Myths, questions, facts about generic drugs in the EU
Published on 2012/02/13
Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(1):34-8.
| Abstract: Many generic drugs are now being prescribed and the trend is increasing. For example, in Austria, the number of all generics prescriptions has more than doubled from 11% in 2000 to 23% in 2010. However, many myths and questions about generic drugs remain and information may be difficult to come by. It is therefore not surprising, as we have discovered in recent years, that even physicians and pharmacists are not always fully up to date in their understanding of generic drugs. Some of their questions centre on issues such as: are generic drugs really as good as the original; are we really dealing with an adequately tested, high quality medicinal product. Today, generic drugs present an equally well-tolerated and efficacious alternative to established medicinal products, which contain well-known, rigorously tested active ingredients. An established originator product undergoes expensive and protracted development (up to 15 years) with inherently high preclinical and clinical research costs in order to be given market approval. The development of generic drugs, on the other hand, is relatively quick and inexpensive, which allows generic drugs to be sold at a distinctly cheaper price. This is due to the waiving of new preclinical and clinical studies, aside from some bioequivalence studies. Their lower price however should not be equated with ‘cheap quality’. In fact, generic medicines undergo the same strict scrutiny by the European or national medicines authorities as reference products. At AGES PharmMed, the Austrian competent authority for marketing authorisation of human medicinal products, generic drugs are subjected to detailed assessment, during which the safety profile and efficacy data of the active substances, as well as the proof of bioequivalence, are thoroughly examined. This is also the case for all other national competent authorities of the EU and EMA. The Austrian Federal Office for Safety in Healthcare issues a marketing authorisation only if all legal and scientific requirements are fulfilled. This marketing authorisation validates the safety, efficacy, and quality of a generic drug. |
Submitted: 20 May 2011; Revised manuscript received: 16 November 2011; Accepted: 25 November 2011
What makes a generic medicinal product generic?
The definition, according to Austrian drug law/the Medicinal Products Act as well as to EU Directive 2001/83/EC, is that a generic medicinal product ‘is a product which has the same qualitative’, i.e. kind of active substance, ‘and quantitative’, i.e. amount of active substance, ‘composition as the reference medicinal product’. For the sake of simplicity, the reference product is often also referred to as the originator. The different salts, esters, or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in their safety and/or efficacy properties. In these cases, the manufacturer of a generic drug has to submit further proof of efficacy and safety.
The pharmaceutical form, which means the distinct way a product is to be administered, of the generic medicinal product has to be the same as for the reference medicinal product [1]. However, remarkably the various types of immediate release oral pharmaceutical forms, e.g. tablets, capsules and dragées, are considered to be one and the same pharmaceutical form. A patient prescribed with a particular medicinal product may therefore be prescribed either a film-coated tablet or a capsule of the same drug by his physician. This in itself does not pose a problem, as the galenic formulation may indeed be different, but the impact on the safety and efficacy profile of the whole product has been judged to be comparable during the approval procedure.
Are different compositions possible?
Differences in composition between the generic and reference medicinal product are possible, but only regarding the excipients, e.g. bulking agents, colouring agents; and not for the active substances. For example, corn starch may be used instead of lactose as an excipient. However, it has to be demonstrated by the applicant of the generic drug that these differences in composition do not influence the therapeutic efficacy and safety or how the drug is absorbed, distributed, metabolised or eliminated by the body, i.e. the drug’s pharmacokinetics must also remain more or less the same.
Bioavailability or bioequivalence trials need to be conducted in order to demonstrate the equivalence between the generic medicinal product and the reference medicinal product. Differences in the manufacturing process compared to the originator are allowed, but the same strict general quality criteria, e.g. controlled production under good manufacturing practice (GMP), apply to the production of the generic medicinal product as well as for the reference product. Also a medicinal product can only be considered as the reference product if it has been granted market approval in at least one Member State of the European Economic Area. However, only the so-called originator can serve as the reference product in bioequivalence testing, but never another generic drug, as this would otherwise mean the allowance of a copy of a copy.
How soon can generic medicinal products appear on the market?
The applicant needs to provide proof that the originator product has been authorised for at least eight years, or that the originator company has issued a written informed consent stating that the generics company is permitted to apply for its generic drugs sooner. As a rule however, the earliest a generic medicinal product is allowed to go on sale is 10 years after the first European originator is granted marketing authorisation. This 10-year market exclusivity can be extended by an additional year if, during the first eight years, the marketing authorisation holder of the originator obtains an additional authorisation for one or more new relevant therapeutic indications [2]. Figure 1 shows the ‘8+2 (+1) Formula’ applicable to generic medicinal products entering the market.
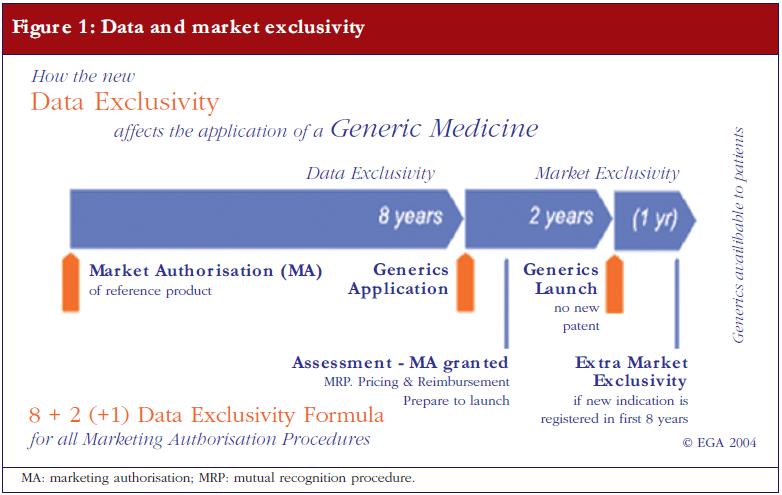
Some originators, however, hold patents—in some cases up to 1,000 patents for one single product were found—which can further postpone the launch of a generic drug. Such delay in market access is therefore possible, even if the marketing authorisation has already been granted to the generic drug. Notably, some misuse of patent strategies was described in the final report of the 2009 sector inquiry of the European Commission for Competition [3]. In a sample of 219 molecules from 2000 to 2007 there were reportedly 1,300 patent-related out-of-court disputes related to the launch of a generics. The number of patent litigations brought to court totalled nearly 700 cases in these seven years and the number of cases increased by a factor of four between 2000 and 2007. The report reached the conclusion that the behaviour and practices of originators contribute to generics delay as well as to difficulties in innovation itself because originators may even block each other.
What is a bioequivalence study?
Bioequivalence studies are often the demanded basis for granting marketing authorisation for a generic medicinal product. They are clinical studies conducted in accordance with Austrian drug law as well as to EU Directive 2001/20/EC and provide data to demonstrate bioequivalence between a test product, i.e. the generic medicinal product, and a reference product, i.e. the originator. The rate and extent of absorption of the medicinal products and therefore the bioavailability of the active substance(s) are determined. It is a widely accepted regulatory assumption, even sometimes challenged by generics disputants, that equivalent plasma concentration time curves represent equivalent efficacy and safety. Therefore, if bioequivalence can be shown after the administration of the same molar dose, equivalence or assumption of so-called essential similarity of the two products in terms of efficacy and safety can be concluded.
How is a bioequivalence study conducted?
The simplest design for a bioequivalence study is the ‘two-way crossover’. In this design, a subject receives one product first (for example, the generic test (T) product) and then, following a sufficiently long wash-out period to ensure that no residual active substance remains in the subject’s body, is given the other product (in this case, the originator reference (R) product). Usually 20–30 healthy volunteer subjects are used for such a study. Subjects can belong to either sex, although the risk to women of childbearing potential should be considered and an existing pregnancy must be ruled out. Repeated blood samples are taken to monitor plasma concentrations of the active substance or its active metabolites, see Figure 2.

More complex designs like bioequivalence studies with a fully-replicate design, meaning a double crossover approach with for example T-R-T-R may be chosen if one wants to evaluate the intra-individual variability of a medicinal product. This becomes necessary when a company wants to apply for an authorisation for a so-called highly-variable drug, which means that the intra-individual variance of a reference product is higher than 30%, which could make the generic drug eligible to somewhat broadened bioequivalence criteria. However, in certain cases of so-called Biopharmaceutics Classification System (BCS) class I drug substances, the bioequivalence study may be waived. These BCS class I substances are highly soluble, completely absorbed and are not considered to have a narrow therapeutic index. Occasionally, BCS class III drugs substances, which have a high solubility but limited absorption can qualify for such waivers but only if they have qualitatively the same excipients at a quantitatively very similar amount as a reference product.
Parameters
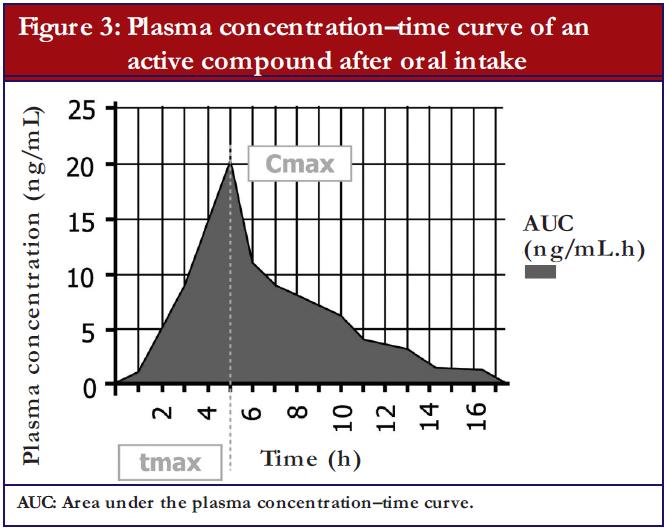
The most important parameters, which are evaluated during a bioequivalence study, are as follows, see also Figure 3:
- Area under the curve (AUC): that is the area under the plasma concentration time curve, which represents the extent of exposure. Usually the AUC0-t is evaluated, which means that a measurement from the time (0) of the drug administration until the last (t) blood sample was drawn. To show that blood samples were drawn for a sufficiently long time period, the measured AUC0-t has to cover at least 80% of the AUC0-inf, which means the AUC extrapolated to infinity. In cases where a drug has a very long half-life, a measurement until a maximum of 72 hours after drug administration (AUC0-72) is considered sufficient, as the absorption phase of immediate release products is fully covered by this approach.
- Maximum plasma concentration of the active substance (Cmax): Cmax provides information on pharmacodynamic and pharmacokinetic properties and is fundamental in the evaluation of adverse events.
- Time to maximum plasma concentration (Tmax): Tmax allows inferences to be drawn, to a certain degree, on the speed of release from the pharmaceutical form and on the absorption from the gastrointestinal tract as well as a raw estimate of the onset of action.
Acceptance limits
The data collected during the bioequivalence study undergo exacting statistical analyses, for which a 90% confidence interval is used. To put it simply, the 90% confidence interval provides a range within which one can be 90% confident the true effect lies. The agreed acceptance limits are valid throughout the EU and can be found in the ‘Guideline on the Investigation of Bioequivalence’ of EMA [1]:
- For the AUC, the 90% confidence interval has to be contained within the acceptance interval of 0.80–1.25. Substances with a narrow therapeutic range, e.g. immunosuppressive drugs such as ciclosporin, may be tightened to an acceptance interval of 0.90–1.11. The need for such tightening is decided case-by-case based on clinical considerations either by the applicant or by the EMA Pharmacokinetic Working Party (PKWP).
- For Cmax the 90% confidence interval also needs to be within the acceptance interval of 0.80–1.25. As before, the acceptance interval may be tightened to 0.90–1.11 for substances with a narrow therapeutic range. However, for drug substances falling under the definition of a highly variable drug, which must always be proven in a fully replicate design bioequivalence study, the interval for Cmax on the other hand can gradually be broadened up to a maximum of 0.70–1.43. This maximum interval is only eligible if intra-individual variance amounts to more than 50%. Intra-individual variance bigger than 30% but smaller than 50% would yield regulatory acceptance intervals between conventional 0.80–1.25 and the maximum tolerable 0.70–1.43 [1].
Why are studies carried out using healthy subjects, rather than patients?
Bioequivalence studies have a special experimental approach: the aim is to enable a comparison of the bioavailability to determine the equivalence of the two drugs, or in other words, to exclude a statistically significant difference between two formulations. Inherently, every study has ‘background noise’, which makes it harder to discern the actual effect. The background noise, also known as ‘bionoise’, is caused by random fluctuations of biological measurements and can be likened to static on the radio. By chance, it is possible that a difference in effect can be masked by this bionoise. The bionoise fluctuations can be caused by the ‘intra-subject variability’ (the physiological variability within a single individual) or can be due to the ‘inter-subject variability’, which describes variability between different subjects. Due to their differences in constitution, co-morbidities and co-medication, patients present as a distinctly heterogeneous population. This heterogeneity makes direct comparisons difficult and is accompanied by profound bionoise. Therefore, subjects for bioequivalence studies are ‘standardised’ as far as possible with the aim of permitting the detection of each and every small difference between the formulations. Healthy volunteers are selected in accordance with strict inclusion criteria, such as being healthy, 18–55 years of age, normal body weight, and non-smokers. The homogeneity of the selected study subjects helps validating the results of the study. Throughout the study, diet, fluid intake, and exercise are standardised and concomitant medication or alcohol is not allowed [4]. The subjects are closely monitored in a clinical setting. All these conditions are maintained to ensure reliable results and to permit the identification of any possible differences between the two pharmaceutical products under test.
Are the acceptance limits strict enough?
The approvable acceptance range from 0.80–1.25, i.e. 80–125%, is sometimes incorrectly thought to represent a difference in efficacy. In fact, acceptance limits define a statistical range and the actual mean lies much closer to 1 (or 100%). Numerous studies have reported that generic drugs’ AUC and Cmax differed only by 3–4% on average from those of the originator [5-8]. They also reported that, in general, the poorer manufactured products, where the bioavailability differed by more than 5–10% from the originator, no longer fulfilled the key criteria for bioequivalence and were not granted marketing authorisation as generic drugs. It should also be noted that most pharmaceuticals rely on the law of mass action. In this non-linear system, an 80% or 125% change in concentration in the range of the dissociation constant—the area where half of all receptors are bound to the active substance—results merely in a +/- 6% change in receptor binding. Moreover, in therapeutics, dosage is sometimes chosen where nearly all receptors, e.g. 90%, are bound. At these dosages, the changes in concentration described above result in an even smaller change, approximately 2%, in receptor binding. A difference in efficacy is therefore close to impossible and will usually not be detectable, either therapeutically or statistically.
What are the rules for conducting bioequivalence studies?
How exactly a bioequivalence study has to be conducted, and which requirements need to be taken into consideration, is laid out in detail in the European bioequivalence guideline, the revised version of which came into effect mid-2010. The guideline clearly specifies the requirements for the design, conduct, and evaluation of bioequivalence studies for all EU countries. Since 2001, when the first bioequivalence guideline was published, many additional aspects were identified which needed to be amended and improved. Minor issues were addressed in interim question and answer documents. After a three-year preparation period, the comprehensively revised version of the guideline came into effect in August 2010. The comments and suggestions of over 50 expert organisations and associations were worked into the 22 draft versions. The revised version therefore now reflects the most up-to-date state of knowledge, which is essential in issuing harmonised and standardised marketing authorisations across Europe. The aim of the guideline was to do away with the ambiguities of the past, which often led to lengthy discussions and differences in professional opinion between the countries and competent authorities of the EU. This also ensures the safety and efficacy of all generic drugs being granted marketing authorisation.
Are bioequivalence studies only used in the development of generic drugs?
Since the task of bioequivalence studies is to detect differences between formulations or pharmaceutical forms, they are indeed not only used as a basis for the licensing of generic medicinal products. In fact, originators may also use bioequivalence studies during their own development since the formulation first used in clinical trials is often not the same which later goes into large-scale market production. Bioequivalence studies are used in these cases to allow bridging of the results obtained in the clinical trials. The same principles in study conduct, data evaluation, and assessment of results by the authority are applied in such originator studies as in the above-described studies for generic drugs. Remarkably, essential similarity between an originator small-scale clinical trial product and a large-scale originator product later to be for sale has never been put in question by anyone. Considering media coverage sometimes casts massive doubt about generics and the way they are authorised, obviously there seems to be an unfounded contrast in the perception dependent on who—the originator company or the generics company—makes use of the bio – equivalence concept for the authorisation of one of their products. Assuming that the bioequivalence concept is valid and trustworthy for authorisation of a new originator product, the same should be applied to the authorisation of a generic drug.
Is the manufacturing quality the same for generic and originator products?
The same quality requirements apply to the manufacturing of generic drugs as for any other medicinal product. Production has to be performed in accordance with GMP and is strictly controlled by evaluating the manufacturing data and by inspections performed not only in Austria and the EU, but also all over the world including countries such as India and South Africa. As for any other medicinal product, quality deficiencies in individual batches are theoretically possible and therefore the Austrian Federal Office for Safety in Health Care as well as the other competent EU authorities closely monitor the quality of all authorised medicinal products on the market. This is achieved by the legal obligation of authorisation holders to inform the authority about every out-of-specification results or other problems in manufacturing and an additional quality-defect notification system involving all healthcare professionals. This guarantees that only high quality medicinal products are available, regardless of whether these products are originators or generics.
When is a generic drug granted market authorisation?
An Austrian or an EU marketing authorisation is only issued when the pharmacokinetic parameters of the generic drug are comparable to those of the reference product and bioequivalence has been successfully demonstrated. Furthermore, the overall benefits of the generic medicine need to outweigh its risks (positive risk–benefit ratio) and its excipients and the manufacturing process must have been demonstrated to not negatively influence its safety and efficacy. Last but not least, all internationally relevant quality standards and legal requirements have to be fulfilled before marketing authorisation can be granted.
Are generic drugs as efficacious as the original?
Since the generic medicinal product contains the same active substance as the originator and comparability has been demonstrated in a successful bioequivalence study, the generic drug is assumed to behave in the same way as the originator. This equality also implies equal efficacy and safety. Austria, as well as other EU Member States, due to its contribution of experts in scientific councils, e.g. the EMA PKWP, also plays a significant role in keeping generics standards high and ensuring that only generic drugs with the same safety and efficacy profiles as their reference products are brought onto the market. A follow-up control is also established by means such as the evaluation of recent scientific literature, clinical trials, pharmacovigilance provisions, manufacturing inspections, quality controls, and an obligatory renewal of the marketing authorisation after a five-year period. Despite these facts there have been repeated attempts by originators and some medical societies to cast doubts on generics efficacy in fields such as osteoporosis medications (bisphosphonates), antipsychotics [9], platelet inhibitors [10] and opioid pain relievers [11]. Taking this into account the final report of the 2009 sector inquiry of the European Commission for Competition states, therefore that any campaigns which put this fact in question (namely that all medicinal products, whether originator or generics, are subject to the same strict requirements of quality, safety and efficacy) ignore the key principles for marketing authorisation in the EU and may mislead the public. The Commission hence urges Member States to take appropriate action if such campaigns are detected in their territory. In Austria, appropriate actions were taken in 2010 when the Austrian Federal Office for Safety in Health Care released a public letter which detailed facts about clopidogrel generics [12], and afterwards published a more detailed, pharmacologically-based article in several physician and pharmacist journals [13] as well as in official journals of the Main Association of Austrian Social Security Institutions and Austrian Health Insurances [14]. Furthermore, recent international scientific publications confirm once more the similar efficacy of generics in this and various other therapeutic fields [15-19].
Do generic drugs antagonise innovation?
It is sometimes said that generic drugs prevent innovation. In fact, the opposite is true. As seen in other areas of research and development, a careful balance must be struck between patent protection and free competition. The current rules regarding patent protection and market exclusivity ensure a sufficiently long period of market protection for originators, while ensuring at the same time a fairly good access to market for generics competitors, at least after the 10-year market exclusivity has expired, and occasionally much more tricky, the patent issues were resolved. Although originator companies repeatedly bemoan a dwindling return of investment due to generics competition [20], data investigating the duration for development and gaining market approval have shown that originators nowadays have, compared to 1990, an additional three and a half years due to faster development and expeditious registration procedures in which to reap the rewards of being the sole market authorisation holder [21, 22]. It is a noteworthy fact that some countries with high rates of generics penetration like Germany or US also have high rates of pharmaceutical innovation and spending in research and development [23]. It would seem, therefore, that generics competition indeed contributes and gives stimulus to the discovery of new medicines. Generic drugs therefore not only help national health services by reducing its costs, they can in fact cause pressure for innovation.
For patients
Many generic drugs are nowadays being prescribed to patients and the trend is increasing. However, some people still feel that myths and questions about generic medicines remain and information may be difficult to come by. In fact, generic drugs are well-tested, high quality medicinal products. They are strictly regulated by the National and European Competent Authorities and they are only granted approval by going through an extensive authorisation process. This process ensures adequate safety and efficacy of generic medicines available on the European market.
References
- European Medicines Agency. CPMP/EWP/QWP/1401/98 Rev.1. Guide line on the investigation on bioequivalence. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
- European Commission. Guidance on elements required to support the significant clinical benefit in comparison with existing therapies of a new therapeutic indication in order to benefit from an extended (11 years) marketing protection period, 2007. Available from: http://ec.europa.eu/health/files/eudralex/vol-2/c/guideline_14-11-2007_en.pdf
- European Commission Competition, EU Sector inquiry, 8 July 2009. Available from: http://ec.europa.eu/competition/sectors/pharmaceuticals/inquiry/communication_en.pdf
- Tschabitscher D, Platzer P, Baumgärtel C, Müllner M. Generic drugs: quality, efficacy, safety and interchangeability. Wien Klin Wochenschr. 2008;120:63-9.
- American Medical Association. Summaries and recommendations of Council on Scientific Affairs Reports. Generic drugs (CSA Rep 6, A-02). 2002 Annual Meeting of the American Medical Association. 2002:13-4.
- Henney JE. Review of generic bioequivalence studies from the food and drug administration. JAMA. 1999;282:1995.
- Nwakama PE. Generic drug products demonstrate small differences in bioavailability relative to brand name counterparts: review of approved ANDAs, FDA. 2005.
- Davit BM, et al. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the FDA. Ann Pharmacother. 2009 Oct; 43(10):1583-97.
- Statement of the Austrian Society of Cardiology (ÖKG) regarding Clopidogrel-Generics. 2010. Available from: http://kardiologie-gefaessmedizin.universimed.com/artikel/stellungnahme-der-%C3%B6sterr-kardiologischen-gesellschaft-%C3%B6kg-zu-clo
- Consensus statement of Austrian Society of Biologic Psychiatry (ÖGBP), Generics and originators in psychiatry, 2008. Available from: http://www.medizin-medien.at/mm/mm011/low-generika.pdf
- Statement of Austrian Society of Pain regarding opioid generic switching due to economic reasons, 2011. Available from: http://www.expertenstatement.com/
- Statement of Austrian Agency for Health and Food Safety, Austrian Medicines and Medical Devices Agency, AGES PharmMed and Austrian Federal Office for Safety in Health Care regarding Clopidogrel Generics. 2010 March 26. Available from: http://www.basg.at/uploads/media/100325_Stellungnahme__Clopidogrel_Generika_1.pdf and http://www.basg.at/uploads/media/100325_Stellungnahme_Clopidogrel_Generika__2.pdf
- Baumgaertel C. Clopidogrel-generics. Austrian Pharmacists Journal, ÖAZ, Ausgabe. 2010;64(11): 666-8. Available from: http://www3.apoverlag.at/pdf/files/OAZ/OAZ-2010/OAZ-2010-11.pdf
- Baumgaertel C. Generics of clopidogrel are equivalent. Publication of Vienna Health Insurance Fund and Austrian Health Insurance, WGKK, Therapie Info. 2010;22(3). Available from: https://www.sozialversicherung.at/mediaDB/692427_therapieinfo_nr3_07_2010.pdf
- Kim SD, et al. Bioequivalence and tolerability of two clopidogrel salt preparations, besylate and bisulfate: A randomized, open-label, crossover study in healthy Korean male subjects. Clin Ther. 2009;31(4):798-803.
- Neubauer H, et al. Comparing the antiplatelet effect of clopidogrel hydrogensulfate and clopidogrel besylate: a crossover study. Clin Res Cardiol. 2009 Sep;98(9):533-40.
- Gagne JJ, et al. Refilling and switching of antiepileptic drugs and seizure-related events. Clin Pharmacol Ther. 2010 Sep;88(3):347-53.
- Kesselheim AS, et al. Seizure outcomes following the use of generic versus brand-name antiepileptic drugs: a systematic review and meta-analysis. Drugs. 2010 Mar 26;70(5):605-21.
- Oluboka O, et al. Does therapeutic equivalence follow bioequivalence? A randomized trial to assess clinical effects after switching from Clozaril to generic clozapine (gen-clozapine). J Clin Pharmacol. 2010 May;50(5):531-5.
- The pharmaceutical industry 2008: current and future trends and strategic issues shaping pharma. ReportLinker. Available from: http://www.reportlinker.com/p087995/The-Pharmaceutical-Industry-2008-Current-and-Future-Trends-and-Strategic-Issues-Shaping-Pharma.html
- Lloyd I. R & D revolution remains just around the corner. Scrip Mag. 2002 Feb;109:72-3.
- Keyhani S, Diener-West M, Powe N. Are development times for pharmaceuticals increasing or decreasing? Health Aff (Millwood). 2006 Mar-Apr;25(2):461-8.
- OECD, Main Science and Technology Indicators, National Science Foundation. Volume 2009/1.
| Author: Christoph Baumgärtel, MD, Department Head, Department Safety and Efficacy Assessment of Medicinal Products, Institute Marketing Authorisation of Medicinal Products & LCM, AGES PharmMed, Austrian Medicines and Medical Devices Agency, 9 Schnirchgasse, AT-1030 Vienna, Austria |
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
A review of patient perspectives on generics substitution: what are the challenges for optimal drug use



Absolutely brilliant write-up.
Hi,
Please update me; can there be more than one marketing authorization holder for an ANDA in the US market?
thanks, Gaurav
Dear Amit Garg,
We very much appreciate your kind feedback.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office
Dear Gaurav Mehta,
Thank you for your interest in GaBI. Please visit GaBI Online for further information.
http://gabionline.net/Guidelines/US-guidelines-for-biosimilars
GaBI Journal Editorial Office
thank you