Author byline as per print journal: Subramanian Venkatesan, MSc; Associate Professor Martine Lamfers, PhD; Professor Sieger Leenstra, MD, PhD; Professor Arnold G Vulto, PharmD, PhD, FCP
|
Abstract: |
Submitted: 9 December 2016; Revised: 2 March 2017; Accepted: 2 March 2017; Published online first: 15 March 2017
Protein kinases are enzymes that add a phosphate group to a protein, and can modulate its function. Protein kinase inhibitors are enzyme inhibitors that block the action of one or more protein kinases. The phosphate groups are usually added to serine, threonine, or tyrosine amino acids on the protein. Most kinases act on both serine and threonine, whereas a smaller proportion acts on tyrosine.
Today’s detailed understanding of cell biology is revealing the fundamental changes that result in cancer. Tyrosine kinase inhibitors (TKIs) are effective in the targeted treatment of various malignancies. Imatinib was the first of this class and was approved in 2001.
A major concern in contemporary health care is the soaring cost of (innovative) medicines. Due to patents and other mechanisms of market protection, innovative medicines benefit from a market monopoly for up to 20 years. When market exclusivity is over, more affordable generic medicines enter the market, as was shown in a previous study [1]. For example, in The Netherlands, temozolomide, an important drug for treating brain cancer, was used by approximately 1,200–1,300 patients at a cost of about 10 million Euros per year to the healthcare system, at the time its patent expired. Before the end of market exclusivity, the annual cost per Defined Daily Dose (DDD), was 23,000 Euros [1]. The DDD is the assumed average maintenance dose per day for a drug used for its main indication in adults. However, when market exclusivity ceased, the cost for almost the same number of patients decreased by 80% to two million Euros per year. It is, therefore, important for healthcare planning decisions and drug purchasing contracts, to be aware of expiring patents. For instance, the UK Medicines Information of the National Health Service periodically publishes a document entitled Prescribing Outlook: New Medicines [2].
When we tried to search a list of patent expiry dates for TKIs, we noticed that such a list was not readily available, in contrast to expiry dates of a selection of biological medicines [3]. For this reason we set out to compile an overview of patent expiry dates of the current clinically approved oral TKIs in the European Union (EU) and in the US. We chose this group of cancer medicines as it is growing quickly and TKIs are often very expensive so great savings can be realized by healthcare systems once their patents have expired.
Protein kinase deregulation is one of the most characteristic traits of cancer biology. Large genomic sequencing studies show that mutations in genes encoding protein kinases are often present [4]. Currently, kinase inhibitors form the largest class of new anticancer drugs [5]. A race between pharmaceutical companies to license new kinase inhibitors has led to a wave of patents contributing to an intricate web of intellectual property rights [6]. The pharmaceutical industry gains exclusivity rights for marketing a drug when the first patents are granted by EU or US patent offices. The protected period is often extended as a reward for reaching certain licensing milestones (e.g. additional six months for paediatric indications).
Competition can be held off by either exclusivity rights or patent rights of the originator drug. Once both have expired, the introduction of generics or, in the case of biological medicines, biosimilars, is allowed and sales of the originator drug usually plummet. Therefore, drug companies try to stretch their exclusivity rights or patent rights in order to keep generics off the market.
By April 2015, 21 TKIs and three serine/threonine kinase inhibitors have been approved for clinical use. In this paper, we will provide an overview of current kinase inhibitors approved for clinical use and their indications. Furthermore, we will address when their patents are due to expire.
An overview of all clinically approved kinase inhibitors, including their indications and date of approval in Europe and the US, was generated by searching the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) databases. The patent expiry dates were collected from various online databases and from various sources in the pharmaceutical industry, see author’s note at the end of this paper.
Targeting breakpoint cluster region-Abelson (BCR-ABL)
The treatment of leukaemia has benefited most from the advent of kinase inhibitors. The first clinically successful TKI was imatinib, which inhibits the product of the Philadelphia chromosome (Ph+) in chronic myelogenous leukaemia (CML), BCR-ABL. Currently, several different TKIs have been developed which target an array of tyrosine kinases of which BCR-ABL is a common denominator. These drugs are often used interchangeably after drug resistance has occurred in CML or acute lymphoblastic leukaemia (ALL).
Bosutinib (Bosulif)
Bosutinib is used in the treatment of CML in which the BCR-ABL fusion gene is present. It is indicated when CML is resistant to treatment with the TKIs imatinib, nilotinib and dasatinib [7, 8]. It was authorized by EMA in March 2013 and the EU patent is expected to expire on September 2024. FDA approved it on September 2012, and the US patent is expected to expire in November 2026. Interestingly, market exclusivity to recoup investment in the US is almost four years longer than in the EU.
Dasatinib (Sprycel)
Dasatinib is used to treat CML and Ph+ ALL patients who do not respond to other treatment [9, 10]. It was authorized by EMA in November 2006 and the EU patent is expected to expire in November 2019. FDA authorized dasatinib in June 2006, and the US patent is expected to expire in October 2025, providing more than 19 years of market exclusivity to recoup the investment.
Imatinib (Gleevec/Glivec)
Imatinib is used in the treatment of multiple diseases, namely CML, Ph+ ALL, myelodysplastic or myeloproliferative diseases (MD/MPD), advanced hypereosinophilic syndrome or chronic eosinophilic leukaemia (HES/CEL), gastrointestinal stromal tumours (GIST) and dermatofibrosarcoma protuberans (DFSP), see EMA Summary of Product Characteristics (SmPC).
It is used against Ph+ CML and ALL since these cancers are mostly dependent on the oncogenic activity of BCR-ABL [11]. In MP/MPD, imatinib is used in patients with platelet-derived growth factor receptor (PDGFR) gene rearrangements [12] and in HES/CEL it is used for patients who have a FIP1L1 and PDGFRα rearrangement [13]. In GIST, imatinib is used in unresectable or metastatic cases and in cases with risk of recurrence after resection [14]. In DFSP, imatinib is used in unresectable or metastatic cases [15]. It was authorized by EMA in November 2001 and the EU patent expired in December 2016. Whereas imatinib was authorized by FDA in May 2001, the same year as in the EU, the US patent is only expected to expire in June 2022. The US patent grants a record market exclusivity of 21 years.
Nilotinib (Tasigna)
Nilotinib is used in CML after the development of resistance to imatinib or as a first-line therapy [16]. It was authorized by EMA in November 2007 and the patent is expected to expire in December 2028, providing a record 21-year EU market exclusivity. Nilotinib was authorized by FDA around the same time (October 2007): the US patent is expected to expire five years earlier than in the EU (July 2023).
Ponatinib (Iclusig)
Ponatinib is used for CML and Ph+ ALL patients who do not respond to dasatinib, nilotinib or imatinib. This situation could, for example, occur due to a resistance-inducing BCR-ABL1 (T315I) point mutation [17]. It was authorized by EMA on July 2013 and the EU patent is expected to expire in June 2028. Ponatinib was authorized by FDA slightly earlier (December 2012) and the US patent is expected to expire in December 2026.
Targeting Bruton’s tyrosine kinase (BTK)
BTK expression is restricted to B cells and has been found to be responsible for constitutively active B cell–receptor signalling in some cases of chronic lymphocytic leukaemia (CLL) and mantle cell lymphoma (MCL).
Ibrutinib (Imbruvica)
Ibrutinib is indicated in relapsed MCL [18] and CLL [19]. In addition, CLL patients who have a 17p deletion or TP53 mutation are also eligible for treatment with this drug. It was authorized by EMA in October 2014 and the EU patent is expected to expire in December 2026. Ibrutinib was authorized by FDA almost a year earlier (November 2013) and has a similar US patent expiry (December 2026).
Targeting epidermal growth factor receptor (EGFR/ERBB1)
EGFR is one of the most intensely investigated oncogenes. Glioblastoma and lung cancer are among the tumours with the most frequent EGFR alterations, ranging from 60% to 15% respectively [20, 21]. Multiple kinase inhibitors have been approved to target EGFR; they are mainly used for the treatment of lung cancer. Despite progress in drug development, resistance to EGFR inhibition remains a major problem, contributing to relapses.
Afatinib (Giotrif)
Afatinib is used against advanced or metastatic non-small-cell lung cancer (NSCLC) with an activating EGFR mutation (exon 19 deletion, L858R, G719X or L861Q) [22–24]. It is only indicated in adult patients who have not been previously treated with TKIs. It was authorized by EMA in September 2013 and the patent is expected to expire in December 2026. Although afatinib was authorized by FDA in the same year, the US patent is expected to expire much earlier (July 2018), with an eight-year shorter market exclusivity.
Erlotinib (Tarceva)
Erlotinib is used against EGFR mutant-driven NSCLC [25]. It is also used against metastatic pancreatic adenocarcinoma [26]. It was authorized by EMA in September 2005 and the patent is expected to expire in March 2020. Erlotinib was authorized by FDA in November 2004 and the US patent is expected to expire in November 2020.
Gefitinib (Iressa)
Similar to erlotinib, gefitinib is used in the treatment of EGFR mutant-driven NSCLC [27]. It was authorized by EMA in June 2009 and the EU patent is expected to expire in September 2019. Gefitinib was authorized by FDA six years earlier (May 2003) but the US patent is only expected to expire a little earlier (May 2017).
Lapatinib (Tyverb)
Lapatinib is indicated in progressive HER2-positive breast cancer [28]. It was authorized by EMA in June 2008 and the EU patent is expected to expire in June 2023. Although lapatinib was authorized by FDA one year earlier (March 2007), the US patent is expected to expire one and a half years earlier (November 2021).
Targeting the hepatocyte growth factor receptor (HGFR/MET), anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1)
NSCLCs contain in approximately 2%, 5% and 20% of the cases genetic alterations in respectively ROS1, ALK and MET [29, 30]. ROS1 and ALK usually undergo gene rearrangements, whereas MET is often amplified.
Crizotinib (Xalkori)
Crizotinib is used against previously treated ALK-positive metastatic NSCLC [31]. It was authorized by EMA in October 2012 and the EU patent is expected to expire in October 2027, whereas crizotinib was authorized by FDA in August 2011 and the US patent is expected to expire in November 2026.
Targeting Janus kinase (JAK)
The JAK2 V617F mutation leads to a persistently active JAK2/STAT3 pathway, which is frequently present in myeloproliferative diseases such as polycythaemia vera (PV).
Ruxolitinib (Jakafi)
Ruxolitinib targets JAK1/2 and is used against the forms of myelofibrosis that cause splenomegaly, namely primary myelofibrosis, post-polycythaemia vera myelofibrosis and post-essential thrombocythaemia myelofibrosis [32]. It was authorized by EMA in August 2012 and the EU patent is expected to expire in August 2027. Ruxolitinib was authorized likewise by FDA in November 2011 and the US patent is expected to expire in December 2027.
Tofacitinib
Tofacitinib inhibits JAK3 and is used in the treatment of patients with moderately to severely active rheumatoid arthritis who have not responded adequately to methotrexate. It has been refused by EMA due to concerns about the risk and type of serious infections seen with its use. However, FDA approved tofacitinib in November 2012 and the US patent is expected to expire in December 2023.
Targeting phosphoinositide 3-kinase delta (PI3K delta)
PI3K delta is one of the four isoforms of this enzyme and is specifically expressed in leukocytes. It is critical for the proliferation and survival of B lymphocytes. Hyperactivation of PI3K delta signalling is one of the main drivers for the malignant transformation of B lymphocytes.
Idelalisib (Zydelig)
Idelalisib is used in combination with rituximab for the treatment of adult patients with CLL who are refractory to therapy, or in patients with a 17p deletion or TP53 mutation. Furthermore, idelalisib is also indicated for the treatment of adult patients with follicular lymphoma (FL) that is refractory to two prior therapeutic lines of treatment, see EMA SmPC. It was authorized by EMA in September 2014 and the EU patent is expected to expire in September 2024. Whereas idelalisib was authorized by FDA around the same time (July 2014), the US patent is expected to expire two years earlier (April 2021).
Targeting vascular endothelial growth factor receptor (VEGFR), PDGFR and Ret Proto-Oncogene (RET)
Mutant RET is believed to mediate migration of the tumour cells whereas VEGFR and PDGFR are thought to mediate angiogenesis. Several multi-targeted tyrosine kinase inhibitors have been developed to simultaneously inhibit these kinases.
Axitinib (Inlyta)
Axitinib is used in advanced renal cell carcinoma (RCC) after resistance to sunitinib or cytokine-based therapy [8, 33, 34]. It was authorized by EMA in September 2012 and the EU patent is expected to expire in June 2025. Although axitinib was authorized by FDA six months earlier (January 2012), the US patent is expected to expire almost three years later (March 2028).
Cabozantinib (Cometriq)
Cabozantinib is used against unresectable or metastatic forms of medullary thyroid cancer. Patients with a RET mutation seem to have more benefit from cabozantinib [35]. It was authorized by EMA in March 2014 and the patent is expected to expire in March 2029, granting 15 years of market exclusivity. Although cabozantinib was authorized by FDA just over a year earlier (November 2012), the US patent is expected to expire almost five years earlier (September 2024), indicating three years shorter market exclusivity in the US.
Nintedanib (Vargatef/Ofev)
Recently, nintedanib, marketed as Vargatef by Boehringer-Ingelheim, was approved for combination therapy with docetaxel for the treatment of adult patients with recurrent NSCLC of adenocarcinoma tumour histology after first-line chemotherapy. Subsequently, the indication was extended to the treatment of adults with idiopathic pulmonary fibrosis (IPF) for which it is marketed as Ofev, see SmPCs, by the same company. It was authorized by EMA in November 2014 and the EU patent is expected to last until October 2025; it is unknown whether this date applies to both brands. Nintedanib was also authorized by FDA in October 2014. Unfortunately, we were not able to retrieve a reliable US patent expiry date for nintedanib in the US.
Pazopanib (Votrient)
Pazopanib is used in the treatment of metastatic RCC [36] and some forms of metastatic soft-tissue sarcoma [37]. It was authorized by EMA in June 2010 and the EU patent is expected to expire in June 2025, allowing 15 years of market exclusivity. Pazopanib was authorized by FDA in October 2009 and the US patent is expected to expire in October 2023, leaving three years less market exclusivity compared with the EU.
Vandetanib (Caprelsa)
Similar to cabozantinib, vandetanib is used against unresectable or metastatic forms of medullary thyroid cancer. Patients with a RET mutation seem to have more benefit from vandetanib [38]. It was authorized by EMA in February 2012 and the patent is expected to expire in August 2028. Although vandetanib was authorized by FDA almost a year earlier (April 2011), the patent is expected to expire in August 2028, leaving a long market exclusivity of more than 17 years.
Regorafenib (Stivarga)
Regorafenib is a tyrosine and serine/threonine kinase inhibitor. In addition to inhibiting several tyrosine kinases, it also inhibits the serine/threonine kinase BRAF. It is used against metastatic colorectal cancer (CRC) which has previously failed treatments, such as fluoropyrimidine-based chemotherapy, anti-VEGF therapy or anti-EGFR therapy, see SmPC. Furthermore, it is also used against metastatic GIST. It was authorized by EMA in August 2013 and the patent is expected to expire in August 2028. Regorafenib was authorized by FDA almost a year earlier (September 2012), but the US patent is expected to expire in July 2024.
Sorafenib (Nexavar)
Sorafenib is used against hepatocellular carcinoma [39], refractory RCC [40] and refractory differentiated thyroid carcinoma. It received EMA authorization in July 2006 and the patent is expected to expire in July 2021. In the US sorafenib was authorized by FDA in December 2005 and the patent is expected to expire in December 2020.
Sunitinib (Sutent)
Sunitinib is used against unresectable or metastatic GIST after failure with imatinib treatment [41]. Furthermore, it is indicated in metastatic RCC [42] and unresectable or metastatic pancreatic neuroendocrine tumours (pNET) [43]. It was authorized by EMA in July 2006 and the patent is expected to expire in July 2021. Likewise, sunitinib was authorized by FDA the same year (January 2006) and the US patent is expected to expire in February 2021.
Targeting BRAF
Cancer cells contain multiple genetic and epigenetic abnormalities. Despite this complexity, their growth and survival can often be impaired by the inactivation of a single oncogene. BRAF is a possible candidate for ‘oncogene addiction’ as it is known and is often mutated in codon 600 in melanoma, making it an attractive drug target.
Dabrafenib (Tafinlar)
Dabrafenib is used against unresectable or metastatic melanoma containing the BRAF V600E mutation, see SmPC. It was authorized by EMA in August 2013 and the EU patent is expected to expire in May 2029. Similarly, dabrafenib was authorized by FDA in May 2013 and the patent is expected to expire in January 2030, leaving almost 17 years of market exclusivity in the US as well as in the EU.
Trametinib (Mekinist)
Like dabrafenib, trametinib is used against melanomas containing the activating BRAF V600E or V600K point mutation [44]. It was authorized by EMA in June 2014 and the EU patent is expected to last until July 2029. However, trametinib was authorized by FDA in May 2013 and the patent is expected to expire in September 2025, resulting in three years less market exclusivity in the US.
Vemurafenib (Zelboraf)
Like the previous two drugs, vemurafenib is used against unresectable or metastatic melanoma containing the BRAF V600E mutation [45]. It was authorized by EMA in February 2012 and the patent is expected to expire in February 2027. Vemurafenib was authorized by FDA in August 2011 and the US patent is expected to expire in June 2029, resulting in an extended almost 18 years of US market exclusivity.
There is no direct link between date of market authorization and date of patent expiry, beyond deducing how much market exclusivity is left. If the date of patent registration and date of submission for market authorization were recorded, one could have made assumptions on the time of product development whether relatively short or very long. The fact that a significant period of patent life is still left for most drugs leads us to conclude that the drug development time was not significantly long. An analysis of the European public assessment reports for a sample of the drugs would give one an idea of the size of the clinical trials for example, and hence, the scope of the drug discovery and development work. We believe that the patenting strategy of the originator company is the most important factor in the market exclusivity periodic sought.
Since market exclusivity for these drugs exceed 10 years, is their high price justified? It would be very difficult to find objective data for this. For some drugs market exclusivity exceeds 15 years. It could be argued that this is more than enough time to recoup their investment and make substantial profits for their investors. However, due to the rapid development of new and better TKIs, it is unlikely that all of these compounds will be in use for the full time of their market exclusivity.
Over the last two decades pharmaceutical companies have raced to introduce and patent promising kinase inhibitors. As a result of market exclusivity, the high price of these branded drugs makes cancer treatment costly and can hamper accessibility to novel cancer drugs. Therefore, it is of interest to know when patents expire and generic versions will allow real market competition.
We have provided a comprehensive overview of the clinically approved kinase inhibitors in the EU and US as of 2015. Most of the clinically approved kinase inhibitors are used to treat types of leukaemia, although indications are being expanded to other types of cancer. The last three years have seen a great rise in the clinical approval of kinase inhibitors.
Market exclusivity for almost all drugs is more than 10 years, averaging around 14–15 years. One drug (vemurafenib) could create a stunning 18 years of market exclusivity and some even beyond the 20-year patent protection limit (Nilotinib in the EU and dasatinib and imatinib in the US). It shows how important patent strategies and early access to the market are for the pharmaceutical industry.
A note of caution is warranted. Patent expiry dates are not as stable as one might expect. A patent only stands as long as it has not been challenged and overruled. The pharmaceutical industry is constantly battling over patents in the legal arena. The data we have presented in this paper is the best we, as clinical researchers, could find using publicly accessible (and sometimes confidential) resources. In addition, it is also possible that later on patents will be extended based on new data. Therefore, our data are presented without any guarantee on accuracy.
There are considerable differences in length of market exclusivity of this class of medicines after they enter the market. In addition, interesting differences exist between the EU and the US, although it is not possible to draw general conclusions about these differences. With the data we have generated, bodies involved in drug reimbursement may be able to better plan their negotiation and access strategies in order to optimize patient access to this important class of medicines.
Editorial assistance by Ms Judith Martin, BSc(Pharm), was highly appreciated.
Stichting STOPhersentumoren.nl
Competing interests: The authors declare no competing financial interests in relation to this paper.
Provenance and peer review: Not commissioned; externally peer reviewed.
This paper is neither a research article (although much research has been invested in allocating and searching sources), nor a formal systematic review. It is an attempt to collect information on duration of market exclusivity of a very cost intensive group of non-biological targeted small molecules, and to make this information available in the public domain. Due to the methodology followed, we are not able to provide 100% transparency on the sources used, because in some cases we could only retrieve the data from pharmaceutical companies on an anonymity basis, and the information is not publicly accessible. We agree this is not ideal, hence the need for this paper, but we believe this is the best you can get. In this respect, the paper is original – you cannot find this compiled information anywhere else in the public domain.
The paper finds its roots in a research project on drug repurposing (looking at new applications for existing drugs), as we noticed that it was extremely difficult to find reliable data on patent expirations for the group of TKIs. From our previous work [1], we know what happens with drug costs if the patent expires, and for the sake of allocating healthcare resources, insight in expiring patents can be relevant for formulary decisions in the hospital.
Subramanian Venkatesan1, MSc, Research Student
Associate Professor Martine Lamfers1, PhD
Professor Sieger Leenstra1, MD, PhD
Professor Arnold G Vulto2, PharmD, PhD, FCP
1Department of Neurosurgery
2Department of Hospital Pharmacy
Erasmus MC, 80 Wytemaweg, NL-3015 CN Rotterdam, The Netherlands
References
1. Dylst P, Vulto A, Simoens S. Societal value of generic medicines beyond cost-saving through reduced prices. Expert Rev Pharmacoecon Outcomes Res. 2015;15(4):701-11.
2. Prescribing outlook – new medicines 2016. Specialist Pharmacy Service. 2016.
3. Derbyshire M. Patent expiry dates for best-selling biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):178-9. doi:10.5639/gabij.2015.0404.040
4. Greenman C, Stephens P, Smith R, Dalgliesh Gl, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153-8.
5. Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10(2):130-7.
6. Akritopoulou-Zanze I, Hajduk PJ. Kinase-targeted libraries: the design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discov Today. 2009;14(5-6):291-7.
7. Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol. 2009;27(3):469-71.
8. Khoury HJ, Cortes JE, Kantarjian HM, Gambacorti-Passerini C, Baccarani M, Kim DW, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119(15):3403-12.
9. Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260-70.
10. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531-41.
11. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the philadelphia chromosome. N Engl J Med. 2015;344(14):1038-42.
12. Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347(7):481-7.
13. Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1 L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201-14.
14. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472-80.
15. McArthur GA, Demetri GD, van oosterom A, Heinrich MC, Debiec-Rychter M, Corless CL, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23(4):866-73.
16. Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251-9.
17. Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075-88.
18. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507-16.
19. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42.
20. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-77.
21. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169-81.
22. Yeh P, Chen H, Andrews J, Naser R, Pao W, Horn L. DNA-mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clin Cancer Res. 2013;19(7):1894-901.
23. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327-34.
24. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98(12):1817-24.
25. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med. 2005;353(2):123-32.
26. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960-6.
27. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129-39.
28. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733-43.
29. Camidge DR, Doebele RC. Treating ALK-positive lung cancer – early successes and future challenges. Nat Rev Clin Oncol. 2012;9(5):268-77.
30. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863-70.
31. Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, AHN Mj, De Pas T, Besse B, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-94.
32. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799-807.
33. Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8(11):975-84.
34. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378(9807):1931-9.
35. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660-6.
36. Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369(8):722-31.
37. Sleijfer S, Ray-Coquard I, Papai Z, Le Cesne A, Scurr M, Schöffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European Organisation For Research And Treatment Of Cancer–Soft Tissue and Bone Sarcoma Group (EORTC Study 62043). J Clin Oncol. 2009;27(19):3126-32.
38. Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134-41.
39. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378-90.
40. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125-34.
41 Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-38.
42 Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115-24.
43. Raymond E, Dahan L, Raoul J-L, Bang Y-J, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501-13.
44. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694-703.
45. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-16.
|
Author for correspondence: Professor Arnold G Vulto, PharmD, PhD, FCP, Department of Hospital Pharmacy, Erasmus MC, 80 Wytemaweg, NL-3015 CN Rotterdam, The Netherlands |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/overview-of-the-patent-expiry-of-non-tyrosine-kinase-inhibitors-approved-for-clinical-use-in-the-eu-and-usa.html
Author byline as per print journal: Niklas Ekman, PhD; Professor Arnold G Vulto, PharmD, PhD; Paul Cornes, MD
|
Abstract: |
Submitted: 19 April 2016; Revised: 1 June 2016; Accepted: 4 June 2016; Published online first: 17 June 2016
The 21st Congress of the European Association of Hospital Pharmacists (EAHP) took place on 16–18 March 2016 in Vienna, Austria. The Biosimilar Medicines Group (formerly EBG) held a satellite symposium entitled ‘The facts about biosimilars’ on 17 March 2016 during the conference.
The Biosimilar Medicines Group represents the leading pharmaceutical companies developing, manufacturing and marketing biosimilars across Europe.
It is a sector group of Medicines for Europe (formerly the European Generic medicines Association).
With 10 years of positive patient treatment experience and 20 products successfully launched, use of biosimilars today offers a huge opportunity to deliver significantly improved access to modern therapies for millions of European patients receiving both chronic and acute care.
Dr Paul Cornes (University Hospitals Bristol, NHS Foundation Trust, Bristol, UK), presenting ‘Biosimilars – can we do without them?’ highlighted the fact that according to the World Health Organization (WHO), cancer has been the world’s top killer since 2010. The disease has the most devastating economic impact of any cause of death in the world. In Europe, 17% of all the ‘healthy’ years lost were due to cancer and 170 million years of ‘healthy life’ were lost due to death and disability from cancer in 2008.
The good news for cancer treatment is that since the 1960s pharmaceutical innovation has enabled the introduction of a whole host of new cancer drugs. The rate of new medicines being introduced is also increasing rapidly. Prior to the 1960s, there were only five cancer medicines available in the US. During the 1960s, only two new cancer medicines were introduced, but in the 30 years between 1970 and 2000 a total of 79 new medicines were introduced and in the five-year period between 2010 and 2015 a staggering 43 new cancer medicines have been introduced. At this rate, more than 100 new cancer medicines could be added in the period between 2010 and 2020, see Figure 1 [1]. This innovation heralds a new era of targeted precision therapy for cancer that could transform the outlook for patients with the world’s most important diseases.
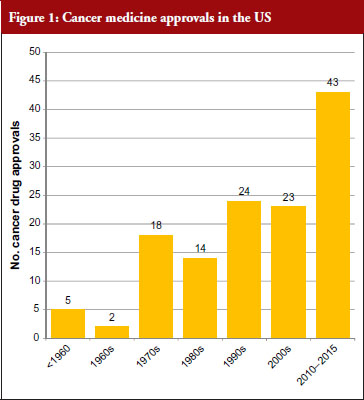
However, there is a widespread belief that this dream may not be affordable – even for the richest nations of the world. Learning howto make this innovation affordable will not just address cancer – but willshow us how to manage the costs of innovation in other diseases as well. One key focus has been on controlling the costs of innovativebiological therapy – as cancer medicine costs are increasing five times faster than any other class of medicine. In fact, eight cancer medicines approved by the US Food and Drug Administration (FDA) in 2015 had six-figure price tags [2].
The problem is not just that these new medicines are expensive, but due to the increasingly ageing population there is ever more cancer to treat. Cancer is often a disease of the elderly, with the peak age for cancer being 70 to 84 years.
The increasing cost of treating the ageing population could therefore lead to healthcare spending growing at faster rate than the gross domestic product (GDP). In the US, healthcare costs were increasing at an annual rate of 7% per year. This rate, if sustained, is forecasted to bankrupt Medicare in nine years and increase the nation’s overall healthcare bill to US$4 trillion in 10 years [3]. Such cost pressures are not just an American issue, as all the world’s developed nations but one have increased health spending in relation to national wealth. It is therefore essential to look at where savings can be made in order to make it possible for the population to age and to still be able to afford innovation in medical treatment.
Simple cuts in the healthcare budget are not the answer. Savings have to be made where they do not put the health of citizens at risk. For example, every 1% decrease in government healthcare spending is associated with a 10.6% rise in maternal mortality in the EU [4]. Debt is therefore a real threat to health.
Given the fact that by 2018, biologicals worth more than US$68 billion in current annual sales will lose patent protection [5], the case for using biosimilars seems clear. Even with only a 20% discount, this could give the world a US$14 billion health innovation fund. Whereas a 30% discount could save US$20 billion and a 40% discount could save US$27 billion.
Dr Cornes highlighted the fact that WHO is clearly an advocate for generics and biosimilars as illustrated by the following WHO statements:
He concluded that ‘we have a common interest between patients, physicians, pharmacists, pharmaceutical industry and payers in the success of biosimilar medicines’. He added that ‘biosimilar medicines offer a reward to world health that will be substantial’. By generating savings from within our existing health budgets, on medicines that are equally safe and effective, we can still afford to invest in healthcare innovation even in times of financial crisis.
Professor Arnold G Vulto’s (Erasmus University, Rotterdam, The Netherlands) presentation entitled ‘Biosimilars: concerns of prescribers and how to address them as a hospital pharmacist’ highlighted the fact that the total medicine bill will grow exponentially with the introduction of so many blockbuster breakthrough medicines. He too pointed to the ‘savings potential of biosimilar medicines’.
Four biologicals approved in 2014: Keytruda (pembrolizumab), Cyramza (ramucirumab), Opdivo (nivolumab) and Sylvant (siltuximab), are anticipated to be blockbusters by 2019, i.e. medicines that generate annual sales of at least US$1 billion, according to sales forecasts from the Thomson Reuters Cortellis database. When considering all new medicines approved in 2014, 12 are expected to become blockbusters and sales of these medicines are forecast to add US$29 billion per year to the cost of drugs.
Five biologicals newly approved in 2015 are predicted to become blockbusters by 2020 [9]. For all new medicines approved in 2015, 16 are expected to become blockbusters and sales of these medicines are forecast to add more than US$36 billion per year to the cost of drugs. These predictions clearly highlight the problem facing governments when it comes to increasing healthcare costs.
One way to modify these increasing costs would be to increase the use of biosimilars. However, as Professor Vulto pointed out, uptake of biosimilars in the EU varies widely between countries and therapeutic areas; and Europe accounts for 80% of global spending on biosimilars [10].
For example, the human growth hormone (HGH) biosimilar Omnitrope (somatropin) was the first product approved in the EU as a biosimilar back in 2006 [11]. Despite this product being available for 10 years, some countries in the EU, e.g. Greece, Ireland and Slovakia, still have little or no uptake of this biosimilar, see Figure 2. Biosimilars penetration in Europe for erythropoietin (EPO) and granulocyte colony-stimulating factor (G-CSF) also varies, from 0% for EPO in countries such as Belgium to 100% for G-CSF in the Czech Republic, Hungary, Romania and Slovakia. In fact, Eastern Europe is leading the way in biosimilar medicines penetration perhaps driven by economic factors.
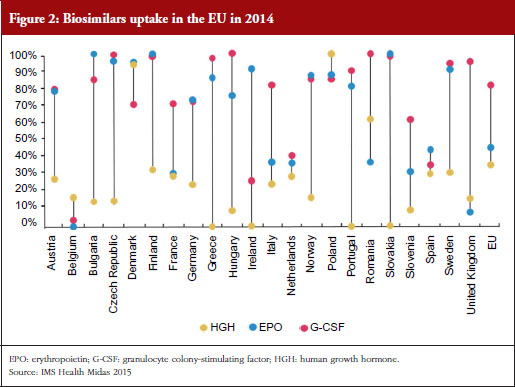
The important issue that needs to be addressed is how to improve physician prescribing of biosimilars. According to Professor Vulto, physicians will prescribe biosimilars when they have sufficient trust in the sameness of the biosimilar. Understanding of the biosimilarity concept is of great importance, especially when there are sufficient incentives to do so.
In the EU, the European Medicines Agency (EMA) assesses the scientific aspects of biosimilars and makes recommendations for a market approval when the biosimilar candidate is assessed to be therapeutically equivalent (comparable quality, safety and efficacy) to the reference biological. Later, based on a scientific appraisal by EMA, decisions on the policy of interchangeability (medical practice) between biosimilars and originator biologicals are made, but not by EMA, rather at the national level. Several EU Member States, such as Finland, Germany and The Netherlands have taken clear positions in support of the interchangeability (a medical practice) of biosimilars, while others, e.g. the UK’s healthcare cost watchdog NICE, have also developed full guidelines [12].
Acceptance of a biosimilar is dependent on the actions of many different stakeholders, such as physicians, patients, pharmacists, third-party payers and policymakers. It is essential to obtain the buy in (‘ownership’) from stakeholders, for example, including prescribers in the production of treatment guidelines. This can help deal with the common misconceptions healthcare professionals may have about biosimilars, such as that they:
A clear information gap exists when it comes to biosimilars. One way Professor Vulto suggested to reduce this gap was for regulators to communicate their knowledge actively to medical professionals. In fact, EMA has urged regulators in EU Member States to provide physicians with more information regarding the concepts of comparability and biosimilarity [13]. He also suggested that regulators should also point out that over the past 10 years, since the introduction of biosimilars, there have not been any new or unexpected safety signals; that the assessment system has worked as expected; and that based on what we have learned from their actual use, the mistrust raised against biosimilars was not justified. The Generics and Biosimilars Initiative (GaBI) was also highlighted as being another source of valuable information to build trust in cost-effective treatments. Professor Vulto added that in order to ‘avoid trouble around switching’ it was essential to convince prescribers of the (financial) advantages for society, without compromising quality of treatment, including increased treatment choices and access to medicines for patients.
Dr Niklas Ekman, a senior researcher at the Finnish Medicines Agency (FIMEA, Helsinki, Finland), gave a presentation on ‘Biosimilars from the perspective of an EU regulator’. He highlighted the fact that batch-to-batch variability is inherent for all biologicals, both for originators and biosimilars. Alterations in the quality profile can be introduced through manufacturing process changes. With all changes, whether for an originator or biosimilar, the pre- and post-change version of the medicinal product needs to be demonstrated to be comparable through a comparability exercise. Manufacturers and regulators therefore have extensive experience in assessing the impact of process changes – including in the case of complex biologicals.
The current EU regulatory definition of biosimilars defines a biosimilar as a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product). A biosimilar demonstrates similarity to the reference medicinal product in terms of quality characteristics, biological activity, pharmacokinetic profile, safety and efficacy based on a comprehensive comparability exercise.
A stepwise approach is used to establish biosimilarity. Comprehensive physicochemical and biological characterization should be followed by non-clinical studies, which should include in vitro functional studies and, if needed, in vivo studies. These should be followed by a comparative pharmacokinetic study in a sensitive and homogeneous study population, such as healthy volunteers (if possible/feasible). Finally, efficacy/safety clinical studies to confirm comparable clinical performance of the biosimilar and the reference product should normally be carried out. These studies should be adequately powered, randomized, parallel group (usually equivalence) trials. The study population should be representative of the approved indication(s) and be sensitive for detecting potential differences and the endpoints should be selected with the aim of investigating possible differences, not demonstrating efficacy per se, see Figure 3.
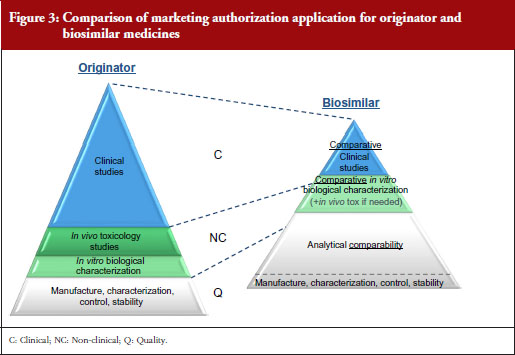
EMA has approved 22 biosimilars to date, although two were withdrawn, leaving a total of 20 biosimilars approved for use in Europe [11]. In March 2016, the agency was also reviewing 12 biosimilars, including adalimumab (2), enoxaparin sodium (2), etanercept (1), infliximab (1), insulin glargine (1), pegfilgrastim (3), rituximab (1) and teriparatide (1).
The success of developing a biosimilar candidate depends on:
In order to prove biosimilarity the amino acid sequence, posology and the route of administration must be the same as the reference biological. In addition, the active substance must be similar in terms of molecular and biological characteristics. Any differences in strength, pharmaceutical form, formulation, excipients or presentation need to be justified. Finally, intended changes to improve efficacy (‘bio-betters’) are not allowed.
Funding of medicines in Finland
The way medicines are funded in Finland results in hospitals leading the adoption of biosimilars due mainly to economic incentives. Prescribers and patients on the other hand have no special interest in biosimilars due to the lack of incentives.
Hospitals
For medicines administered in public hospitals the cost is borne by the community as a whole.
Pharmacies
When prescription medicines are dispensed by pharmacies the costs are covered by the Social Insurance Institution (state). Patients pay an annual maximum of Euros 610 for reimbursed medicines. There are three different levels of reimbursement; 40%, 64% and 100%. Biosimilars have the same reimbursement level as their reference biological. Due to the high price of biologicals, in practice, reimbursement is a prerequisite for the use of any biologicals outside hospitals.
The Finnish Medicines Agency, FIMEA, announced in May 2015 that it considers EU biosimilars interchangeable with their reference biologicals. Automatic substitution at the pharmacy level, however, is not included in the current FIMEA recommendation [14, 15].
The FIMEA position paper on the interchangeability of biosimilars concludes the following:
The position of FIMEA is therefore that biosimilars licensed in the EU are interchangeable with their reference products under the supervision of a healthcare professional. As with any biological products, the switch should be documented (including brand name and batch number).
Interchangeability recommendations in other European countries
According to Dr Ekman, similar positions have been adopted by other EU national authorities, including the Medicines Evaluation Board (MEB) in The Netherlands; the Paul Ehrlich Institute (PEI) in Germany; and the Health Products Regulatory Authority (HPRA) in Ireland.
Dutch MEB recommendation
Exchange between biological medicines (regardless of whether they are originator biological products or biosimilar medicinal products) is permitted, but only if adequate clinical monitoring is performed and the patient is properly informed.
German PEI recommendation
Biosimilars can be used in the same way as their reference products to which they have shown equivalence. This implicitly covers both patients who have not yet received biological therapy as well as patients who previously received the originator product.
Irish HPRA recommendation
If it is planned to change the medicine a patient receives from a reference to a biosimilar medicine or vice versa, the treating physician should be involved; this should involve discussion between the prescriber/patient, and prescriber/dispensing pharmacist.
At the end of the session, Medicines for Europe’s Market Access Director Maarten van Baelen concluded that biosimilars today provide a huge opportunity to deliver significantly improved access to existing and future innovative therapies for millions of European patients in both chronic and acute care while supporting the sustainability of our healthcare systems.
Medicare is a national social insurance programme, administered by the US federal government since 1966. It provides health insurance for Americans aged 65 and older who have worked and paid into the system. It also provides health insurance to younger people with disabilities.
Minor wording changes have been made to the presentations to clarify the points made.
The authors wish to thank Michelle Derbyshire, PhD, GaBI Online Editor, in preparing this meeting report.
Disclosure of financial and competing interests: The satellite symposium was organized by the Biosimilar Medicines Group, a sector group of Medicines for Europe. Fees were paid to the organization for the conference slot, however, none of the speakers received direct payments from Medicines for Europe.
Dr Paul Cornes has received honoraria from Accord Healthcare, Amgen, Bernstein, British Medical Journal, European Generic medicines Association, Hospira, Janssen, Lilly, Merck Serono, Napp, Pharmaceutical Association of Malaysia, Pfizer, Roche, Sandoz and Teva.
Professor Arnold G Vulto declares no personal financial interest in any pharmaceutical business. Any honoraria resulting from his participation in advisory boards or as a speaker at scientific or commercial meetings and any consulting fees received are given to Erasmus University Hospital. Companies/organizations involved are: AbbVie, Amgen, Biogen, European Generic medicines Association, Mundipharma, Pfizer/Hospira, Roche, Sandoz.
Dr Niklas Ekman is employed by a regulatory agency, and has nothing to disclose.
Provenance and peer review: Not commissioned; internally peer reviewed.
Niklas Ekman, PhD
Senior Researcher
Quality Assessor for biological medicinal products
Member of the Biosimilar Working Party (BMWP), European Medicines Agency (EMA)
Finnish Medicines Agency (FIMEA), 103b Mannerheimintie, PO Box 55, FI-00034 FIMEA, Finland
Professor Arnold G Vulto, PharmD, PhD
Deputy Head Hospital Pharmacy
Professor of Hospital Pharmacy and Practical Therapeutics
Erasmus University Medical Center, PO Box 2040, 230 Gravendijkwal, NL-3015 CE Rotterdam, The Netherlands
Paul Cornes, MD
Clinical Outcomes Group, Bristol Oncology Center, University Hospital Bristol, UK
References
1. Cornes P. Pictogram created from data in – Savage P.Development and economic trends in cancer therapeutic drugs: analysis of modern and historical treatment costs compared to the contemporary GDP per capita. J Clin Oncol. 2014;33(suppl; abstract 17535).
2. Jarvis LM. The year in new drugs. Chemical and Engineering News. 2016;94(5):12-7.
3. Langreth R. Will health costs bankrupt America? Forbes. 23 Feb 2011.
4. Maruthappu M, Ng KY, Williams C, Atun R, Agrawal P, Zeltner T. The association between government healthcare spending and maternal mortality in the European Union, 1981–2010: a retrospective study. BJOG. 2015;122(9):1216-24.
5. Derbyshire M. Patent expiry dates for best-selling biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):178-9. doi:10.5639/gabij.2015.0404.040
6. World Health Organization. The World Health Report 2010. Chapter 4: More health for the money [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://www.who.int/whr/2010/10_chap04_en.pdf
7. World Health Organization. Expert Committee on Biological Standardization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 23 October 2009 [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
8. World Health Organization. The World Medicines Situation Report, 2011 [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://apps.who.int/medicinedocs/documents/s20054en/s20054en.pdf?ua=1
9. Mullard A. 2015 FDA drug approvals. Nat Rev Drug Discovery. 2016;15(2):73-6.
10. GaBI Online – Generics and Biosimilars Initiative. European uptake of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Jun 1]. Available from: www.gabionline.net/Reports/European-uptake-of-biosimilars
11. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Jun 1]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
12. National Institute for Health and Care Excellence. Introducing biosimilar versions of infliximab: Inflectra and Remsima [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://publications.nice.org.uk/introducing-biosimilar-versions-of-infliximab-inflectra-and-remsima-htta329/insights-from-the-nhs-managing-the-introduction-of-biosimilar-medicines
13. Kurki P. Biosimilars for prescribers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):33-5. doi:10.5639/gabij.2015.0401.008
14. GaBI Online – Generics and Biosimilars Initiative. Finnish drug regulator recommends interchange-ability of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Jun 1]. Available from: www.gabionline.net/Policies-Legislation/Finnish-drug-regulator-recommends-interchangeability-of-biosimilars
15. FIMEA. Interchangeability of biosimilars – position of Finnish Medicines Agency Fimea. 22 May 2015 [homepage on the Internet]. [cited 2016 Jun 1]. Available from: www.fimea.fi/documents/542809/838272/29197_Biosimilaarien_vaihtokelpoisuus_EN.pdf
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/reducing-healthcare-costs-and-building-trust-in-biosimilar-medicines.html
Author byline as per print journal: Professor Stefan Mühlebach, PhD, Professor Arnold Vulto, PharmD, PhD, Jon SB de Vlieger, PhD, Vera Weinstein, PhD, Beat Flühmann, PhD, Vinod P Shah, PhD
|
Introduction: Besides biologicals, a new class of complex drugs – non-biological complex drugs (NBCDs), e.g. liposomes, iron carbohydrate products and glatiramoids – has emerged. Originator NBCD products have been approved by established regulatory rules. However, their follow-on products comprise a challenge to the regulators, manufacturers, physicians and pharmacists. |
Submitted: 12 August 2013; Revised: 24 September 2013; Accepted: 27 September 2013; Published online first: 11 October 2013
The progress in pharmaceutical sciences and manufacturing techniques together with more targeted and better-tolerated pharmaceutical formulations gave rise to a new class of medicinal products with complex macromolecular (nanoparticulate) structures, the so-called non-biological complex drugs (NBCDs) [1, 2]. Amongst others, NBCDs comprise liposomes, iron carbohydrate products and glatiramoids. The complexity of these synthetic NBCDs may even exceed that of biologicals. Originator NBCDs have received regulatory approval based on proven quality, efficacy and safety and have been established for many years. Some NBCDs have already been introduced before the growing awareness about the specific issues related to the production and characterization of nanostructures. Accordingly, regulatory approval of NBCD follow-on products is subject to a lively discussion about the requirements to prove therapeutic equivalence and being eligible as substitute for the reference product.
The Non-Biological Complex Drugs Working Group at Dutch Top Institute Pharma, a public–private partnership in The Netherlands, is a network of scientific and clinical experts from academia, industry, regulatory bodies, and knowledge institutes to discuss specific aspects of the development and evaluation of NBCDs and give its expert opinion. This paper briefly summarizes the key discussion points of a closed workshop at the FIP (International Pharmaceutical Federation) Centennial Congress in 2012, organized by the FIP Board of Pharmaceutical Sciences, in order to assist the understanding of the regulatory challenges associated with NBCD follow-on products such as the definition of science-based policies for interchange and substitution. The closed workshop on invitation only was followed by an open discussion symposium the following day at the FIP Centennial Congress about the proposed terminology and a planned paper on points to consider for regulation [3, 4].
NBCD are defined as a medicinal product of non-biological origin with an active substance that is not a homo-molecular structure, but consists of different closely related and mostly nanoparticulate structures. Accordingly, there is not a single substance that can be isolated, quantitated and fully characterized or described by state-of-the-art physicochemical analytical means [4]. Changes in the composition and morphology of an NBCD can substantially influence the quality, biological properties and therapeutic profile of the medicinal product and result from minute variations in the manufacturing process [1, 2, 5]. However, not all structural changes and mechanisms that affect the therapeutic profile are fully understood. Notably, the complexity of NBCD prevents establishing full proof of pharmaceutical equivalence by state-of-the-art analytical means, which comprises one of the two pillars in the evaluation of a generic medicinal product, see Figure 1.
In contrast to the mainly direct and systemic drug-target interaction of small molecules with defined receptors in a concentration-dependent manner, most NBCDs comprise nanoparticles from which the active ingredient has to be released or formed and then transported to certain biological compartments where the intended activity should be performed. Even a slightly different release or formation rate of the active ingredient, e.g. due to differences in higher order structure of the precursor molecules, can negatively affect the safety and efficacy profile of an NBCD or its follow-on product. For example, in products for intravenous iron therapy such as iron carbohydrate nanoparticles, the highly reactive iron is bound in a polynuclear core, which in turn is stabilized by a carbohydrate shell. After intravenous administration, this complex is first phagocytized by monocytes where the iron is released and transiently stored before it is bound to transferrin and transported to the target tissue where it exerts its action, e.g. incorporation into newly synthesized erythrocytes in the bone marrow; or is stored in an accessible compartment for later physiological use [6, 7]. Hence, the biological activity of an NBCD is not necessarily correlated to its serum pharmacokinetics (central compartment), the generics pathway’s second pillar to show bioequivalence.
In the case of glatiramoids, products that comprise a complex mixture of polypeptides for the treatment of relapsing-remitting multiple sclerosis, even no pharmacokinetic profile and no validated biomarker for efficacy are available. However, although the originator, Copaxone®, and a follow-on product are similar in many physiochemical properties, e.g. size distribution, molar ratio of amino acids; sensitive chemical and biological analyses demonstrated differences between those products, e.g. gene expression patterns of glatiramoid-primed murine splenocytes [8].
In contrast to follow-on compounds of biologicals that are evaluated according to distinct biosimilar guidelines (originally established in the EU), some NBCD follow-on products are classified as generics although the two prerequisites for the generics approach cannot be fulfilled. As outlined above, the inability to fully characterize an NBCD prevents proof of pharmaceutical equivalence or clinically meaningful differences between a follow-on NBCD and its reference product. Moreover, bioequivalence assessment in healthy volunteers does not necessarily reflect the biological fate of and therapeutic response to an NBCD as outlined by the example of the iron sucrose complex below. Notably, this lack in proof of therapeutic equivalence and potential differences in tolerability or safety of NBCD follow-on products can easily become a concern since these products are often used as chronic treatment and in patients with already poor health states, e.g. iron carbohydrate products for haemodialysis patients, liposomal formulations of cytotoxic agents for cancer patients and glatiramoids for multiple sclerosis patients [1, 9, 10]. In such patients, even slight but clinically meaningful differences between the products may interfere with clinical success and thus the feasibility for interchange with the innovator’s product.
The potential clinical consequences of the above-mentioned differences could be illustrated by comparative clinical and non-clinical studies of a well-established NBCD (iron sucrose, Venofer®) and different follow-on preparations (iron sucrose similars, ISS). One study evaluated the effects of switching iron treatment from the iron sucrose originator to an ISS in 75 consecutive stable, haemodialysis-dependent chronic kidney disease patients who underwent at least 60 dialysis sessions before and after the switch at a French dialysis centre [11]. After the switch to an ISS, haemoglobin levels decreased rapidly and anaemia medication had to be increased to return to targeted haemoglobin levels after quite a lengthy re-adjustment period. In addition to this apparent lack of therapeutic equivalence of an ISS, other ISS were associated with an increased risk of adverse events (658 patients at a South Korean centre who had been treated with the iron sucrose originator or an ISS) [12], even if the originator iron sucrose has been well tolerated before (three case reports in Germany) [13]. Furthermore, non-clinical studies showed not only differences between ISS and the originator product [14] but also among different ISS [15], particularly with respect to off-target iron disposition from ISS and induction of oxidative stress and inflammation.
In many countries generics approval of follow-on products allows automatic substitution at the pharmacy level. Since the International Nonproprietary Names of the innovator’s and follow-on products are the same, clinicians, caregivers and patients are often not aware of the change in medication. In contrast to the substitutability and interchangeability of fully characterized small molecule generics with well-established therapeutic equivalence, approval for substitution or interchange of NBCD products should only be granted on the basis of appropriate non-clinical and/or clinical comparisons. Together with the comparability of physicochemical quality this would allow to exclude clinically meaningful differences between the NBCD follow-on products and the reference product [1, 2]. Lacking information in case of insufficient response or intolerance can lead to unnecessary diagnostic tests and use of potentially more invasive and more expensive treatment options [8, 10]. Overall, drug product replacement that is guided by acquisition cost only may increase other costs and not be cost-effective from the patient’s and payer’s perspective.
At the NBCD workshop in the course of the FIP Centennial Congress 2012, experts from academia, industry, regulatory bodies and knowledge institutes agreed that there is a need for a globally harmonized approach to authorize NBCD follow-on products. This approach should be linked to an accepted common terminology [4]. Also the requirements for an abbreviated procedure showing comparability between different types of NBCD follow-on products and feasible reference products should be clear. In order to approve an NBCD follow-on product that will be interchangeable with the innovator’s product, relevant and comparative clinical and/or non-clinical trials should be performed. Aims of these studies will be an appropriate characterization of the NBCD with up-to-date analytical techniques and to identify the extent of similarity with the originator product. Clinical trials should be sufficiently powered, conducted in patients rather than healthy volunteers to cover for disease-associated changes in pharmacokinetics (PK) and pharmacodynamics (PD). Furthermore, trials should include suitable biological tests to evaluate the similarity of PK, PD and safety/tolerability in populations with similar aetiology of the disease as for the aimed indication. For these means also surrogate efficacy and safety markers may be used. Based on the degree of similarity that could be confirmed by the results of such trials and markers, approval of an NBCD follow-on product can be gradually extended to allow for interchange or even automatic substitution in newly diagnosed patients or those on existing chronic treatment. Such a stepwise similarity approach towards totality of evidence can help manufacturers of follow-on products in the development of safe and effective products and to make a realistic prediction of development costs and timelines.
Furthermore, the expert panel indicated that post-approval pharmacovigilance for NBCD and follow-on products should be based on specific brand names as already proposed earlier [16]. An information exchange among treating healthcare professionals and eventually the patient is mandatory to allow for appropriate treatment and drug product traceability in the individual patient.
An increasing number of NBCDs including nanomedicines become target for development and introduction of follow-on products. Recent clinical data with NBCD follow-on products (iron sucrose) that were approved like small molecule generics revealed significant differences in efficacy and tolerability compared to the originator product. Accordingly, regulators are prompted to establish a global regulatory framework that considers the structural complexity and specific biological properties of NBCD and provides clear guidance for the development and documentation of safe and effective follow-on products. The experts from academia, industry, regulatory bodies and knowledge institutes at the FIP 2012 workshop suggest a stepwise similarity approach that includes appropriate clinical and/or non-clinical studies that evaluate markers of PK, PD (if applicable) and safety/efficacy in relevant patient populations. As long as proof of therapeutic equivalence and similar safety profiles by appropriate studies is missing, interchange and automatic substitution between NBCDs and their follow-on products should be discouraged. Overall, a critical review of the current and emerging regulation of NBCDs and NBCD follow-on products encourage further multidisciplinary research and consensus discussions among all stakeholders to develop guidance towards the definition of an NBCD and the development of NBCD follow-on products [3].
Medical writing support was provided by Mr Walter Fürst, SFL Regulatory Affairs and Scientific Communication, Switzerland, and funded by Vifor Pharma Ltd.
Disclosure of financial and competing interests: This manuscript was written within the framework of the Non-Biological Complex Drugs Working Group, hosted at Dutch Top Institute Pharma. The NBCD Working Group is currently supported by Sanofi, Teva Pharmaceutical Industries Ltd and Vifor Pharma International Inc. Professor Stefan Mühlebach is an employee of Vifor Pharma Ltd, Dr Vera Weinstein is an employee of Teva Pharmaceutical Industries, and Dr Beat Flühmann is an employee of Vifor Fresenius Medical Care Renal Pharma.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor Stefan Mühlebach1,2, PhD
Professor Arnold Vulto3, PharmD, PhD
Jon SB de Vlieger4, PhD
Vera Weinstein1,5, PhD
Beat Flühmann1,6, PhD
Vinod P Shah1, PhD
1Steering Committee member, NBCD Working Group, TI Pharma, Leiden, The Netherlands
2Vifor Pharma Ltd, Glattbrugg, Switzerland
3Erasmus University Medical Center, Hospital Pharmacy, Rotterdam, The Netherlands
4TI Pharma, PO Box 142, NL-2300 AC Leiden, The Netherlands
5Teva Pharmaceutical Industries, Petach Tikva, Israel
6Vifor Fresenius Medical Care Renal Pharma, St Gallen, Switzerland
References
1. Borchard G, Fluhmann B, Muhlebach S. Nanoparticle iron medicinal products—requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studies. Regul Toxicol Pharmacol. 2012;64(2):324-8.
2. Schellekens H, Klinger E, Mühlebach S, et al. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
3. Schellekens H, Stegemann S, Weinstein V, et al. How to regulate nonbiological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2013 Sep 25. doi:10.1208/s12248-013-9533-z
4. Crommelin DJ, de Vlieger JS, Weinstein V, et al. Different pharmaceutical products need similar terminology. AAPS J. 2013 Sep 25. doi:10.1208/s12248-013-9532-0
5. Ehmann F, Sakai-Kato K, Duncan R, et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine (Lond). 2013;8(5):849-56.
6. Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61(6):933-52.
7. Geisser P, Burckhardt S. The pharmacokinetics and pharmacodynamics of iron preparations. Pharmaceutics. 2011;3(1):12-33.
8. Bakshi S, Chalifa-Caspi V, Plaschkes I, et al. Gene expression analysis reveals functional pathways of glatiramer acetate activation. Expert Opin Ther Targets. 2013;17(4):351-62.
9. Mamidi RN, Weng S, Stellar S, et al. Pharmacokinetics, efficacy and toxicity of different pegylated liposomal doxorubicin formulations in preclinical models: is a conventional bioequivalence approach sufficient to ensure therapeutic equivalence of pegylated liposomal doxorubicin products? Cancer Chemother Pharmacol. 2010;66(6):1173-84.
10. Varkony H, Weinstein V, Klinger E, et al. The glatiramoid class of immunomodulator drugs. Expert Opin Pharmacother. 2009;10(4):657-68.
11. Rottembourg J, Kadri A, Leonard E, et al. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10):3262-7.
12. Lee ES, Park BR, Kim JS, et al. Comparison of adverse event profile of intravenous iron sucrose and iron sucrose similar in postpartum and gynecologic operative patients. Curr Med Res Opin. 2013;29(2):141-7.
13. Stein J, Dignass A, Chow KU. Clinical case reports raise doubts about the therapeutic equivalence of an iron sucrose similar preparation compared with iron sucrose originator. Curr Med Res Opin. 2012;28(2):241-3.
14. Toblli JE, Cao G, Oliveri L, et al. Comparison of oxidative stress and inflammation induced by different intravenous iron sucrose similar preparations in a rat model. Inflamm Allergy Drug Targets. 2012;11(1):66-78.
15. Toblli JE, Cao G, Giani J, et al. Different effects of European iron sucrose similar preparations and originator iron sucrose on nitrosative stress, apoptosis, oxidative stress, biochemical and inflammatory markers in rats. NDT Plus 2011;4 [abstract]:SuO028.
16. Wysowski DK, Swartz L, Borders-Hemphill BV, et al. Use of parenteral iron products and serious anaphylactic-type reactions. Am J Hematol. 2010;85(9):650-4.
|
Author for correspondence: Professor Stefan Mühlebach, PhD, Vifor Pharma Ltd, CH-8152 Glattbrugg, Switzerland |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/the-authorization-of-non-biological-complex-drugs-nbcds-follow-on-versions-specific-regulatory-and-interchangeability-rules-ahead.html
Author byline as per print journal: Pieter Dylst, PharmD; Professor Steven Simoens, MSc, PhD; Professor Arnold G Vulto, PharmD, PhD
|
Introduction: A reference pricing system is a system that establishes a common reimbursement level or reference price for a group of interchangeable medicines, i.e. the reference group. This article provides an overview of the different characteristics of the different reference pricing systems in Europe. Additionally, the impact of reference pricing on price competition, generic medicine use, pharmaceutical expenditure and health outcome will be discussed. |
Submitted: 16 May 2012; Revised: 8 August 2012; Accepted: 15 August 2012; Published online first: 17 August 2012
A reference pricing system is a system that establishes a reimbursement level or reference price for a group of interchangeable medicines. If a medicine is priced above the reference price, the patient pays the difference between the price of the medicine and the reference price, in addition to any other co-payments, e.g. prescription fee, percentage co-payment [1].
Unlike its name suggests, a reference pricing system is not a pricing system, but in fact a reimbursement system. A reference pricing system sets a common reimbursement level, i.e. reference price, for a group of medicines, thereby generating savings for the third-party payer. Manufacturers are in principle free to set prices, although medicines priced above the reference price incur an additional patient co-payment and generic medicines in some countries, e.g. Belgium, need to be priced below the reference price in order to be reimbursed.
Reference pricing can help governments to contain public pharmaceutical expenditure as it controls the reimbursement level of medicines. A reference pricing system may also promote generic medicine use because originator medicines priced above the level of the reference price are likely to lose market share as a result of the additional patient co-payment.
Many European countries have already installed a reference pricing system, see Table 1. Sweden had adopted a reference pricing system in 1993 but abandoned this in 2002 [2]. In Norway, reference pricing applied from 1993 until the end of 2000. In 2003, the Norwegian government installed a system called ‘index pricing’ to a set of off-patent medicines, which has many resemblances with a reference pricing system [3, 4]. Reference pricing is in many European countries combined with other policies such as prescribing by international non-proprietary name or generics substitution, as this combination of policies seems to positively influence each other [5].
This article describes the characteristics of the different reference pricing systems in Europe. Also, the impact of reference pricing on price competition, generic medicine use, pharmaceutical expenditure and health outcome will be discussed. To this effect, a literature review and survey were carried out. The survey was used to document the current status of reference pricing systems in Europe. Survey data were collected from member associations of the European Generic medicines Association in the context of their 2011 survey of European drug retail markets [6].
Table 2 gives an overview of the different methods used by European countries to set reference prices. A country may employ one or a combination of method(s) to establish reference prices. The reference price is generally calculated as a function of market prices of medicines. The medicines which are taken into account for the calculation of these reference prices can differ between countries, see Table 2.
The different methods of setting reference prices need to be considered in the context of price competition and the volume of generic medicine use. Some European countries set the reference price at the average price level of (generic) medicines in the reference group. If accompanied by incentives to stimulate demand for generic medicines, generic medicine companies have an incentive to compete, thereby driving down (reference) prices of medicines.
Once the generic medicines market reaches a minimum level of development—for instance, a generic drug market share by volume of 40%—the reference price can be further reduced and set at the level of the lowest priced (generic) medicine. For instance, the high generic medicines market share in combination with reference prices based on the lowest priced medicines in Poland support price competition between companies and maximize savings from generic medicines use. However, other countries such as Italy and Spain have set the reference price based on the lowest priced medicine in the absence of strong incentives to stimulate demand for generic medicines, thereby undermining the development of the generic medicines market [9].
Conversely, in countries with developing generic medicines markets, setting the reference price at a higher level to encourage generic medicines market entry can be introduced as a temporary measure to boost the generic medicines market until it reaches a more mature level of development [9]. For instance, when the reference pricing system was first introduced in Portugal in 2003, the reference price was established at the level of the most expensive generic medicine. This approach increased the number of generics competitors and competition in the generic medicines market [10].
This recommendation reflects the approach used in Germany, where the reference price is calculated as a function of medicine prices and the number of generics competitors. Higher reference prices are awarded in reference groups with fewer generics competitors, thereby stimulating market entry of generic medicines companies. Conversely, reference prices are reduced and price competition is stimulated in established markets, but not to the extent that it becomes economically unviable for generic medicines companies to remain on the market [11].
In Belgium, the reference price is set at 69% (imposed by the government) of the price of the originator medicine on the day that the patent expires. This method has the benefit of guaranteeing savings to health insurance funds, but has in general not generated price reductions of generic medicines below the reference price [12].
In a reference pricing system, equivalent medicines are put together in a reference group as defined by:
The term ‘generic reference pricing’ refers to a level 1 reference group, whereas ‘therapeutic reference pricing’ relates to level 2 and 3 reference groups [13].
The methods for establishing reference groups in European countries are outlined in Table 3. Countries can implement a combination of these methods to group medicines in reference groups.
 [6]
[6]
The majority of countries group medicines by active substance, see Table 2. Such a method may lead to ‘re-allocation of demand’ away from off-patent medicines included in the reference pricing system towards patented medicines with a similar therapeutic indication that do not fall under the reference pricing system [9].
Re-allocation of demand occurred in Italy in the case of ranitidine: the falling market share of originator ranitidine following the advent of generic medicines was offset by increasing sales of patented medicines with the same therapeutic indication, e.g. omeprazole and its derivatives. Estimates suggest that re-allocation of demand was responsible for an increase in public pharmaceutical expenditure by 3.1% in 2003 [14].
Following the patent expiry of omeprazole in The Netherlands, a higher percentage of patients switched to another proton pump inhibitor (such as pantoprazole and esomeprazole, which are not included in the reference pricing system) than before the patent expiry of omeprazole [15].
In the Valencian region of Spain, the inclusion of fluoxetine in the reference pricing system and the market entry of generics competitors resulted in a shift away from fluoxetine to other antidepressants, e.g. escitalopram, venlafaxine, which were not included in the reference pricing system [16].
In countries that define reference groups by pharmacological class or by therapeutic class, see Table 3, the heterogeneity of medicines within the reference group increases. As a result, physicians may be incited to prescribe a specific medicine for financial reasons, i.e. avoidance of patient co-payment, rather than for clinical reasons, i.e. effectiveness, safety, drug–drug interaction profile [9].
Differences exist between European countries with respect to the consideration of dosage equivalence when establishing reference groups [13]. If there is no differentiation between dosages, e.g. 10 mg, 20 mg and 40 mg of the same active substance are included in the same reference group; 1 mg of active substance will be the cheapest in the highest dosage formulation. Thus, patients have an incentive to switch to the highest dosage of the medicine with the lowest co-payment. Alternatively, reference groups may be defined based on the defined daily dose of medicines. However, one defined daily dose of a specific medicine may not be therapeutically equivalent with one defined daily dose of another medicine in the same reference group. Defined daily doses of medicines also might change over time. World Health Organization therefore stresses that the defined daily dose methodology should not be misused for pricing and reimbursement decisions [17].
Hungary already applied a reference pricing system by active substance since 1997 and introduced a reference pricing system by therapeutic class for, amongst other medicine classes, statins in 2003. Statins were put together in one reference group based on their mechanism of action, without taking into account differences in pharmacologic profile, safety and effectiveness between individual products. As the reference price was determined based on the price per mg, the reference pricing system financially promoted the use of higher-dosed statins such as atorvastatin 40 mg and fluvastatin 80 mg. As a result, the majority of patients switched to higher-dosed statins and the anticipated reduction in the average price of prescribed statins did not materialize [18].
What is the impact of a reference pricing system on price competition? The literature suggests that the introduction of a reference pricing system reduces prices of all medicines that are included in the system [19, 20]. Obviously, price reductions tend to be larger for originator medicines than for generic medicines. Also, greater price reductions have been witnessed in markets where generic medicine competition already occurred prior to the introduction of a reference pricing system [19].
The European experience with respect to price reductions below the reference price is mixed: such price competition has occurred in some countries, e.g. Lithuania [20], but not in others, e.g. Slovenia, Spain [21]. Conversely, some medicines that were originally priced below the reference price actually increased their prices to the level of the reference price following the implementation of a reference pricing system in The Netherlands [22] and in Spain [23]. Therefore, in addition to setting reference prices as a function of the prices of existing medicines, some countries have established a fixed minimum price difference between generic and originator medicines, e.g. Finland, Portugal, Spain [6].
The number of generic medicine competitors within a reference pricing system appears to be associated with the extent of competition, although this impact occurs mainly through discounting to the distribution chain rather than through price competition. Larger discounts have been observed when there are more generic medicine competitors [20, 24].
Finally, as compared with other mechanisms to regulate prices such as price caps i.e. a system where the regulator sets a maximum price that can be charged for a medicine, a Norwegian study reports that a reference pricing system stimulates generics competition to a greater extent and leads to lower prices than price cap regulation [3].
In Germany, medicine prices dropped by 10–26% in the first years following the introduction of the reference pricing system in 1989. This price reduction was greater for originator medicines that faced more generic medicines competition. However, price reductions were counterbalanced by an increase in prices of medicines that were not included in the reference pricing system [25, 26]. In Norway, the implementation of a system called ‘index pricing’ to a set of off-patent medicines in 2003, which has many resemblances with reference pricing, reduced prices of originator medicines by 18% and prices of generic medicines by 8% [3]. In Sweden, medicines covered by the reference pricing system saw a drop in prices by 19% following the introduction of the system [27].
In general, the implementation of a reference pricing system has been accompanied by an increase in the use of medicines priced at or below the reference price [13]. However, a literature review indicated that a reference pricing system does not aid generic medicines use if:
In Germany, therapeutic reference pricing applies to the group of statins since 2005. As the manufacturer of atorvastatin claimed that atorvastatin was superior to other statins in terms of side effects and drug interaction profile, the manufacturer kept the price of atorvastatin above the reference price. As a result, the market share of atorvastatin declined from 33.3% prior to the reference pricing system to 4.8% in 2006. This volume shifted mainly towards simvastatin to which no additional patient co-payment was applicable as its price was below the reference price [28].
A literature review has indicated that reference pricing systems generate savings in the short term, but that savings are probably limited to the one-off impact of the introduction of the reference pricing system and that savings may be substantial at the level of individual medicines, but not necessarily at the level of total pharmaceutical expenditure [19]. Figure 1 shows how the introduction of a reference pricing system may impact long-term pharmaceutical expenditure [29].
 [29]
[29]
The limited scope for savings is to be expected for at least two reasons [29]. First, the coverage of reference pricing systems tends to be limited to off-patent medicines. Patented medicines are included in the reference pricing system by therapeutic class in some countries only. Second, although a reference pricing system may reduce medicine prices and may affect the substitution of new more expensive medicines for older less expensive medicines, it does not affect other drivers of increasing pharmaceutical expenditure such as medicines use by ageing populations, the introduction of expensive biotechnology medicines and orphan medicines, and the transformation from acute to chronic diseases.
A potential concern of therapeutic reference pricing is that switches in medicine use in response to the reference pricing system may adversely affect patient health outcomes and thereby increase the use (and costs) of other healthcare services. There are few studies that explore the impact of reference pricing systems on health and they suffer from methodological limitations. Nonetheless, the available evidence suggests that there is no association between reference pricing systems and health outcomes [13, 19].
A related issue is whether a reference pricing system influences access to health (care) of patient groups with a different socio-economic status. For instance, are patients with a lower socio-economic status more likely to buy costly originator medicines (incurring an additional patient co-payment) in a reference pricing system, thereby placing a financial burden on patient groups with a lower ability to pay? Very few studies have investigated this issue, but a recent Belgian study showed that patients with a lower socio-economic status tended to buy the cheapest (generic) medicines within a reference group and incurred lower medication costs than patients with a higher socio-economic status, thereby laying to rest potential socio-economic equity concerns related to reference pricing systems [8].
Reference pricing is used by many European countries as one element of governments’ strategies to contain public healthcare expenditure. It puts pressure on pharmaceutical companies to compete with the reference priced product but also reduces competition beyond this reference price. It also makes patients sensitive of drug prices, as an increased use of drugs priced at or below the reference price level was observed. The introduction of reference pricing generates a once-only setback of expenditures but does not affect the overall growth rate of health expenditure in the long term. No association between reference pricing and health outcome has been observed.
Over the last decades, European healthcare budgets have been increasing. Therefore, governments have implemented reference pricing, amongst other measures, in order to contain the pharmaceutical budget. In the reference pricing system, a common reimbursement level is set for a group of comparable and interchangeable medicines. The difference between this reimbursement level and the actual price of the medicine is paid by the patient, a so-called co-payment. This provides patients with a financial incentive to opt for the least expensive medicine. Reference pricing seems to reduce prices of medicines, increases the use of cheaper medicines, i.e. without co-payment, generates savings for healthcare budgets in the short term and does not seem to have a negative influence on the health of patients.
Competing interests: Professor Steven Simoens holds the EGA Chair ‘European policy towards generic medicines’. The authors have no conflicts of interest that are directly relevant to the content of this manuscript. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Provenance and peer review: Commissioned; externally peer reviewed.
The paper is abstracted based on the presentation at the 24th Annual EuroMeeting, Drug Information Association, 28 March 2012, Copenhagen, Denmark.
Professor Steven Simoens, MSc, PhD, Research Centre for Pharmaceutical Care and Pharmacoeconomics, Katholieke Universiteit Leuven, Onderwijs en Navorsing 2, PO Box 521, 49 Herestraat, BE-3000 Leuven, Belgium
Professor Arnold G Vulto, PharmD, PhD, Deputy Head Hospital Pharmacy, Professor of Hospital Pharmacy and Practical Therapeutics, Erasmus University Medical Center, PO Box 2040, NL-3000 CA Rotterdam, The Netherlands
References
1. Folino-Gallo P, Muscolo L, Vogler S, Morak S. PHIS Glossary: Glossary for pharmaceutical policies/systems developed in the Pharmaceutical Health Information Systems (PHIS) Project. PHIS/AIFA/GÖG; July 2009 [update 2011 Apr].
2. Vogler S, Habl C, Leopold C, Rosian-Schikuta I, de Joncheere K, Thomsen TL. PPRI Report. Vienna, Austria: Commissioned by European Commission, Directorate-General Health and Consumer Protection and Austrian Federal Ministry of Health, Family and Youth; 2008.
3. Brekke K, Grasdal A, Holmås TH. Regulation and pricing of pharmaceuticals: Reference pricing of price cap regulation? Eur Econ Rev. 2009;53:170-85.
4. Håkonsen H, Horn AM, Toverud E-L. Price control as a strategy for pharmaceutical cost containment – What has been achieved in Norway in the period 1994–2004? Health Policy. 2009;90(2):277-85.
5. Vogler S. The impact of pharmaceutical pricing and reimbursement policies on generics uptake: implementation of policy options on generics in 29 European countries—an overview. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(2):93-100. doi:10.5639/gabij.2012.0102.020
6. European Generic medicines Association. 2011 Market Review. Brussels, Belgium: European Generic medicines Association; 2011.
7. Vogler S, Habl C, Bogut M, Voncina L. Comparing pharmaceutical pricing and reimbursement policies in Croatia to the European Union Member States. Croat Med J. 2011;52:183-97.
8. Vrijens F, Van de Voorde C, Farfan-Portet M-I, le Polain M, Lohest O. The reference price system and socio-economic differences in the use of cheap medicines. Brussels, Belgium: KCE; 2010. Report No.: 126A.
9. Simoens S, De Coster S. Sustaining generic medicines markets in Europe. Journal of Generic Medicines. 2006;3(4):257-68.
10. Portela C. Reference pricing system and competition: case study from Portugal. Croat Med J. 2009;50(5):429-39.
11. Stargardt T, Schreyögg J, Busse R. [Pharmaceutical reference pricing in Germany: definition of therapeutic groups, price setting through regression procedure and effect]. Gesundheitswesen. 2005;67(7):468-77.
12. Simoens S, De Bruyn K, Bogaert M, Laekeman G. Pharmaceutical policy regarding generic drugs in Belgium. Pharmacoeconomics. 2005;23(8):755-66.
13. Dylst P, Vulto A, Simoens S. The impact of reference-pricing systems in Europe: a literature review and case studies. Expert Rev Pharmacoecon Outcomes Res. 2011;11(6):729-37.
14. Garattini L, Ghislandi S. Off-patent drugs in Italy. A short-sighted view? Eur J Health Econ. 2006;7:79-83.
15. Klok RM, Boersma C, Oosterhuis I, Visser ST, De Jong-Van den Berg LTW, Postma MJ. Switch patterns before and after patent expiry of omeprazole: a case study in The Netherlands. Aliment Pharmacol Ther. 2006;23(11):1595-600.
16. Ubeda A, Cardo E, Sellés N, Broseta R, Trillo JL, Fernández-Llimós F. Antidepressant utilization in primary care in a Spanish region. Impact of generic and reference-based pricing policy (2000–2004). Soc Psychiatry Psychiatr Epidemiol. 2007;42(3):181-8.
17. World Health Organization Collaborating Centre for Drug Statistics and Methodology. Use of ATC/DDD. Oslo, Norway: WHOCC; 2012.
18. Kaló Z, Muszbek N, Bodrogi J, Bidló J. Does therapeutic reference pricing always result in cost-containment? The Hungarian evidence. Health Policy. 2007;80(3):402-12.
19. Galizzi MM, Ghislandi S, Miraldo M. Effects of reference pricing in pharmaceutical markets: a review. Pharmacoeconomics. 2011;29(1):17-33.
20. Garuolienè K, Godman B, Gulbinovic J, Wettermark B, Haycox A. European countries with small populations can obtain low prices for drugs: Lithuania as a case history. Expert Rev Pharmacoecon Outcomes Res. 2011;11(3):343-9.
21. Puig-Junoy J. Impact of European pharmaceutical price regulation on generic price competition: a review. Pharmacoeconomics. 2010;28(8):649-63.
22. Danzon P, Ketcham J. Reference pricing of pharmaceuticals for Medicare: evidence from Germany, the Netherlands and New Zealand. Cambridge, MA: National Bureau of Economic Research; 2004.
23. Puig-Junoy J. The impact of generic reference pricing interventions in the statin market. Health Policy. 2007;84(1):14-29.
24. Puig-Junoy J. Do higher-priced generic medicines enjoy a competitive advantage under reference pricing? Appl Health Econ Health Policy. 2012; in press.
25. Kanavos P, Costa-Font J, Seeley E. Competition in off-patent drug markets: Issues, regulation and evidence. Economic Policy. 2008;23(55):499-544.
26. Pavcnik N. Do pharmaceutical prices respond to potential patient out-of-pockets expenses? Rand J Econ. 2002;33(3):469-87.
27. Andersson K, Petzold M, Sonesson C, Lönnroth K, Carlsten A. Do policy changes in the pharmaceutical reimbursement schedule affect drug expenditure? Interrupted time series analysis of cost, volume and cost per volume trends in Sweden 1986–2002. Health Policy. 2006;79(2):231-43.
28. Stargardt T. The impact of reference pricing on switching behaviour and healthcare utilization: the case of statins in Germany. Eur J Health Econ. 2010;11:267-77.
29. Dylst P. The impact of reference-pricing systems in Europe. 24th Annual EuroMeeting, DIA, 2012 March 28, Copenhagen, Denmark.
|
Author for correspondence: Pieter Dylst, PharmD, Research Centre for Pharmaceutical Care and Pharmacoeconomics, Katholieke Universiteit Leuven, Onderwijs en Navorsing 2, PO Box 521, 49 Herestraat, BE-3000 Leuven, Belgium |
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
Reference price systems: stakeholder dialogue and involvement
Source URL: https://gabi-journal.net/reference-pricing-systems-in-europe-characteristics-and-consequences.html
Copyright ©2025 GaBI Journal unless otherwise noted.



 [
[ [
[