shao meta-analysis method [9]; and (b) the z-distribution method with no assumption on homogeneity of variances [10].
We concluded that although the differences were minor, the homoscedastic method is recommended, unless there are clear differences in variances, because it is the most conservative approach for estimating the confidence intervals for adjusted indirect comparisons.
Application of adjusted indirect treatment comparison for generics
We investigated the differences in bioavailability between generics prequalified by the World Health Organization (WHO) using adjusted indirect comparisons [7, 11]. These studies investigated a diverse group of products from the antimalarials artemether/lumefantrine [7], first-line antituberculosis drugs, [11] and first-line antiretroviral drugs with a total of 394 indirect comparisons. In contrast to the ±20% acceptance range used for direct comparisons, a ±30% acceptance range is proposed for adjusted indirect comparisons [7, 11], due to the limited precision of indirect comparisons [1, 4].
First, these studies demonstrated the utility of adjusted indirect treatment comparison to compare the bioavailabilities between generic drug products that had been compared with the same reference product in direct comparisons. Second, the outcome of these comparisons indicate that antimalarial artemether/lumefantrine, first-line anti-tuberculosis, and first-line antiretroviral generics prequalified by WHO can be interchanged without any safety and efficacy concerns in clinical settings. Although some comparisons were outside the conventional acceptance limits of ±20% for direct comparisons, there were no generic–generic comparisons outside the ±30% for indirect comparisons, except one comparison for efavirenz Cmax. Failure to show equivalence within a ±30% acceptance range in one out of 394 adjusted indirect comparisons should be interpreted as insignificant number since it is less than 0.3% of the comparisons.
The results obtained with the prequalified generics are consistent with the outcomes reported elsewhere using data from other regulatory authorities [12–14]. Herranz et al. showed that exposures obtained with generic tacrolimus formulations in the Spanish market were within the ±20% acceptance range based on adjusted indirect treatment comparisons [12]. In addition, results from adjusted indirect comparisons were consistent with those from direct comparisons for multiple generic formulations of gabapentin products marketed in The Netherlands [13]. Using data from bioequivalence studies submitted to the Dutch Medicines Evaluation Board (CBG-MEB) for atorvastatin, bicalutamide, naratriptan, olanzapine, perindopril, venlafaxine, cyclosporine, tacrolimus and mycophenolate mofetil, Yu and colleagues showed that in 80% of the cases the indirect comparisons between generics fulfilled the conventional acceptance limit of ±20%, while the remainder were within ±30% [14]. A point estimate constraint in the bioequivalence studies may be relevant for drugs with a narrow therapeutic index, e.g. cyclosporine and tacrolimus, where switching between generics of these drugs is not restricted. Generally, narrow therapeutic drugs are usually assessed with a narrowed acceptance range, e.g. 90.00–111.11%, to ensure interchangeability with the reference [15–17].
We observed that assurance regarding interchangeability of two generic drug products is reduced when either the point estimate ratios in the original studies are shifted from unity by more than 5% or when the width of the 90% confidence interval is large in the direct comparisons [11]. Therefore, we investigated the influence of point estimate, variability of the pharmacokinetic parameters (Cmax and AUC), and the sample size in the original studies on the ability to demonstrate bioequivalence between generics in the adjusted indirect comparisons [18]. However, sample size and variability are not independent since the sample size is calculated based on the expected variability and the desired statistical power. Thus, statistical power is the most relevant parameter for consideration in the computations.
We calculated the outcome of adjusted indirect comparisons for 14,592 scenarios using 57 possible differences between point estimates from 0% to 14% and 16 possible study powers from 50% to 99.99%. The study results illustrated that demonstrating bioequivalence within the conventional acceptance limits of 80–125% by means of adjusted indirect comparisons is only possible if the difference between the point estimate is small (< 5%) for any sufficiently powered study (> 80%). Furthermore, even when both studies are overpowered, the difference cannot be larger than 14%. This study showed that in cases where generic–generic switching maybe of concern, regulators might consider a point estimate constraint in the original studies.
The variance for the adjusted indirect comparison is additive, 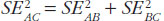 , thus, the major limitation of adjusted indirect comparisons of bioequivalence studies is the reduced precision. On one hand, the inability to show bioequivalence by means of indirect comparisons is not proof of inequivalence, but it may be simply that there is not enough statistical power to make this conclusion. On the other hand, when bioequivalence is shown within the conventional acceptance limits for indirect comparisons despite the reduced statistical power, we can consider not only that the generic drug products are bioequivalent but also very similar.
, thus, the major limitation of adjusted indirect comparisons of bioequivalence studies is the reduced precision. On one hand, the inability to show bioequivalence by means of indirect comparisons is not proof of inequivalence, but it may be simply that there is not enough statistical power to make this conclusion. On the other hand, when bioequivalence is shown within the conventional acceptance limits for indirect comparisons despite the reduced statistical power, we can consider not only that the generic drug products are bioequivalent but also very similar.
The validity of indirect comparisons is dependent on the methodological quality and assumptions. Similarity of trials involved in adjusted indirect comparisons should be carefully assessed to ensure that there are no important differences between the trials under comparison in aspects that could bias the estimated formulation effect. Although standard requirements are applied on the design, conduct and analysis of results of the bioequivalence studies submitted for the prequalification of generics [15], in some cases different study designs might be employed. For example, metabolite versus parent as the analyte, or plasma versus urine as the biological fluid collected for analysis, or multiple versus single dose studies. Studies with these different study designs cannot be compared because the formulation effect cannot be expected to be the same between them. However, we consider results from conventional 2 × 2 crossover designs and replicate designs as combinable. There is no consensus on whether parallel and crossover trials should be combined in indirect comparisons [2], however, this may be possible if the participants and interventions are comparable. In all the analyses performed, all the studies were crossover trials.
In contrast to adjusted indirect comparisons of efficacy trials, confidence in the methodological quality and similarity of adjusted indirect comparisons of bioequivalence studies is assured because of the general consistency in the basic design of these studies. For instance, the characteristics of participants in bioequivalence studies are commonly defined, i.e. usually healthy adult male and/or female volunteers within 18–55 years of age, which controls for the differences in baseline characteristics between treatment groups, whereas differences in disease state in efficacy trials is of concern. The objective of bioequivalence studies is to evaluate formulation differences and external validity of the results is based on the assumption that the effect of the drug in the target populations, i.e. patients would be the same for the test and reference. Nevertheless, in some cases, subject–by–formulation interaction could occur, e.g. when one formulation has excipients that are not tolerated by specific subgroups of patients that are not present in the reference formulation. This is often mitigated by the regulatory requirement to declare such excipients on the product label. Non-randomization of the trials can confound the results if other differences between the treatment groups are linked to the outcomes. Randomization ensures that like is compared with like, i.e. that there are no differences between the groups in any factors other than the intervention itself, in this case, formulation effects. In the bioequivalence studies considered in the analysis of the prequalified generics, subjects were randomized in the allocation of sequence.
Despite the utility of the indirect comparisons, the evidence from such analyses should be interpreted with caution. The internal validity of direct comparisons should be carefully evaluated to reduce bias. In the analysis of the prequalified generics, methodological quality of the studies was not assessed as part of the adjusted indirect comparisons since only the generics that were prequalified were included in the analysis. The prequalification process entails stringent assessment including inspection of the contract research organizations at which the studies were performed, thus providing assurance of the quality of the prequalified product. Therapeutic doses are usually standardized as highest available strength, although in some cases the studies used lower doses. Nonetheless, the results are reported as mean ratios, thus the effect of dose on the outcomes is negligible. The studies were all single dose studies with the same outcome measure of pharmacokinetic parameters Cmax and AUC in all the studies, estimated using validated software. The parent compound in plasma was analysed using validated bioanalytical methods. Despite the general consistency with the bioequivalence studies used in the adjusted indirect comparisons, changes in the requirements over time encompassing several revisions of the guidelines could potentially impact on the methodological quality between studies conducted at different time points; this may be corrected by the use of the same reference product in the different bioequivalence studies. Only the studies using a common reference product as listed by the WHO Prequalification of Medicines Programme were compared in the adjusted indirect comparisons. Though US and European reference products are both accepted in the WHO Prequalification of Medicines Programme, and no distinction was made in the analysis, in some cases these are not the same due to different manufacturing sites and different excipients. However, it is assumed that these products are bioequivalent to the pivotal clinical batch used for gaining marketing authorizations in both jurisdictions.
In conclusion, adjusted indirect comparison is a useful tool to compare relative bioavailabilities between generics that have been compared with a common reference in direct comparison to ensure interchangeability between the generics. The investigated antimalarial – artemether/lumefantrine, first-line antituberculosis and antiretroviral generic drug products prequalified by WHO were considered interchangeable without safety and efficacy concerns. We have also demonstrated that the ability to show bioequivalence between generic drug products by means of indirect comparisons depends on the difference between the point estimates of the bioequivalence studies, which is the point estimate of the indirect comparison, and the power of the bioequivalence studies that are combined. In this respect, concluding equivalence in the indirect comparison within the conventional acceptance limits of 80–125% is only possible when: (a) point estimate difference between generics are low (< 5%) for any sufficiently powered study (> 80%); or (b) the differences do not exceed 14% when both studies are overpowered. Therefore, in cases where it is important to ensure generics interchangeability, the regulatory authorities may consider a point estimate constraint in the original bioequivalence studies. In the general case, due to the reduced precision of indirect comparison, a slightly wider acceptance limits (± 30%) is proposed for indirect comparisons.
Competing interests: The authors declare no conflict of interest.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Luther Gwaza1,2, BPharm, MPhil; John Gordon3, PhD; Henrike Potthast4, PhD; Marc Maliepaard5, PhD; Jan Welink5, Hubert Leufkens1,5, PhD; Matthias Stahl6, MD; Alfredo García-Arieta7, PhD
1Utrecht Institute for Pharmaceutical Sciences (UIPS), Utrecht, The Netherlands
2Medicines Control Authority of Zimbabwe, 106 Baines Avenue, Harare, Zimbabwe
3Division of Biopharmaceutics Evaluation 2, Bureau of Pharmaceutical Sciences, Health Canada, Locator 0201C, 101 Tunney’s Pasture Driveway, Ottawa, ON K1A 0K9, Canada
4Federal Institute of Drugs and Medical Devices, 3 Kurt-Georg-Kiesinger-Allee, DE-53175 Bonn, Germany
5Medicines Evaluation Board, 500 Graadt van Roggenweg, NL-3531 AH Utrecht, The Netherlands
6Group Leader Medicines Assessment Prequalification Team – Medicines, HIS/EMP/RHT, World Health Organization, 20 Avenue Appia, CH-1211 Geneva 27, Switzerland
7División de Farmacología y Evaluación Clínica, Departamento de Medicamentos de Uso Humano, Agencia Española de Medicamentos y Productos Sanitarios, 1 Calle Campezo, Edificio 8, Planta 2 Oeste, ES-28022 Madrid, Spain
References
1. Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683-91.
2. Glenny AM, Altman DG, Song F, Sakarovitch C, Deeks JJ, D’Amico R, Bradburn M, Eastwood AJ; International Stroke Trial Collaborative Group. Indirect comparisons of competing interventions. Health Technol Assess. 2005 Jul;9(26):1-134, iii-iv.
3. Song F. What is indirect comparison? What is …? series. February 2009.
4. Song F, Altman DG, Glenny AM, Deeks JJ. Validity of indirect comparison for estimating efficacy of competing interventions: empirical evidence from published meta-analyses. BMJ.2003; 326(7387):472.
5. Sutton AJ, Higgins JP. Recent developments in meta-analysis. Stat Med. 2008;27(5):625-50.
6. Schöttker B, Lühmann D, Boulkhemair D, Raspe H. Indirect comparisons of therapeutic interventions. GMS Health Technol Assess. 2009;5.
7. Gwaza L, Gordon J, Welink J, Potthast H, Hansson H, Stahl MM, García-Arieta A. Statistical approaches to indirectly compare bioequivalence between generics: a comparison of methodologies employing artemether/lumefantrine 20/120 mg tablets as prequalified by WHO. Eur J Clin Pharmacol. 2012;68(12):1611-8.
8. Chow SC, Liu J. Meta-analysis for bioequivalence review. J. Biopharm Stat. 1997;7(1):97-111.
9. Chow SC, Shao J. Bioequivalence review for drug interchangeability. J Biopharm Stat. 1999;9(3):485-97.
10. Krauss GL, Caffo B, Chang YT, Hendrix CW, Chuang K. Assessing bioequivalence of generic antiepilepsy drugs. Ann Neurol. 2011;70(2):221-8.
11. Gwaza L, Gordon J, Welink J, Potthast H, Leufkens H, Stahl M, et al. Adjusted indirect treatment comparison of the bioavailability of WHO-prequalified first-line generic antituberculosis medicines. Clin Pharmacol Ther. 2014;96(5):580-8.
12. Herranz M, Morales-Alcelay S, Corredera-Hernández MT, de la Torre-Alvarado JM, Blázquez-Pérez A, Suárez-Gea ML, et al. Bioequivalence between generic tacrolimus products marketed in Spain by adjusted indirect comparison. Eur J Clin Pharmacol. 2013;69(5):1157-62.
13. Yu Y, Teerenstra S, Vanmolkot F, Neef C, Burger D, Maliepaard M. Interchangeability of gabapentin generic formulations in the Netherlands: a comparative bioavailability study. Clin Pharmacol Ther. 2013;94(4):519-24.
14. Yu Y, Teerenstra S, Vanmolkot F, Neef C, Burger D, Maliepaard M. Investigation into the interchangeability of generic formulations using immunosuppressants and a broad selection of medicines. Eur J Clin Pharmacol. 2015;71(8):979-90.
15. World Health Organization. WHO Expert Committee on Pharmaceutical Preparations. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. in WHO Technical Report Series, No. 992. 2015, Annex 7 [homepage on the Internet]. [cited 2016 Apr 11]. Available from: http://apps.who.int/medicinedocs/documents/s21898en/s21898en.pdf
16. European Medicines Agency. Committee on Medicinal Products for Human Use (CHMP). Guideline on the investigation of bioequivalence. 20 January 2010. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr ** [homepage on the Internet]. [cited 2016 Apr 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
17. U.S. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Guidance for industry. Bioavailability and Bioequivalence studies for orally administered drug products — general considerations. March 2003 homepage on the Internet]. [cited 2016 Apr 11]. Available from: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3995B1_07_GFI-BioAvail-BioEquiv.pdf
18. Gwaza L, Gordon J, Potthast H, Welink J, Leufkens H, Stahl M, et al. Influence of point estimates and study power of bioequivalence studies on establishing bioequivalence between generics by adjusted indirect comparisons. Eur J Clin Pharmacol. 2015; 71(9):1083-9.
|
Author for correspondence: Alfredo García-Arieta, PhD, División de Farmacología y Evaluación Clínica, Subdirección General de Medicamentos de Uso Humano, Agencia Española de Medicamentos y Productos Sanitarios, 1 Calle Campezo, Edificio 8, Planta 2 Oeste, ES-28022 Madrid, Spain
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/assessment-of-the-interchangeability-between-generics.html
Equivalence of generic medicines in general and immunosuppressants in particular – a regulatory opinion on switching of ciclosporin, tacrolimus and mycophenolate mofetil
|
Abstract:
This position paper deals with our regulatory opinion on registered generic immunosuppressants such as ciclosporin, tacrolimus and mycophenolate mofetil, and provides arguments why these medicines are considered equally safe and effective as the branded drug based on demonstrated bioequivalence. Though regulators acknowledge the worries from the field, we are of the opinion that there are no compelling pharmacological arguments to date against the sensible use of generic immunosuppressants in clinical practice, under the shared and mutual care by prescribers and pharmacists.
|
Submitted: 18 February 2013; Revised: 27 March 2013; Accepted: 28 March 2013; Published online first: 9 April 2013
Background
As soon as the protection period of 10 years of a branded drug has expired, it is possible to seek a marketing authorization for a generic form of this drug. This gives rise to the situation that patients may no longer be treated with the original product (proprietary, branded drug), but with a generic medicine. In that case generic substitution takes place, where the branded drug is exchanged with a product with an identical active ingredient. In the last 30 years we have gained extensive experience with such generics substitution, not only in The Netherlands but also in other parts of Europe and the US. For example, in The Netherlands, treatment with generics has now become the standard for drugs such as statins, proton pump inhibitors and antihypertensive drugs.
Though the Dutch Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen, MEB) is not directly involved in the actual substitution strategy in The Netherlands, registration of generic medicines will only take place if the MEB is convinced that the generic medicine has the same efficacy and safety profile as the innovator medicine. As our contribution to the discussion on generics substitution, we explain why the MEB considers this to be the case also for immunosuppressants, based on the quality of the medicine and bioequivalence testing.
Generic facts
What are the facts on generic medicines? A generic medicine is a product with the same active ingredient, the same strength and the same pharmaceutical form as the branded drug (in other words, is pharmaceutically equivalent). If the manufacturer of the generic drug product demonstrates that its exposure in time (which, for products with immediate release characteristics, is determined by area under the curve (AUC) and Cmax) is equal to that of the branded medicine – so the two products are bioequivalent – the generic and branded medicines are considered to be therapeutically equivalent. This assumption is logical, because when a drug is absorbed in the same way (as demonstrated by the bioequivalence study), its further pharmacological behaviour only depends on the characteristics of the molecular active ingredient. The potential differences in inactive excipients between branded and generic drug formulation are then no longer relevant. For generic and branded drugs, the molecular active substance is qualitatively and quantitatively the same. Therefore, once bioequivalence is demonstrated, the company that manufactures the generic drug can refer to the clinical studies performed with the branded drug for the efficacy and safety of the generic drug product, with no need for additional clinical trials prior to registration.
In most cases, bioequivalence is demonstrated in healthy volunteers [1]. It is well known that the exposure in healthy volunteers may be different than that in patients, due to comorbities of the patient. However, it is important to realize that this will affect branded and generic medicines equally. In addition, the actual exposure in a healthy volunteer is the result of a combination of endogenous factors, including renal and hepatic function, metabolizer status, e.g. poor or extensive metabolizer; ethnic background, and gastric pH, which affect a drug’s absorption, metabolism and elimination. When comparable exposure between a branded and generic medicine has been demonstrated in a healthy volunteer, relative exposure in patients, determined by a different mix of these endogenous characteristics, also is expected to yield comparable exposure. Versantvoort et al. [2] illustrated this principle with a bioequivalence study in which a poor metabolizer was present among extensive metabolizers: though the exposure in the poor metabolizer was dramatically higher – probably even requiring a dose adjustment in clinical practice – the relative exposure of the branded and generic drug within this subject remained comparable. The same principle will hold for other comorbidities, like renal or hepatic impairment: when bioequivalence has been demonstrated in healthy volunteers, the relative exposure change will be the same for the branded and generic medicine. Therefore, bioequivalence demonstrated in healthy volunteers will be valid for the patient population.
In most cases, excipients are inactive, and a single-dose bioequivalence study is considered sufficient to obtain registration of a generic drug. If active excipients, such as in a gastro-resistant coating, are present in the drug formulation, additional data specifically relevant to this active excipient are required in order to demonstrate that the excipient’s behaviour is comparable with that of the branded medicine. For example, in case of a gastro-resistant coating, comparable pH dependent dissolution should be demonstrated, and an additional bioequivalence study with food (resulting in increased gastric pH) should be provided [3]. For other specific formulations, e.g. liposomal, sorbitol, cyclodextrin or microemulsion containing formulations, other specific additional requirements are needed [3, 4].
Of note, bioequivalence studies are not only used for registration of generic medicines, but also in drug development of a newly invented medicine where appropriate [5], or a line extension after registration of a branded medicine. For instance, registration of the 0.5 mg Prograf strength was based on a bioequivalence study under single-dose conditions [6]. It is therefore clear that identical regulations are used both for branded and generic drug products, and thus, these medicines undergo the same rigorous scrutiny upon admission.
Normally, bioequivalence is considered to have been demonstrated when the 90% confidence intervals of the generic:branded ratios for AUC as well as Cmax are within 80–125%. These acceptance criteria are strict, and are outlined in the Guideline on the Investigation of Bioequivalence [1]. Additional stringent requirements are placed on the actual analytical assay that is used in such bioequivalence studies to quantify the plasma or blood concentrations [7]. With respect to generic immunosuppressants, additional care has been taken by the regulatory authorities, by narrowing the acceptance range for some immunosuppressants, in order to further reduce the likelihood of obtaining clinically relevant differences in exposure when switching to and from generic medicines. The option to narrow the acceptance range is given in the current (2010) as well as the previous version (2001) of the Guideline on the Investigation of Bioequivalence, for medicines with a narrow therapeutic index (NTI). Since a worldwide definition of an NTI is lacking, this is considered by the European Medicines Agency (EMA) on a case-by-case basis. Indeed, the acceptance criteria for generic immunosuppressants have been adjusted, i.e. to 90–111% for ciclosporin AUC and Cmax, and to 90–111% and 80–125% for tacrolimus AUC and Cmax, respectively [3]. Due to the microemulsion formulation applied in Neoral, which has led to a pronounced increase in predictability of the ciclosporin exposure and reduced food effect as compared to its precursor Sandimmune, bioequivalence for ciclosporin generics should be demonstrated under fasted as well as fed conditions. With regard to tacrolimus, only the 90% confidence intervals for AUC was narrowed, since due to accumulation of tacrolimus upon repeated dosing, a potential difference between formulations in Cmax after single dosing can be expected to be less at steady state, if AUC is the same for the two formulations. Therefore, the normal acceptance criteria for Cmax can be used in single-dose bioequivalence studies for tacrolimus [3]. For mycophenolate mofetil, for which bioequivalence is demonstrated based on exposure of the mycophenolic acid metabolite, no narrowing of the criteria was considered necessary by EMA [3].
Overall, the strict requirements for demonstrating bioequivalence are equally valid for branded and generic drug products. Thus, the demonstration of bioequivalence is strong evidence to secure the substitution of a generic product for the branded medicine.
Generic doubts
Nevertheless, questions arise from a number of clinical disciplines that, due to claimed specific characteristics within their patient population, some patients are not suitable for generics substitution. One of these disciplines is transplantation medicine. Concerns regarding the substitution of immunosuppressants by generic drug products are understandable from the recipient’s perspective: the impact of failing immunosuppressant therapy following transplantation can be dramatic. In the scientific literature, some publications support generics substitution, e.g. by suggesting comparable efficacy and safety with ciclosporin generic formulations as with branded equivalents [8–12]. Similar support comes from demonstrations of bioequivalence of generic and branded tacrolimus in kidney transplant patients [13] and comparable clinical outcomes with branded or generic tacrolimus in kidney and liver transplant patients [14, 15], with the routinely applied therapeutic dose monitoring for tacrolimus being advised as a safeguard [16, 17]. Conversely, over the past few years, a number of reviews and clinical guidelines raise concerns about generics substitution [18, 19]. In Europe, the European Society for Organ Transplantation (ESOT) published recommendations on generics substitution of immunosuppressive drugs, which were based on the guideline drafted by the Dutch Renal Transplant Society [18]. Although some concerns in ESOT recommendations are acknowledged, overall, in our opinion there appears to be an overemphasis on assumed shortages of pharmacokinetic (PK) or clinical data relating to generic drug product registration. Examples of such assumptions include those relating to the Ctrough/Cmin, multiple dose conditions, or the fact that bioequivalence between different generics is not formally tested. These topics are discussed below. Although the requirements posed by the regulatory authorities on the description of PK of generics are limited (i.e. almost equal AUC and Cmax, and similar quality), these requirements are well thought over. Many of the concerns raised regarding generics substitution are not deducible to scientific facts or studies, but often involve a number of recurring arguments which are demonstrably incorrect. We discuss a number of those related to immunosuppressants below.
One incorrect assumption often expressed is that, though AUC and Cmax obtained with a branded immunosuppressive drug and its generic may be the same, there may still be differences in certain critical points of the plasma concentration–time curves. This argument has been expressed for ciclosporin, where plasma concentrations two hours after administration (C2) or trough levels (Ctrough) are used to monitor and adjust ciclosporin exposure and dose [18]. However, for an immediate-release product like ciclosporin the PK after the initial absorption from the gastrointestinal tract is essentially governed by the molecular active substance only. Since this substance is identical for the branded and generic ciclosporin formulations, differences in C2 or Ctrough, despite comparable AUC and Cmax in the case of demonstrated bioequivalence, will be an extremely unlikely event. In a field that is so familiar with therapeutic drug monitoring, it is remarkable that this is seen as a possibility.
Another argument raised is that bioequivalence for immunosuppressants should be demonstrated under steady-state conditions instead of the currently required single-dose conditions only, since in clinical practice steady-state conditions may be more important [18]. It is agreed that in clinical practice steady-state conditions are important, and it is acknowledged that for certain medicinal products the absolute exposure under steady-state may be different from that after a single dose, due to accumulation upon multiple dose administration. However, there is no reason to assume that the relative exposure obtained under single-dose conditions will be different from that under steady-state conditions. It is well known that the sensitivity of detecting a difference in exposure between two different formulations under steady-state conditions is less than after a single dose [1]. Viewed from the opposite perspective, assessment of bioequivalence under steady-state conditions for ciclosporin would lead to a less stringent assessment of bioequivalence. Applying lower standards for generics is certainly not acceptable to authorities as the Dutch MEB and EMA. After absorption of a medicinal product, its PK is only determined by the molecular active substance. Therefore, there is no reason to assume that the PK behaviour will be different for an immediate-release generic drug product compared with the branded drug under steady-state conditions, when a comparable absorption has been demonstrated under the most sensitive condition, i.e., after single-dose administration.
In certain cases, therapeutic substitution (the exchange of two different types of formulations or two different active ingredients for the same indication) appears to be used to indicate that presumed problems with generics substitution are plausible [18]. This is exemplified by the reported reference to the product description (Summary of Product Characteristics, SmPC) of tacrolimus formulations, which contain a warning that patients must remain on the same formulation. This warning makes sense, and it is clear that the underlying reason for this warning is the fact that there are different types of branded tacrolimus formulations with different release characteristics and therefore different pharmacokinetics on the market, namely Prograf, being an immediate release formulation given twice daily and Advagraf, a prolonged release formulation for once daily administration. Everyone would agree that these different formulations, which are intended for either once daily or BID (twice a day) dosing, should not be interchanged, and indeed issues upon accidentally interchanging these two branded tacrolimus formulations have been reported. However, it is unjust to extrapolate founded warnings in the tacrolimus SmPC against substitution between different types of tacrolimus formulations to substitution between equivalent types of tacrolimus formulations, as in the case of generics substitution, where the release characteristics are equivalent.
The suspicion that generic–generic substitution leads to increased, potentially clinically relevant variability in exposure, which is also used as an argument against generics substitution [18], has not been demonstrated. The occurrence of greater, possibly clinically significant, differences in exposure is a theoretical possibility, which would occur when 90% confidence intervals of different generics would be in the opposite part of the 80–125% criterion. However, given the small observed difference in mean exposure between an arbitrary generic and branded drug [20], the occurrence of great differences in exposure upon generic–generic substitution seems unlikely, though formally it cannot be excluded. For the antiepileptic drugs gabapentin and topiramate, which are registered in The Netherlands, the absence of increased differences in exposure when different generics were exchanged was shown by research conducted at the MEB using bioequivalence data obtained from registration files at the MEB. These data were used to estimate 90% confidence intervals following the substitution of different generic formulations of gabapentin or topiramate [21]. Research towards such simulated generic–generic substitution data for immunosuppressants is currently ongoing at the MEB. In that respect, it is important to note that ciclosporin generics in The Netherlands were registered prior to the narrowed acceptance criteria of 90–111% for this product, both under fasting and fed conditions, implemented by EMA. MEB closely monitors any signs of unacceptable efficacy or safety reports related to these drugs.
Despite the arguments provided above, it cannot be disputed that in certain isolated cases, issues with generics are reported. However, these are considered as exceptional cases, e.g. sometimes related to intolerance to certain excipients like lactose, fructose or galactose, which may be present in generics and not in branded products (and vice versa). However, in the vast majority of switches, substitution proceeds without problems. It is acknowledged that factors other than differences in exposure may play a role in the perception of generics and the outcome of generics substitution in patients, for example, with differences in shape and colour of generics, which may lead to distrust, mistakes or reduced compliance among patients. The consequences of such differences may even increase when the branded and generic drug are frequently changed, which is a realistic scenario in The Netherlands, where the frequency of switching has increased over the years due to the current pricing and reimbursement policy of the Dutch health insurance companies. Frequent switching to other generics may be expected to negatively affect compliance and confidence, could potentially increase the chance of errors, and should therefore be avoided as much as possible.
It is the joint responsibility of the pharmacist and prescriber to monitor this switching and to provide satisfactory communication for the benefit of the patient, in case generics substitution takes place. In our opinion, inadequate communication between pharmacist and prescriber cannot be used as an argument against the use of generics [18], but should better lead to incentives to solve this issue.
Uncertainty about the underlying principles and legislation of generics, combined with otherwise well appreciated and valued patient care, appear to be leading in the frequently provided arguments against generics substitution, rather than solid evidence for the occurrence of problems. It is reasonable to assume that a well-informed prescriber is able to play a major role in the perception of generic immunosuppressants by the patient, and in that respect, MEB should also take a part in this discussion and education.
Regulatory agencies like MEB are actively involved in governing the safe use of generic immunosuppressants. Pharmacovigilance structures are in place, and adverse events reported related to immunosuppressants, as well as other medicinal products, are taken very seriously. In case there are signs of unexpected disproportional adverse events or inefficacy with any drug – be it a generic or branded – MEB is obliged to take action. For the pharmacovigilance system to work, it is essential to report issues to the relevant pharmacovigilance centres in the different EU Member States, in order to be able to keep a close eye on the actual quality of generics, and to reduce the time before a signal can be picked up. In that sense, regulations have recently been amended with a more pronounced place for reporting adverse events by patients, who are considered ‘hands-on’ experts.
Conclusion
From a regulatory point of view, generic immunosuppressants like ciclosporin, tacrolimus and mycophenolate mofetil are considered as safe and effective as the branded drug based on demonstrated bioequivalence, and therefore considered interchangeable. Though we are aware of worries expressed in the field, we are of the opinion that there are no compelling pharmacological arguments to date against the sensible use of generic immunosuppressants in clinical practice, under the shared and mutual care of prescribers and pharmacists.
For Patients
Generic drugs are prescribed more and more. Sometimes, the change of prescription from branded to a generic medicine leads to unrest and doubts among patients, e.g. on whether generic drugs are equally safe and work equally well as the branded medicines. These doubts are acknowledged and understood. In this paper, we aim to clarify what is done by regulators to safeguard the use of generics as much as possible. From the prescriber’s and pharmacist’s perspectives, we expect and promote a professional and adequate collaboration to take appropriate action in isolated cases when a generic drug does not meet its expectation in an individual patient.
Editor’s comments
This paper is based on and expanded from a pro-con discussion that appeared in the Nederlands Tijdschrift voor Nefrologie (Journal of the Dutch Renal Transplant Society) in February 2012.
Disclaimer
The views expressed in this opinion are the personal views of the participating authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Dutch Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen, MEB).
Competing interests: None.
Provenance and peer review: Commissioned; externally peer reviewed.
Authors
Marc Maliepaard1, PhD
Yang Yu1,2, PharmD
Professor Hubert GM Leufkens1,3, PharmD, PhD
1Dutch Medicines Evaluation Board, Utrecht, The Netherlands
2Department of Pharmacology, Maastricht University Medical Centre, Maastricht, The Netherlands
3Utrecht Institute for Pharmaceutical Sciences, Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht University, Utrecht, The Netherlands
References
1. European Medicines Agency. Guideline on the investigation of bioequivalence [homepage on the Internet]. 2010 [cited 2013 Mar 27]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
2. Versantvoort C, Maliepaard M, Lekkerkerker F. Generics: what is the role of registration authorities. Neth J Med. 2008;66:62-6.
3. European Medicines Agency. Questions & Answers: positions on specific questions addressed to the pharmacokinetics working party [homepage on the Internet]. 2013 [cited 2013 Mar 27]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002963.pdf
4. Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product [homepage on the Internet]. 2013 [cited 2013 Mar 27]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf
5. Baumgärtel C. Myths, questions, facts about generic drugs in the EU. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1:34-8. doi:10.5639/gabij.2012.0101.009
6. Bekersky I, Dressler D, Boswell GW, Fergen B, Tracewell W, Mekki Q. Bioequivalence of a new strength tacrolimus capsule under development. Transplant Proc 1998;30(4):1457-9.
7. European Medicines Agency. Guideline on bioanalytical method validation [homepage on the Internet]. 2011 [cited 2013 Mar 27]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf
8. Kahn D, Muller E, Pascoe M. Safe conversion to cicloral, a generic cylosporine, in both stable and de novo renal transplant recipients. Saudi J Kidney Dis Transpl. 2010;21(3):426-32.
9. Diarra DA, Riegersperger M, Säemann MD, Sunder-Plassmann G. Maintenance immunosuppressive therapy and generic cyclosporine A use in adult renal transplantation: a single center analysis. Kidney Int Suppl. 2010 Mar;115:S8-11.
10. Tarek BO, Nadia BF, Anis K, Saloua L, Abderrahmane A, Lamia T, Amel L, Mohamed L. Assessment of bioequivalence of a generic cyclosporine (Equoral) by a prospective randomized controlled trial on allogeneic stem cell transplant recipients. Transplant Proc. 2010;42(9):3735-8.
11. Niemczyk M, Paczek L. Generic formulation of cyclosporine A, Equoral®, in de novo kidney transplant recipients: five-year follow-up. Ann Transplant. 2011;16(2):59-62.
12. Durlik M, Paczek L, Rutkowski B, Lewandowska D, Debska-Slizien A, Chamienia A, Wyzgal J, Ognista-Gajda A, Niemczyk M. The efficacy and safety of ciclosporin (Equoral®) capsules after renal transplantation: a multicentre, open-label, phase IV clinical trial. Ann Transplant. 2010;15(3):51-9.
13. Alloway RR, Sadaka B, Trofe-Clark J, Wiland A, Bloom RD. A randomized pharmacokinetic study of generic tacrolimus versus reference tacrolimus in kidney transplant recipients. Am J Transplant. 2012;12(10):2825-31.
14. Spence MM, Nguyen LM, Hui RL, Chan J. Evaluation of clinical and safety outcomes associated with conversion from brand-name to generic tacrolimus in transplant recipients enrolled in an integrated health care system. Pharmacotherapy. 2012;32(11):981-7.
15. Ensor CR, Trofe-Clark J, Gabardi S, McDevitt-Potter LM, Shullo MA. Generic maintenance immunosuppression in solid organ transplant recipients. Pharmacotherapy. 2011;31(11):1111-29.
16. McDevitt-Potter LM, Sadaka B, Tichy EM, Rogers CC, Gabardi S. A multicenter experience with generic tacrolimus conversion. Transplantation. 2011;92(6):653-7.
17. Momper JD, Ridenour TA, Schonder KS, Shapiro R, Humar A, Venkataramanan R. The impact of conversion from prograf to generic tacrolimus in liver and kidney transplant recipients with stable graft function. Am J Transplant. 2011;11(9):1861-7.
18. van Gelder T. European Society for Organ Transplantation Advisory Committee recommendations on generic substitution of immunosuppressive drugs. Transpl Int. 2011;24(12):1135-41.
19. Harrison JJ, Schiff JR, Coursol CJ, Daley CJ, Dipchand AI, Heywood NM, et al. Generic immunosuppression in solid organ transplantation: a Canadian perspective. Transplantation. 2012;93(7):657-65.
20. Davit BM, Nwakama PE, Buehler GJ, Conner DP, Haidar SH, Patel DT, et al. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the United States Food and Drug Administration. Ann Pharmacother. 2009;43(10):1583-97.
21. Maliepaard M, Banishki N, Gispen-de Wied CC, Teerenstra S, Elferink AJ. Interchangeability of generic anti-epileptic drugs: a quantitative analysis of topiramate and gabapentin. Eur J Clin Pharmacol. 2011;67(10):1007-16.
|
Author for correspondence: Marc Maliepaard, PhD, Senior Clinical Assessor, Clinical Pharmacologist, Pharmacotherapeutic Group 3, College ter Beoordeling van Geneesmiddelen/Dutch Medicines Evaluation Board, PO Box 8275, NL-3503 RG Utrecht, The Netherlands
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
Why bioequivalence and unconditional interchangeability of generic drugs are not the same
Source URL: https://gabi-journal.net/equivalence-of-generic-medicines-in-general-and-immunosuppressants-in-particular-a-regulatory-opinion-on-switching-of-ciclosporin-tacrolimus-and-mycophenolate-mofetil.html
Copyright ©2025 GaBI Journal unless otherwise noted.
Generics and Biosimilars Initiative (GaBI)
Tel: +32 474989572 | Fax: +32 14 583 048