Biosimilars naming, label transparency and authority of choice – survey findings among European physicians
|
Introduction: A survey of the views of European physicians on familiarity of biosimilar medicines has demonstrated the need for distinguishable non-proprietary names to be given to all biologicals.
Methods: The Alliance for Safe Biologic Medicines recruited 470 prescribers with clinical experience of biologicals in France, Germany, Italy, Spain and the UK to answer questions relating to their experience with these medicines in a 15-minute web-based survey which was carried out in the last quarter of 2013.
Results: Of the physicians surveyed, 53% mistakenly felt that an identical non-proprietary name implies identical structure; 61% said that identical non-proprietary names imply that the medicines are approved for the same indications, which they may not be, and 24% said they recorded only the non-proprietary name of the biological product in the patient record.
Conclusion: The responses of the European physicians demonstrate the need for distinguishable non-proprietary names to be given for all biologicals. Biosimilars, in contrast to generic drugs, have different structures, may have a different therapeutic profile, and may not be approved for all the indications for which the reference product has been approved.
|
Submitted: 11 March 2014; Revised: 15 May 2014; Accepted: 19 May 2014; Published online first: 2 June 2014
Introduction
With the growing number of biosimilar medicines on the European market [1], the Alliance for Safe Biologic Medicines (ASBM) has completed a survey of European physicians to:
- examine attitudes of European physicians on biosimilar naming and substitution
- assess physician knowledge, sources of information and need for further education on biosimilars and
- provide data to put policy developments at EU and national level into perspective and inform policy recommendations.
Responses from 470 prescribers located in France, Germany, Italy, Spain and the UK were collected and analysed. Respondents were all specialists who prescribe biologicals, including nephrologists, rheumatologists, dermatologists, neurologists, endocrinologists and oncologists. The perspective of European physicians reflects hands-on clinical experience with biologicals in a therapeutic setting and highlights the point that non-proprietary names matter to patient safety.
The findings point to some confusion among physicians in Europe in the area of biological and biosimilar medicines, which indicates the need of further education [2] with proper information such as the Consensus Information Paper 2013 published by the European Commission [3]. Physicians were not in agreement on where the authority lies over selecting the most suitable biological medicine for a patient – with the physician and patient or with the pharmacist.
The absence of a Europe-wide agreement on how biological and biosimilar medicines are recorded was also identified. This will need to be rectified in order to achieve effective pharmacovigilance, limiting possible adverse events in the future [4].
Methods
By the last quarter of 2013, a total of 4,324 survey invitations were sent to prescribers in France, Germany, Italy, Spain and the UK. Participants were selected from a large global market research panel of prescribers; 1,002 responded, giving a total response rate of 23.1%. 62 of the 1,002 screened out. 470 prescribers (20% from each of the five European countries) completed the survey. Oncologists were paid the US equivalent of $32.00 to complete the survey. All other participants were paid the US equivalent of US$25.00. All surveys were presented in the local language (English, French, German, Italian and Spanish). Prescribers answered questions in a 15-minute web-based survey.
Prescribers included nephrologists (18%), rheumatologists (17%), dermatologists (17%), neurologists (16%), endocrinologists (16%) and oncologists (16%). They were based in hospitals (58%); academic medical centres (24%); private, family practices (8%); community settings (8%); multi-specialty clinics (2%); or other settings (1%).
Most physicians (46%) had 11–20 years’ experience, while 18% had 6–10 years, 28% had 21–30 years, 7% had more than 30 years and only 1% had 5 years’ or less experience. Nearly three quarters (70%) conducted more than 50 patient appointments a week, while a third (29%) conducted 20–50 appointments, and 1% conducted fewer than 20. Of these, the vast majority (92%) prescribed biological medicines. Three quarters of physicians (76%) said they knew that their patients were treated with biological medicines by other healthcare providers; while 12% knew that their patients were not treated with biological medicines elsewhere and the remaining 12% were not sure.
Only 19% of the surveyed physicians said they always used the European Public Assessment Report (EPAR) to learn about the details of a medicine for prescribing and monitoring, while 43% used it occasionally and a similar proportion (38%) never used it at all. Over 80% of physicians used the summary of product characteristics (SmPC) and the label to learn about the medicine either all of the time (43%) or some of the time (43%). Transparent information in the SmPC and the label are relevant for appropriate physician information. Other sources of information used are shown in Table 1.
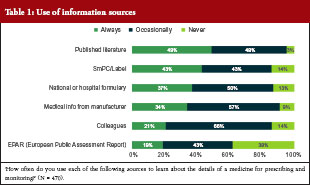
Results
Prescriber’s knowledge of biosimilars
The prescribers’ overall knowledge of biologicals and biosimilars was ascertained on the basis of questions related to their understanding of these medicines coupled with information on where they had gained this understanding (at meetings, in journals or from biological or biosimilar companies, etc).
Most physicians (46%) responded that they had only a basic understanding of biological medicines, while 43% said they had a complete understanding. Only 1% of all physicians surveyed had never heard of biological medicines, while 11% were not able to define them. These results varied by country, a relatively high proportion of physicians surveyed from Spain (62%) were ‘very familiar’ with biological medicines, while this figure was only 30% in France. Results from the remaining countries stood at around 40%: Germany (39%), Italy (42%) and the UK (40%).
Over half (54%) of those surveyed reported that they are ‘familiar’, but with only a basic understanding, with biosimilar medicines, while 20% were unable to define biosimilars and 4% had never heard of them. Only 22% of physicians surveyed were ‘very familiar’ with a complete understanding of biosimilar medicines. As before, this varied by country; a higher proportion of physicians in both Germany (59%) and the UK (59%) were ‘familiar with a basic understanding’ with biosimilar medicines compared with physicians in France (44%).
The findings highlight an urgent need for further education of physicians and others related to the prescribing of these medicines. Further dialogue and collaboration between physicians, authorities and the healthcare biotech industry continues to be a priority. With this in mind, it is important to ascertain where physicians currently find out information about biosimilars.
The question of how physicians had become familiar with biosimilars was answered by only 357 of the 470 recruits (76%). Most of these gained familiarity through attending conferences and seminars (47%), while 35% learnt through self-study, 11% through studies sponsored by biosimilar companies and the remaining 6% split equally between studies sponsored by innovator companies, clinical trial participation and other routes. Prescribers in Spain were relatively more likely to have learnt about biosimilars through biosimilar company-sponsored study (21%) and relatively less likely to have learnt through self-study (20%). Self-study was more likely among physicians in France (37%), Germany (44%), Italy (31%) and the UK (45%), where the importance of scientific publishing is apparent. On this note, although only 20% of physicians in Spain learnt about biosimilars through self-study, 38% said they would prefer to learn through scientific publications. The preference for learning through scientific publications was shared by physicians in Germany (37%), Italy (44%) and Spain (39%).
Biosimilar approval awareness
Asked whether they were aware that a biosimilar might be approved for several or all indications of the innovator product on the basis of clinical trials in only one of these indications, over a third (37%) of all 470 physicians in the study believed that all indications have been clinically tested. The finding highlights a worrying lack of understanding in this area, making the case for further education and improved, more informative, and transparent labelling. There was a higher level of awareness of indication extrapolation among physicians in Italy, where 78% responded that biosimilars could indeed be approved for several or all indications of the innovator product on the basis of clinical trials in only one indication. 63% of prescribers in France responded that biosimilars could be approved for several or all indications, alongside 52% of physicians in Germany, 65% in Spain, and 55% in the UK.
Recording biologicals
Accurate recording is the linchpin of effective pharmacovigilance. In this section of the survey, physicians were asked how biological medicines were prescribed and recorded, and how adverse events were reported.
The vast majority (95%) of all physicians surveyed would identify any prescribed medicine, including biologicals, in the patient record, although this was slightly less likely in Germany (87%) and Spain (92%). If a patient was receiving a biological medicine prescribed by another healthcare provider, this was not identified in the patient record in 11% of cases. Again, this varied by country: a higher proportion of physicians from the UK (12%) and Germany (33%) do not record this information.
The question of how biological medicines were identified for prescription or in a patient record illuminated a worrying lack of Europe-wide convention. Based on answers from 417 of the physicians questioned, there was a three-way split in the way that biological medicines for prescription or recording in a patient record were identified between: (i) brand name and non-proprietary name (32%); (ii) brand name (30%); and (iii) non-proprietary/generic name (24%). A sizeable proportion (14%) answered that identification varies by medicine. The brand name was used most widely in France (53%) and Germany (40%), while brand name and non-proprietary name were more likely to be used in Italy (42%). The non-proprietary name/generic name was most likely to be used in the UK (37%).
Reporting adverse events
Alongside questions on how biological medicines were identified, physicians were asked how medicines were identified when reporting an adverse event (AE). Medicines were identified by both brand name and non-proprietary name by 54% of the 470 physicians questioned, by brand name by 29% of physicians, and by non-proprietary/generic name by 17% of physicians. Products were most likely to be recorded by brand name by physicians in France (58%) and Germany (36%).
Batch number inclusion when reporting an AE varied widely: from always (40%); to sometimes (33%); to never (27%). Batch number was always included by 57% of physicians in Italy and 45% of physician in Germany, but never included by 38% of physicians in France and 43% of physicians in Spain.
Physicians who did not routinely include batch numbers when reporting AEs (answering ‘Sometimes’ or ‘Never‘ to this question) were questioned further, highlighting areas that will need to be addressed before this data, essential to successful pharmacovigilance, can be included routinely, see Table 2. Of 281 physicians (60% of all those questioned) who did not routinely include batch number, a relatively high proportion of physicians from Germany (62%) said they did not have the number available when they reported the AE. Nearly a third of physicians in France (29%) and a quarter of physicians in the UK (24%) did not know where to find the information.
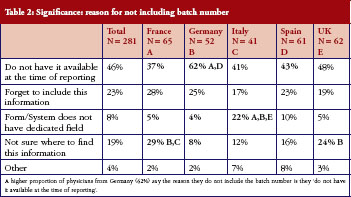
Non-proprietary name implications
The fact that biosimilars, in contrast to generic drugs, can have different structures and therapeutic profiles, and can be approved for less than all indications of the reference product, may be lost if two distinct medicines have the same non-proprietary scientific name. Clearly, it is potentially unsafe to assume these products are identical and approved for the same indications.
Answers to the question ‘If two medicines have the same non-proprietary scientific name, does this suggest to you or imply that the medicines are structurally identical’ highlight considerable confusion among physicians. Over half (53%) of physicians questioned held the incorrect assumption that these products are structurally identical. A surprising 15% had no opinion either way, which could suggest that the respondents did not understand the question. Among those more likely to believe that these products are structurally identical were physicians from France (59%), Germany (68%) and the UK (59%).
In turn, this leads to confusion over whether different medicines with the same non-proprietary name can be safely switched (between treatments or during a treatment course) or substituted. Asked whether two medicines with the same non-proprietary scientific name could be given safely to a patient with the same result, 40% said no, 47% said yes, and the remaining 13% had no opinion. There is no clear pattern here, prescribers in France (57%) were slightly more likely to believe their products could be safely switched than those in Italy (40%) and Spain (38%), but there were sizeable proportions of ‘yes’ and ‘no’ answers in every country surveyed, see Table 3.
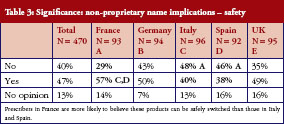
The complexity is further illustrated by answers to the question that ‘if two medicines have the same non-proprietary scientific name, does this suggest to you or imply that a patient could be safely switched between the products during a course of treatment and expect the same result as treatment with only one of the products’. Thirty-nine per cent of physicians believed patients could be safely and effectively switched during a course of treatment, 45% believed they could not, and 16% had no opinion. Nearly half (49%) of prescribers in France believed switching was safe, while only 34% of physicians in Spain believed this.
Similarly, the physicians questioned were unclear whether two medicines with the same non-proprietary scientific name are approved for the same indications: 61% said they were; 31% said they were not; and 9% had no opinion. In other words – and these findings did not vary between countries – two thirds of prescribers do not have an understanding of the complexity and sensitivities surrounding biosimilars.
Clear naming and labelling is paramount. If a patient has an adverse reaction, which can occur months after receiving a biological medicine, the medicine needs to have been properly identified from the start. A clear naming system is essential in order to make identification possible, thereby enhancing access to these life-changing therapies, while also protecting patient safety.
Pharmacy substitution
The question of authority over selecting the most suitable biological medicine for a patient was posed in this survey, and physicians’ responses revealed a range of opinions across Europe. Of 470 physicians questioned, 24% thought it was critically important to have the sole authority to decide, together with their patients, the most suitable biological medicine for their disease. 48% thought it was ‘very important’, 23% thought it was ‘somewhat important’, 4% thought it was ‘slightly important’ and 1% thought it was ‘not important’.
These responses varied by country, with the highest proportion of physicians in Italy (34%) and Spain (33%) considering sole authority ‘critical’. Sole authority was considered only ‘very important’ or ‘somewhat important’ by physicians in Germany (38% and 37% respectively).
The importance of ‘dispense as written’ (DAW) or ‘do not substitute’ showed a similar pattern. Overall, 27% considered it critical, 47% thought it was ‘very important’, 20% thought it was ‘somewhat important’, 5% thought it was ‘slightly important’ and 1% thought it was ‘not important’. In Spain, 41% of physicians considered DAW critical, while only 13% of physicians in Germany considered it critical.
Most physicians among the 470 questioned felt it was important that pharmacists provide notification that their patient had received a biological other than the one prescribed if the patient was receiving chronic (repeated) treatment: 30% considered notification critical, 47% considered it very important, 16% somewhat important, 6% slightly important and 1% considered it not important.
Similarly, 62% of all physicians considered it not acceptable if the pharmacist decided which biological (innovator or biosimilar) to dispense, 35% considered it acceptable and 3% considered it totally acceptable. Unilateral decision making at the pharmacy was not considered acceptable by most prescribers, particularly in Italy where 77% of physicians considered it unacceptable.
Asked to define ‘bio-naïve’, 76% of physicians in the survey believed that this meant ‘a patient who has never received any biological treatment of this class.’ This was equally accepted across the countries studied, although slightly less likely in Germany, where only 66% of physicians agreed with this definition, see Table 4.

Conclusion
Biological medicines have had a profound effect in many medical fields, from oncology to neurology and across a host of other debilitating diseases, but the complexity of the molecule and its manufacturing process result in significantly higher cost than that of the small-molecule medicine. As the patents on biologicals begin to expire, it is hoped that the arrival of biosimilars – drugs that are similar, but not identical, to these innovator biologicals – will reduce the financial burden on healthcare systems [5]. But to benefit from these medicines it is crucial that prescribing physicians understand what these medicines are, and what these medicines are not.
The responses of European physicians recorded in this study reflect serious gaps in what is known about biological drugs in general and biosimilars in particular. There are several misconceptions regarding biologicals, and considerable education is needed in the area of differences between generic products and biosimilar products.
Unlike generic drugs, biosimilars are not identical to the innovator biological on which they are based; they have different structures, may have a different therapeutic profile, and may not be approved for all the indications for which the reference product was approved.
There is a clear need for distinguishable non-proprietary names to be given to all biological medicines to ensure intended prescribing as well as to support product identification when reporting and tracing adverse events [6–8].
Increasing numbers of biological medicines, both originator and biosimilar, are being approved around the world. According to the findings in this study, most physicians use the SmPC and the label to learn about a medicine, illustrating how important it is that clear information, and informative labelling, is provided for every biological and biosimilar.
With the co-existence of different biosimilars from different manufacturers, an effective pharmacovigilance system is urgently needed in order to allow accurate adverse events reporting. How these products are named will clearly play a central role in facilitating pharmacovigilance worldwide, supporting the safe use of these medicines [4, 8].
Alongside the prescribing physician, the pharmacist also plays a key role in effective pharmacovigilance. Switching between brand name pharmaceutical drugs and their generics presents little or no threat to patient safety since they should be identical, but switching between a biological drug and its biosimilar, or between different biosimilars, is not the same. Across Europe, this study revealed a range of opinions among prescribing physicians as to where the authority should lie when deciding on the most appropriate biological or biosimilar, with most insisting that physicians and their patients – not pharmacists – should have sole authority when making these decisions. It is important for most physicians to retain the authority to use ‘do not substitute’ to ensure the patient receives the correct medicine.
In addition to a clear need for further education for physicians who prescribe these medicines, distinguishable non-proprietary names are important to practising physicians, and we hope this system will be used by the World Health Organization in crafting a global standard that will improve patient safety worldwide [1].
The findings of this study echo, and reinforce, concerns already raised by the medical community. Effective Europe-wide, and by extrapolation worldwide, pharmacovigilance is urgently needed in order to benefit fully from the considerable advances offered by biological drugs and biosimilars, while limiting future adverse events.
|
Key points of the 2014 European prescribers’ survey
- 53% of physicians surveyed felt that an identical non-proprietary name implies identical structure – which is not the case for biosimilar medicines. 15% of physicians had no idea whether this was the case or not, highlighting further confusion or lack of understanding
- 61% of physicians said that identical non-proprietary names imply that the medicines are approved for the same indications, 9% had no idea if this was the case or not.
- 17% of physicians record only the non-proprietary name/generic name of the biological product when recording an adverse event. 27% of physicians never include the batch number when reporting an adverse event and 33% do so only sometimes.
- 86% of physicians used the SmPC/label to learn about the medicine either all of the time (43%) or some of the time (43%). Only 19% of physicians said they always use the European Public Assessment Report (EPAR) to learn about the details of a medicine for prescribing and monitoring, with 43% using it occasionally and 38% never using it at all.
- 70% of physicians consider it either critical or very important that they together with their patients have the sole authority to decide on the most suitable biological medicine.
|
Acknowledgement
The authors wish to thank the medical writing and editorial support by Dr Bea Perks, GaBI Journal Editor.
Funding sources
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners who contribute to ASBM’s activities. Visit www.SafeBiologics.org for more information.
Disclosure of financial and competing interests: Dr Richard O Dolinar, Chairman of the Alliance for Safe Biologic Medicines (ASBM), and Mr Michael S Reilly, Executive Director; are employed by ASBM.
This paper is funded by ASBM and represents the policies of the organization.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Richard O Dolinar, MD, Chairman
Michael S Reilly, Executive Director
Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
References
1. Rovira J, et al. Biosimilars in the European market. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1):30-5. doi: 10.5639/gabij.2013.0201.012
2. The future of biological therapy: a pathway forward for biosimilars Generics and Biosimilars Initiative Journal, 2013;2(1):36-40. doi: 10.5639/gabij.2013.0201.014
3. GaBI Online – Generics and Biosimilars Initiative. Biosimilars: what physicians need to know [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2014 May 15]. Available from: www.gabionline.net/Reports/Biosimilars-what-physicians-need-to-know
4. Clayton J. Tighter EU rules on pharmacovigilance for biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(2):56-7. doi:10.5639/gabij.2012.0102.015
5. Haustein, et al. Saving money in the European healthcare systems with biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4). 120-6. doi:10.5639/gabij.2012.0103-4.036
6. Thorpe R, Wadhwa M. Terminology for biosimilars–a confusing minefield, Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):132-4. doi:10.5639/gabij.2012.0103-4.023
7. Alexander E. The biosimilar name debate: what’s at stake for public health, Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):10-2. doi:10.5639/gabij.2014.0301.005
8. Dolinar R. WHO leadership in public safety on biosimilars to be commendedGenerics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):167. doi:10.5639/gabij.2013.0204.047
| Author for correspondence: Michael S Reilly, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2014 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/biosimilars-naming-label-transparency-and-authority-of-choice-survey-findings-among-european-physicians.html
WHO leadership in public safety on biosimilars to be commended
|
Abstract:
As a practicing endocrinologist and Chairman of the Alliance for Safe Biologic Medicines, I am writing to commend the World Health Organization for its attention to and upcoming action on the important issue of non-proprietary names for biotech medicines.
|
Submitted: 20 September 2013; Revised: 21 September 2013; Accepted: 21 September 2013; Published online first: 27 September 2013
More than 50 years ago, World Health Organization (WHO) established the International Nonproprietary Name (INN) expert group to assign non-proprietary names to medicinal substances so that each could be recognized globally by a unique name. Although initially intended to facilitate prescribing, non-proprietary names have come to play an essential role in tracking and tracing, and attributing adverse events to the right product – an important aspect of ensuring medicine safety once a product is on the market. Distinguishable names are particularly important for biologicals.
WHO has shown thoughtful leadership in facilitating the further development of effective biological identification via a unique and universally available designated name for each pharmaceutical substance. This action is very timely in light of the growing number of biosimilars and non-biocomparables arriving on the market. The urgency of this action is apparent with the first monoclonal antibody biosimilar approved in Europe by the European Commission in September 2013 bearing the identical INN as the innovator product.
The practicality of WHO’s action is evidenced in Australia, where the Therapeutic Goods Administration (TGA) has already acted on WHO’s vision for biological and biosimilar naming conventions. Indeed, TGA’s action demonstrates both the importance and the functionality of distinguishable INNs. Under the new regulation, TGA requires the non-proprietary name to include a biosimilar identifier, consisting of the prefix ‘sim(a)’, and a three-letter code issued by WHO’s INN Programme Committee according to its draft policy. The TGA regulation explains, ‘As small differences between biosimilars can give rise to differences in clinical behaviour, in particular in immunogenic effects, certain additional nomenclature provisions are necessary to ensure that it is possible to distinguish between biosimilars and clearly identify the reference product.’
Since the active substance of biologicals is made by or derived from living organisms and most biologicals are very large molecules, biologicals can be sensitive to very minor changes in the manufacturing process. Unlike chemically based drugs, small differences in the manufacturing process and handling, etc.; can significantly affect the nature of the finished biological and the way it functions in the body. Therefore, no two biologicals made from different cell lines and/or using different manufacturing processes are ever identical to the reference product they aim to replicate and, hence, can have a significant impact in a patient’s body.
Knowing specifically which product, produced by which manufacturer a patient received is essential to keeping medicines and patients safe. If a patient develops an unwanted immune response over time, the doctor and regulators will know exactly which product to evaluate. For that reason, product naming is one of the key elements of biological product safety. In fact, the Alliance for Safe Biologic Medicines (ASBM) conducted a survey of more than 350 specialists who prescribe or treat patients with biologicals. We found that non-proprietary product names are the primary means by which physicians identify products.
The vast majority of physicians (99 per cent) refer to biological medicines by INN and not by the ‘national drug code’ number assigned to each product in the United States – for both recording in charts and for reporting adverse events. If distinct non-proprietary names are not given to products, we cannot appropriately track and trace adverse events nationally and even more so globally and may waste valuable treatment time trying to identify the root cause.
ASBM has worked for nearly three years to support health agencies including the US Food and Drug Administration and WHO, in the mission to safely bring biosimilars to patients. We believe all biological policies must be guided by the recognition that biologicals are scientifically different than traditional chemical drugs and the laws governing their approval and regulation must reflect that scientific reality. ASBM will soon be attending the WHO’s upcoming Consultation on International Nonproprietary Names (INN) for Pharmaceutical Substances meeting in October 2013, to further build on the emerging consensus that distinct names are the only way to keep patients safe.
Funding sources
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals – from patients to physicians, innovative medical biotechnology companies and others who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners, who contribute to ASBM’s activities, with the primary funding provided by the Steering Committee, funds the ASBM’s efforts. Visit www.SafeBiologics.org for more information.
Disclosure of financial and competing interests: Dr Richard O Dolinar, Chairman of the Alliance for Safe Biologic Medicines (ASBM), is employed by ASBM.
This paper is funded by ASBM and represents the policies of the organization.
Provenance and peer review: Not commissioned; internally peer reviewed.
|
Author: Richard O Dolinar, MD, Chairman, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/who-leadership-in-public-safety-on-biosimilars-to-be-commended.html
The future of biological therapy: a pathway forward for biosimilars
Author byline as per print journal: Richard O Dolinar, MD; Michael S Reilly
|
Abstract
Biologicals are advanced prescription drugs to treat cancer, rheumatoid arthritis HIV/AIDS, multiple sclerosis and other debilitating diseases. In November 2010, the US Food and Drug Administration (FDA) began consultation with patient groups, physicians and industry on how to approve the first copies of these drugs, known as follow-on biologics in the US or biosimilars. As FDA moves forward in implementing this pathway, it is essential that all stakeholders work together to ensure patient safety remains the top priority to achieve safe and efficacious patient care.
|
Submitted: 15 October 2012; Revised: 4 March 2013; Accepted: 29 March 2013; Published online first: 8 April 2013
Introduction
Biologicals are large, complex molecule drugs that treat serious illnesses. They are created using proprietary and unique processes involving living cells, and range from sugars and proteins to tissues and nucleic acids [1]. Examples of biologicals can be found in vaccines, certain blood treatments, and gene therapy [2]. Biological medicines are unique because, unlike more established drugs, they are not chemically synthesized. According to the US Food and Drug Administration (FDA), biologicals ‘often represent the cutting-edge of biomedical research and, in time, may offer the most effective means to treat a variety of medical illnesses and conditions that presently have no other treatments available’ [2]. In fact, it is estimated that by 2016, biological medicines will comprise 48 per cent of the top 100 best-selling drugs [3].
Continued improvements in manufacturing efficiency, increasing access to these drugs and reducing costs are as important as new discoveries. Most importantly, with FDA outlining its latest guidance at the end of 2012 for the future of biological medicines, patient safety must be the primary focus of all stakeholders.
Biologicals meet biosimilars
Although the breakthroughs in biologicals have been groundbreaking, the future of health care is not just about new molecules. It is also about the new and exciting frontier of biosimilars, also known as ‘follow-on biologics’ in the US; biosimilars are products that enter the marketplace after the patent of an ‘innovator drug’ expires, in this case a certain biological.
Biosimilars have similar properties to existing biological products (hence the term ‘biosimilar’), therefore, patients can rest assure of the efficacy of these biological therapies. As the Generics and Biosimilars Initiative (GaBI) points out, FDA states that there are ‘no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product’ [4].
Importantly, however, due to the complexity of biologicals, a ‘follow-on biologic’ can only be made similar, rather than identical. Most of the discussion between biologicals and biosimilars contained herein this paper, moreover, is about switching, rather than the initial selection of these medicines.
It is always important to frame a discussion of biosimilars by remembering that there is no such thing as a generic biological. ‘Big’ molecules (biologicals) are more than just a larger version of ‘small’ chemically synthesized ones. Biologicals are created from living organisms and are not as simple to replicate as traditional drugs, such as aspirin and antihistamines. A biosimilar is not a generic drug, and that is more than just a detail.
Because of the complexity of manufacturing a biosimilar, it is imperative that FDA implements a system – a pathway – which provides complete transparency to patients and their physicians. An example of this patient-centered pathway is encouraging US States to require pharmacists to secure a patient’s consent prior to substituting an interchangeable biological product for the one prescribed, or at a minimum to ensure that treating physicians are notified when a substitution has occurred. Undoubtedly, it is in the patient’s best interest to have the pharmacist and physician work together.
The prescribing physician should be notified of the switch in a timely manner. Additionally, a strong track and trace system is needed to detect side effects, both to determine the rate at which rare events occur and to identify those not known at the time of market entry. Keeping patients and physicians in the healthcare decision-making process ameliorates potential harm to the patient in the event a product is found to have adverse side effects after it enters the market.
Today’s biologicals defined
Novel biologicals act by novel targets, technology platforms and/or mechanisms of action compared with previously approved biologicals.
Next-generation biologicals (‘biobetters’) have the same target or mechanism of action as a previously approved biological but include structural changes, bi-functional targeting (with or without a biosimilar core) or an improved formulation that may result in an expected improvement in clinical profile.
Because biological therapeutics are highly complex and large protein molecules, they require a wide variety of analytical methods to ensure consistent quality. Indeed, given the complexity of biologicals and their manufacture (in which living cells produce the core molecule and make post-translational modifications), the innovator product has inherent lot-to-lot variability. In light of this inherent variability and the diverse and complex analytical methodologies required to characterize the molecules, it is not realistic to exactly replicate an innovator molecule. Therefore, the concept of a biosimilar is to make a molecule that is as similar to the innovator as possible. Accordingly, follow-on molecules in the biological space are termed biosimilars, rather than biogenerics.
Biosimilars are structurally highly similar versions of marketed biological medicines. They will have been evaluated and approved by a regulatory authority on the basis of analytical and clinical comparison to the already marketed product.
Across the globe, with the goal of making biologicals more accessible, regulators either have adopted or are considering legislation or regulations to establish pathways for the approval of biosimilars. Because biosimilars are never exact copies of the innovator medicine, establishing appropriate standards for biosimilarity remains an important area for scientific, legislative and regulatory debate [5].
It is important for legislators and regulators to promote safety and science when crafting new policy pertaining to these life-saving medicines. That is why the Alliance for Safe Biologic Medicines (ASBM) works to promote four key principles as US regulators implement an approval pathway for biosimilars:
- prioritizing patient safety
- leveraging what we know
- promoting pharmacovigilance
- keeping physicians engaged.
Prioritizing patient safety
Biotechnology companies that seek FDA approval for an interchangeable biosimilar need to demonstrate, through de novo clinical trials, that switches from the biosimilar to the reference innovator product (and vice versa) have no negative effect on safety and/or effectiveness, particularly as a result of immunogenicity. It is also important to realize that for one product, the risk of immunogenicity may differ depending on the therapeutic indication [6].
In fact, when it comes to biosimilars, the most important issues facing global drug regulators are the scientific and technical factors related to a determination of biosimilarity. Minor differences in manufacturing processes, such as different host cells, cell culture and purification methods, can have a clinically significant impact on a biological’s safety and effectiveness. It is not just whether or not a biosimilar ‘works’, it is about whether or not it works as well or in the same way once it is administered to a patient. Just as no two patients are exactly alike, neither are biosimilars identical to the innovator product.
The age of ‘personalized medicine’ is predicated on the ‘four rights’—being able to administer the right medicine to the right patient in the right dose at the right time [7].
One issue at the forefront of the biosimilars debate is that of ‘switching’—changing a patient’s therapy from the innovator product to a biosimilar. Since biosimilars are not identical to their innovator products, many nations around the world have clearly stated that automatic substitution is inappropriate. Because biosimilars generally cost less than innovator drugs, there is significant economic pressure to switch patients to lower cost biosimilars. But when cost-centric concerns are allowed to trump the practice of patient-centric medicine, there is a potential negative impact on both patient safety and clinical effectiveness. The decision to switch a patient must be a clinical one made by the treating physician rather than a legislator, regulator, or insurance provider.
The United States is the only nation that requires its regulatory authority to evaluate whether a biosimilar is similar enough to the already marketed ‘reference’ product that it can be substituted without physician intervention, permission or authorization. Under US law, FDA would designate such products ‘interchangeable’. In addition to scientific challenges associated with making such a designation, there are substantial administrative hurdles that, if not effectively addressed, could jeopardize patient safety.
Leveraging what we know
Regardless of whether you live in the EU, US or Canada, people might see the prescription drug approval process as mysterious and arcane and have never heard of biosimilars, follow-on biologics or subsequent entry biologics (SEBs). In the US, the regulatory pathway for approval of biosimilars is still in its very early stages, lagging behind the regulatory bodies in the EU and Canada. There are many contentious issues surrounding the ‘biosimilar pathway’. Some are economic and focus on legal concerns such as patents and data exclusivity, but these are not within the purview of FDA. What rightly concerns the US drug regulator is safety and efficacy.
The Patient Protection and Affordable Care Act empowered FDA to develop a biosimilar pathway. Having the authority is one thing, getting it done in a timely, suitable, and scientifically robust manner is something else altogether. While FDA retains many of the best regulatory scientists in the world, its human and capital resources are severely limited. Rather than creating a biosimilar pathway from scratch, the US needs not reinvent the wheel. The European Medicines Agency (EMA) began to establish the first formal regulatory pathway for biosimilars in 2003, and that process can serve as a baseline model upon which the US can build.
In February 2012, FDA issued three highly anticipated documents providing guidance on its biosimilar approval pathway. This guidance set forth FDA’s current thoughts on ‘key scientific and regulatory factors involved in submitting an application for biosimilar products’ [8]. The documents help clarify application processes mandated under the new abbreviated regulatory pathway that was included in the Patient Protection and Affordable Care Act.
According to the agency, ‘Healthcare professionals and consumers can be assured that the FDA will require licensed biosimilar and interchangeable biological products to meet the Agency’s exacting standards of safety and efficacy’ [9].
Any biosimilar approval should be based upon the overall assessment of biosimilarity to the innovator through robust analytical, non-clinical and clinical data. For some biological molecules, certain studies may not be necessary. In the event that a biosimilar manufacturer is seeking approval for multiple indications, extrapolation of data should be scientifically justified.
By pioneering in this regulatory area for the last eight years, the EU has gathered much data, which can, at a minimum, help inform policymakers. US policymakers should take advantage of this opportunity to learn from the experiences of their counterparts, both the positive and negative.
Promoting pharmacovigilance
Pharmacovigilance is the surveillance of a drug’s performance, particularly of adverse reactions, after it has been released for marketing. As biosimilars may be approved based on less data, pharmacovigilance plays an even more important role. Before biosimilars are thoroughly introduced into the US marketplace, a robust traceability system – including distinctive labels, distinguishable names, product tracking codes, and a way to report adverse events – must be in place to facilitate accurate surveillance.
Through enhanced, 21st century pharmacovigilance, the US can do a better job analyzing data and drawing conclusions relative to many unknown differences between innovator products and biosimilars. The forces of globalization have enabled many other nations to work together to promote pharmacovigilance. As of 2010, 134 nations are party to the World Health Organization (WHO) pharmacovigilance programme [10]. This WHO pharmacovigilance initiative ‘aims … to enhance patient care and patient safety in relation to the use of medicines; and to support public health programmes by providing reliable, balanced information for the effective assessment of the risk–benefit profile of medicines’ [10]. It is only by embracing this type of understanding of how biosimilars impact patient health in this ‘real world’ that patients will be able to use them in the most appropriate way to achieve the best outcomes for their personalized needs. As former Eli Lilly & Co CEO, Mr Sidney Taurel stated at the Cleveland Clinic, ‘The time is ripe for FDA, the healthcare industry, and the medical community to collaborate on a reform of our nation’s pharmacovigilance system. Such reform will allow us to speed up the recognition of safety signals and understand the true efficacy of new medicines more quickly’ [11].
Automatic substitution complicates the pharmacovigilance that is needed for all biological and biosimilar medicines to ensure safety for patients. Pharmacovigilance is facilitated when physicians make the decision to substitute a biosimilar product. Automatic substitution makes biosimilar pharmacovigilance significantly more difficult. There should be greater clarity and transparency in state substitution laws, and attention must be paid to any prospective policies that infringe physicians’ freedom to prescribe the medicines that they deem most appropriate for a particular patient. The need to improve and expand pharmacovigilance systems must also be applied to any changes made in originator biologicals since they may also cause unintended outcomes. To quote Dr Woodcock, ‘Our improving the use of marketed drugs, to a great extent, is going to involve partnering with the growing patient safety movement. The vast majority of harm from approved drugs comes from misuse, inappropriate use … failure to use, abuse and medical mix-ups’ [8].
When it comes to biosimilars—primum non nocere (first do no harm).
Complex and extreme challenges often require creative solutions. An excellent example of this is the development of biobetters. Biobetters are to biosimilars what Apple’s iPod Touch is to its iPod Shuffle. Where a biosimilar will be a mere structural imitation, a biobetter will possess some molecular or chemical modification that constitutes an improvement over the originator drug. As such, it must be evaluated and approved through the traditional pathway.
Such enhancements may range from a longer half-life, allowing for less frequent dosing, to more potency or less toxicity. That is innovation driven by the new reality of biosimilar competition. And it should not be surprising since, among other things, competition drives innovation.
Keeping physicians engaged
Sir William Osler, widely regarded as the father of modern medicine, wrote ‘If you listen carefully to the patient they will tell you the diagnosis.’ Arriving at a diagnosis and appropriate treatment plan has always represented intimate collaboration between patients and physicians.
But today, physicians are increasingly seeing the decisions that they and their patients reach about specific treatment plans second guessed by distant ‘third parties’, working for government agencies or insurance providers, who may not be aware of the unique individual circumstances of a particular patient.
Physicians must practice both the art and science of medicine, but the issue of cost threatens to interpose itself between physicians and patients. While cost is certainly a crucial topic when it comes to health care, it cannot trump patient safety.
And nowhere is this debate more immediate, urgent, or profound than when it comes to the issue of therapeutic switching, that is, switching patients between products not considered interchangeable. Physicians carefully collaborate with their patients to choose the most appropriate treatment, considering the patient’s disease state, ability to tolerate side effects, and stage of life. The crucial differences between biosimilars and small molecule generics are that biosimilars are difficult and expensive to get approved, complicated and challenging to manufacture, and generally have a short shelf life. The savings are expected to track the experience in Europe to date and reflect a 10–30% discount from the originator product rather than 90%, as with chemical drugs.
Reformers need to recognize that policies giving healthcare administrators control over treatment regimes are hazardous to patient health, and actually inflate overall costs.
In Europe, regulatory authorities understand that the successful adoption of biosimilars requires physician buy-in and for that reason EMA advises that the physician should be in charge of the decision to switch between the reference product and biosimilar, or vice versa [12]. Physician confidence translates into increased utilization and in fact, biosimilar medicines have gained a foothold in some European countries as a result of a strategy to persuade physicians to start new patients on a biosimilar rather than switch existing patients [13].
There is a need of enhancing the ‘biological experience’ for physicians/prescribers. Adequate physician education, sufficient clinical data and appropriate reimbursement services for physicians will result in greater use of biologicals and biosimilars [14].
The repercussions of choosing short-term savings over long-term results, of cost-based choices over patient-centric care, of ‘fail first’ policies over the right treatment for the right patient at the right time—are pernicious to both the public purse and the public health.
Conclusion
The explosive growth of biological medicines, and the emergence of biosimilars as revolutionary tools to fight the most difficult of diseases, is cause for great celebration in the fight to provide advanced health care to patients worldwide. By abiding by the general principles of prioritizing patient safety, leveraging the information we know, promoting pharmacovigilance and keeping physicians engaged, a golden age for innovative and affordable health care is within reach.
As indicated in an FDA hearing on its biosimilar guidelines in Washington DC, USA, in May 2012, the debate over the future of the biosimilar approval pathway is far from over [15]. Issues in need of further consideration include, among others, defining proteins, stricter methodology in labelling and naming biosimilars.
It is important to note that, although beyond the scope of this paper – which focused on switching between biological products – biosimilars can sometimes be used as the initial and only therapy. Also, the manufacture of innovator products can involve changes that require tracking of outcomes.
In fact, biosimilar medicines have gained a foothold in some European countries as a result of a strategy to persuade physicians to start new patients on a biosimilar rather than switch existing patients [14].
The future of biological medicines will be bright if patients, physicians, biotechnology companies, and other stakeholders work together to ensure patient safety is the foremost priority of the biosimilar policy discussion. Then, the future of healthcare debate can move beyond partisan discussion over healthcare access and cost, to a discussion of the diseases that biological medicines can successfully conquer next.
For patients
Biologicals and biosimilars are not covered under the 1984 Hatch-Waxman Act, which created an abbreviated approval process for generic versions of conventional drugs. On 23 March 2010, however, the Patient Protection and Affordable Care Act was signed in to law and included a pathway for the approval of biosimilars (also referred to as the Biologics Price Competition and Innovation Act). This abbreviated approval pathway for biosimilars gives FDA the authority to define the implementation process. The law also gives FDA the authority to further define the detail regarding scientific standards and the extent of analytical, preclinical and clinical data necessary for the approval biosimilars to ensure patient safety and the effectiveness of the biosimilar.
As FDA moves forward with the process it is important for patients, physicians, pharmacists, and all other stakeholders to be engaged in these efforts to ensure that the top priority for US regulators is patient safety. Our hope is that the principles outlined in this paper will help them to achieve this worthy goal for patients.
Acknowledgement
Dr Brett Johnson with the International Cancer Advocacy Network (ICAN) also contributed to the production of this paper.
Funding sources
The Alliance for Safe Biologic Medicines (ASBM) is an organization composed of diverse healthcare groups and individuals—from patients to physicians, innovative medical biotechnology companies and others who are working together to ensure patient safety is at the forefront of the biosimilars policy discussion. The activities of ASBM are funded by its member partners, who contribute to ASBM’s activities, with the primary funding provided by the Steering Committee, funds the ASBM’s efforts. ASBM’s Steering Committee is comprised of Alliance for Patient Access, American Academy of Dermatology, American Association of People with Disabilities, Association of Clinical Research Organizations, Colon Cancer Alliance, Genentech, Global Healthy Living Foundation, Health HIV and Kidney Cancer Association. It is the mission of the ASBM to serve as an authoritative resource center of information for the public, medical community, FDA and other government staff during the implementation of the biosimilars approval pathway and beyond.
Disclosure of financial and competing interests: Dr Richard O Dolinar, Chairman of the Alliance for Safe Biologic Medicines (ASBM) and Mr Michael S Reilly, Executive Director of the Alliance for Safe Biologic Medicines; are the primary authors of the paper. Both authors are employed by ASBM.
This paper was funded by the Alliance for Safe Biologic Medicines and represents the policies of the organization.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Richard O Dolinar, MD, Chairman
Michael S Reilly, Executive Director
Alliance for Safe Biologic Medicines, PO Box 3691 Arlington, VA 22203, USA
References
1. U.S. Department of Health and Human Services (HHS) [homepage on the Internet]. What is a biologic? [cited 2013 Mar 4]. Available from: http://answers.hhs.gov/questions/3262
2. U.S. Food and Drug Administration (FDA) [homepage on the Internet]. Center for Biologics Evaluation and Research. What is a biological product? 2013 [cited 2013 Mar 4]. Available from: http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194516.htm
3. World Preview 2016. [cited 2013 Mar 4]. Available from: http://www.azcentral.com/news/election/azelections/azfactcheck/fact-story.php?id=199.
4. GaBI Online – Generics and Biosimilars Initiative. FDA definitions of generics and biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Mar 4]. Available from: www.gabionline.net/Biosimilars/General/FDA-definitions-of-generics-and-biosimilars
5. Chow SC, Ju C. Assessing biosimilarity and interchangeability of biosimilar products under the Biologics Price Competition and Innovation Act. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1). Epub ahead of print. doi: 10.5639/gabij.2013.0201.004
6. Declerck PJ. Biologicals and biosimilars: a review of the science and its implications. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(1):13-6. doi:10.5639/gabij.2012.0101.005
7. Peter P. A problem diagnosed. Center for Medicine in the Public Interest. [cited 2013 Mar 4]. Available from: http://www.cmpi.org/in-the-news/in-the-news/a-problem-diagnosed
8. Eisenberg C, Kupchyk A. Nixon Peabody [homepage on the Internet]. FDA issues biosimilar draft guidance. FDA Alert. 2012 Feb 28 [cited 2013 Mar 4]. Available from: http://www.nixonpeabody.com/FDA_issues_biosimilar
9. U.S. Food and Drug Administration [homepage on the Internet]. Biosimilars. [cited 2013 Mar 4]. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm
10. World Health Organization [homepage on the Internet]. Pharmacovigilance. [cited 2013 Mar 4]. Available from: http://www.who.int/medicines/areas/quality_safety/safety_efficacy/pharmvigi/en
11. Eli Lilly & Co. Lilly’s CEO calls for reform of nation’s drug safety system. [homepage on the Internet]. 2007 Oct 2 [cited 2013 Mar 4]. Available from: http://newsroom.lilly.com/ReleaseDetail.cfm?ReleaseID=266917.
12. European Medicines Agency [homepage on the Internet]. Questions and answers on biosimilar medicines (similar biological medicinal products). 2012 Sep 27 [cited 2013 Mar 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
13. Declerck PJ, Simoens S. A European perspective on the market accessibility of biosimilars. Biosimilars. 2012;2:33-40.
14. Class JN, Langis L. A patient-centered paradigm for the biosimilars market. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(1):17-21. doi:10.5639/gabij.2012.0101.006
15. GaBI Online – Generics and Biosimilars Initiative. FDA’s public hearing on biosimilars draft guidances [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Mar 4]. Available from: www.gabionline.net/Biosimilars/General/FDA-s-public-hearing-on-biosimilars-draft-guidances
|
Author for correspondence: Michael S Reilly, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691 Arlington, VA 22203, USA
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related articles
Promoting access to biosimilars: a public−private partnership model for biosimilar development in underserved populations
A patient-centred paradigm for the biosimilars market
Source URL: https://gabi-journal.net/the-future-of-biological-therapy-a-pathway-forward-for-biosimilars.html
Copyright ©2025 GaBI Journal unless otherwise noted.
Generics and Biosimilars Initiative (GaBI)
Tel: +32 474989572 | Fax: +32 14 583 048