Author byline as per print journal: Professor John-Joseph Borg, PhD; Yolanda Elias Gramajo, MD; Professor Andrea Laslop, MD; Robin Thorpe, PhD, FRCPath; Jian Wang, MD, PhD
|
Introduction: Biosimilars have the potential to improve access to medicines for many across the globe. However, work is required to ensure adequate regulation, pharmacovigilance and education about biosimilars. Colombia implemented biosimilars regulation in 2017 and a 3rd Colombian Educational Workshop was organized by GaBI and the Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) in 2019 to follow up on progress and provide a forum for further discussion. |
Submitted: 9 April 2020; Revised: 8 June 2020; Accepted: 15 June 2020;; Published online first: 29 June 2020
Biosimilars or similar biotherapeutic products (SBPs), are a relatively new class of biotherapeutic agent. With appropriate regulatory and pharmacovigilance procedures in place, these products have the potential to improve access to medicines worldwide. These drug products are being developed and are entering markets across the globe. With sufficient trust in and uptake of these products, healthcare costs could be reduced dramatically.
Europe has been at the forefront of developing biosimilars regulation in recent years and now an internationally agreed system of regulatory approval is needed. Similarly, pharmacovigilance of biosimilars needs to be done on a global level. There are also questions regarding the interchangeability of biosimilars and a global understanding of this should be laid out. To achieve consensus on these points, open discussion across national borders and between different stakeholder groups is key.
To facilitate discussion concerning quality assessment of biologicals/biosimilars in Colombia, in 2016 and 2017, an educational workshop [1] and a follow-up meeting [2], were organised by the Generics and Biosimilars Initiative (GaBI) in collaboration with the National Food and Drug Surveillance Institute of Colombia (Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA). Together, these meetings provided a forum to exchange knowledge on best practice and enabled the status of biosimilars quality assessment in Colombia to be viewed with clarity [3].
Following the implementation of biosimilar regulation in Colombia in 2017, a third meeting on the regulatory assessment of biosimilars in Colombia was held in 2019. This interactive workshop provided a forum for regulators, clinicians, pharmacists, academics and healthcare professionals from Colombia who are involved in biological/biosimilar medicines evaluation, to share knowledge and exchange information with expert speakers from Canada, Europe and the US. They engaged in active discussions that concerned the current status of biosimilars regulation in Colombia; standards and the stepwise approach of quality and immunogenicity assessment of biologicals/biosimilars; structure–function relationship of biological medicines; switching; pharmacological (pharmacokinetic (PK)/pharmacodynamic (PD)) and clinical studies of biosimilars; extrapolation of indications; post-marketing pharmacovigilance practices of biologicals/biosimilars to ensure patient safety; and future educational needs and how to improve the uptake of biosimilars.
Overall, the meeting aimed to bring experts together to clarify biosimilars regulatory concepts and concerns, in order to improve biosimilar regulation and increase their uptake in Colombia.
On 30 April 2019, GaBI held the 3rd Colombian Educational Workshop on Regulatory Assessment of Biosimilars. As with previous workshops [1–3], the aim was to review and explain the current status of biosimilars regulation in Colombia. The format was similar to that followed in prior scientific meetings and educational workshops as reported in GaBI Journal [4–6]. There were a number of expert speaker presentations followed by Q&A and an in-depth panel discussion. The presentations are downloadable from the GaBI website [7]. All participants were encouraged to engage in active discussion about the presentations given by expert speakers and to discuss selected case studies.
Presentations were in English or Spanish with simultaneous translations into Spanish or English, respectively.
The workshop began with a welcoming speech from Dr Lucia Ayala, Director of Medical Devices and Technologies at INVIMA. This included a brief introduction to the Colombian regulatory approach to biosimilars. The Austrian Medicines and Medical Devices Agency’s (AGES MEA) Head of Scientific Office, Professor Andrea Laslop, then provided a second welcome address. She celebrated the longstanding collaboration between INVIMA and GaBI and briefly outlined the workshop objectives (as summarized in the Introduction).
Biosimilars in Colombia: a year after regulation implementation
Mr Aurelio Enrique Mejía Mejía, Director of Medicines and Health Technologies at Colombia’s Ministerio de Salud y Protección Social, and Johanna Andrea García Cortes, Professional Specialist at INVIMA, gave presentations which discussed the implementation of Colombia’s Biotechnology Decree 1782/2014. Their presentations entitled ‘Colombia’s biological/biosimilar regulation: a year after its implementation’ and &lsqup;Biosimilar regulation in Colombia: one year later’, respectively, can be found on GaBI website [7].
In a brief summary, the two presentations outlined details of the 2014 Biotechnology Decree 1782. This laid out the requirements and procedures of INVIMA to evaluate the quality, safety and efficiency of biological medications and aimed to enable the appropriate approval and commercialization of such products in Colombia in the future. The two presentations discussed the decree and its impact following implementation in August 2017. In the first year of implementation, it has led to more therapeutic options being available to patients and an increase in competition in Colombia’s biological drug market. At present, 23 biosimilars are under review in Colombia, of which trastuzumab has undergone all regulatory processes and achieved marketing authorization. In addition, there are regulations in place to ensure that biosimilars enter the market meeting global standards of safety and efficacy. The next step is to improve the education of healthcare professionals to help increase the uptake of biosimilar products across the country.
Quality assessment of biologicals/biosimilars – most relevant quality attributes: case study on monoclonal antibody
Professor John-Joseph Borg, Malta Medicines Agency’s Director of Post-Licensing, discussed the quality assessment of biologicals/biosimilars in the European Union (EU) [8].
Professor Borg outlined the basic concept of a biosimilar:
He noted that, due to the complexity of biosimilars, it is not appropriate to follow the same approach as with simple generic drugs when addressing bringing biosimilars to market. As such, comparability exercises are required for biosimilars to be authorized in the EU.
In the EU, a complete and appropriate quality dossier is required for a biological product to be approved (Module 3). For a biosimilar, this must also include a comprehensive comparability exercise (quality, non-clinical, clinical), see figure 1. He added that establishing comparability is a stepwise approach and non-clinical and clinical data are required before product developers should move on to address the next step and address any residual uncertainty. Licensing decisions are then made based on the entire data package which includes quality, non-clinical and clinical parameters that demonstrate similarity to a reference product. All licensing is done on a case-by-case basis.
So, to build an application dossier for a biosimilar, a product developer will need a full Module 3, plus the comparability exercises (there should be multiple data points with all data being recorded over a period of time), followed by non-clinical studies and then clinical studies. If a product meets the requirements and is approved, conditions can then be attached to approval in the post-authorization phase.
Professor Borg further discussed the comparability evaluation and noted that all aspects mentioned above must be taken into account. This evaluation includes details of the nature and level of knowledge of the product, i.e. its complexity, structure-activity relationship, relationship between therapeutic and endogenous proteins and the mode of action. He also discussed aspects of immunogenicity and highlighted that this is an issue for all biological products. If a reference causes immunogenicity, so will a biosimilar. The risk factors for immunogenicity are both product and patient related.
Another key issue when considering biosimilars is that any variation in process can cause a different product to be produced, see figure 2. These variations can occur at any point during processing. Therefore, a regulator needs to know about every change made during production/processing and any effects that changes may, or may not, have. The sourcing and testing of the reference product must also be done over a period of time to account for any differences in the reference.
Overall for biologicals, the quality is highly dependent on the manufacturing process which is very complex. For a product to be accepted, it must meet quality criteria and be validated with safety and efficacy data. Professor Borg noted that there are additional critical quality aspects for a biosimilar and he went through a non-exhaustive list of these, see Table 1.
The EU Committee for Medicinal Products for Human Use (CHMP) guideline for comparability was discussed, see figure 3. This included notes on the origin of the reference product and how, when comparing a reference to a biosimilar, the same reference product (from the same country of origin) should be used over time.
Professor Borg gave an overview of the chemistry, manufacturing and controls (CMC) documents for monoclonal antibodies (mAbs) derived from a monoclonal cell line. This included a description of the development and production of mAbs and their characterization (physicochemical characterization; immunochemical properties; biological activity; information on purity, impurity and contaminants; and on quantity). The talk was finalized with an outline of Remsima biosimilar comparability where he included a comprehensive list of the studies comparing the physicochemical and biological activity of the Remsima biosimilar and the Remicade originator.
Pharmacological studies (PK/PD) to assess biosimilar medicinal products
Dr Jian Wang, Division Chief of the Clinical Evaluation Division, Biologics and Genetic Therapies Directorate, Health Canada, discussed biosimilar pharmacological (PK/PD) studies.
Biosimilars are similar to their brand-name reference products and not the same due to being made from different cell lines and by different manufacturing processes. To support regulatory approval, guidance/policy requests that comparisons between the biosimilar and its reference are made. These include, see Figure 4:
Clinical studies are needed to ensure that residual uncertainty from quality assessment does not cause clinically meaningful differences in efficacy, safety and/or immunogenicity in the sensitive population. Dr Wang stated that the main goal of clinical PK/PD studies is to rule out unacceptable PK/PD differences that could indicate the presence of significant structural and functional differences between a biosimilar and a reference product.
Regarding comparative PK studies, Dr Wang noted that these comparative clinical PK studies should be conducted in a setting that is reflective of the clinical situation and/or is sensitive to detect differences between the biosimilar and the reference. They should also be planned based on the characteristics of the reference, including its mode of action, safety profile and PK properties. In general, PK studies can be done on healthy volunteers. However, these individuals may not always reflect the PK parameters of patients. As such, comparative PK studies can also be conducted in the patient population.
The same principles of study design, statistical methods and criteria of acceptance for small molecules are used as a general guidance for biologicals. When the route of administration is intravenous (IV) which does not include an absorption phase, some additional parameters from elimination phase will be included for equivalence assessment.
In some aspects of study design, the single dose cross-over design is the most sensitive PK study design, for short half-life biologicals. However, this design can be limited by the properties of the biological such as having a long half-life or following the formation of antidrug antibody (ADA). In specific cases, parallel and/or multi-dose designs can be considered.
Many biologicals, including mAbs, cytokines, and growth factors, display target mediated drug disposition (TMDD), see figure 5. For such biologicals, three considerations must be made when conducting comparative PK/PD studies:
Critical quality attributes can influence the PK of a mAb and therefore, may have a direct impact on biosimilarity, see Table 2. In terms of immunogenicity, most biologicals induce some level of ADAs and these may have undesirable clinical effects on PK, efficacy and/or safety, including immunogenicity. It was also noted that PopPK studies are being used in demonstrating comparability for biosimilar mAbs as supportive studies.
Finally, on comparative PD studies, Dr Wang stated that PD studies are desirable, if feasible, and can help reduce residual uncertainty. PD parameters should be investigated as part of the phase III trial. He then discussed PD surrogates and aspects of PD sensitivity (including clinical, assay and dosing). Also, it is necessary to measure the baseline endogenous levels of a biological in blood plasma if the biological product is produced endogenously. In conclusion, for products with a reliable PD marker, a high quality and sensitive PD study (usually combined with PK) may be better than an efficacy study in terms of detecting differences in efficacy between a biosimilar and its reference product.
Head-to-head clinical studies and biosimilarity studies to assess biosimilar medicinal products
Dr Yolanda Elias Gramajo, Senior Clinical Evaluator, Clinical Trials Division, Health Canada, discussed head-to-head clinical studies and biosimilarity studies to assess biosimilar medicinal products.
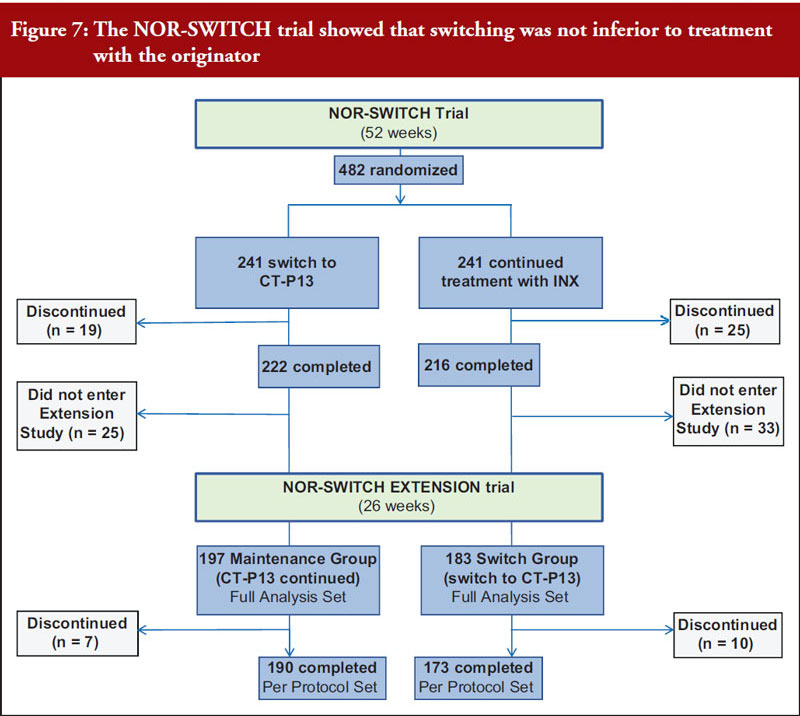
Dr Elias Gramajo began by outlining that the pathway for biosimilar development and approval is designed to demonstrate similarity to its reference product with respect to quality, safety and efficacy. This uses a stepwise approach that is often depicted as a pyramid, see figure 6. This includes analytical, non-clinical and clinical studies. She gave a brief overview of the general regulatory requirements and the requirements for biosimilar development, see figure 7.
As part of the clinical programme for biosimilar development, comparability exercises are required. The comparative exercise objective is to demonstrate similarity in PK/PD, efficacy, safety and immunogenicity between the biosimilar and the reference product. In general, clinical trials are required (phase III for at least one indication) but in some cases PK/PD studies can suffice.
When it comes to the clinical development programme for biosimilar products, it is preferred that clinical trials follow an equivalence design. Here, a trial has the primary objective of showing that the response to two or more treatments differs by an amount which is clinically unimportant. This is usually demonstrated by showing that the true treatment difference is likely to lie between a lower and upper equivalence margin of clinically acceptable differences. She outlined some key considerations for comparative clinical trials including that generally 90%–95% confidence interval (CI) equivalence margin in the PP population (per-protocol) is acceptable, randomized design, double-blind/adequately powered, power and sample size sufficient to detect difference, dose and route consistent with reference.
Sensitive clinical study populations were then discussed, and that the comparative clinical study should be conducted in a sufficiently sensitive population that is representative of the authorized indications to detect differences between the biosimilar and its reference. The endpoint should be considered to improve the detection of potential differences between the biosimilar and the reference within the sensitive population.
When it comes to safety, immunogenicity is the most important aspect. Dr Elias Gramajo presented a number of points relevant to this including ADA formation. She also outlined the immunogenicity assessment strategy which includes screening assays, confirmatory assays, neutralizing assays and PK/clinical impact assessment. In all cases a biosimilar should never be more immunogenic than its reference in terms of ADA incidence or concentration.
In conclusion, when a biosimilar is authorized it can be granted all therapeutic indications held by the reference based on the totality of evidence obtained from all comparative analyses, see figure 8.
Extrapolation of biosimilars
Professor Andrea Laslop, Chair of the workshop, delivered a presentation on the principles of extrapolation of indications in the EU. Biosimilar extrapolation is when a biosimilar can be used for any indication that the reference product is approved for, even if the biosimilar itself has not been directly studied in a comparative clinical trial for that indication.
Professor Laslop outlined some general considerations for extrapolation, which is an important feature of the biosimilar approval process but that it continues to cause contention. Many EU biosimilar guidelines have been set up to address the issue and to date, extrapolation has been implemented in all biosimilar product approvals. She highlighted the fact that the concept is not new and has been used for biosimilars and generics and also in paediatric indications and other special populations. In addition, extrapolation can occur when there have been changes to the manufacturing processes of biologicals.
Concerning changes in the manufacturing process, comparability exercises are carried out to ensure the product efficacy and safety is not altered. When such changes are made, the product will be different and thus by definition, it will be a biosimilar (typically clinical data are not required to approve manufacturing changes).
Mechanism of action (active site) is key to extrapolation, see Table 3. This is mediated by functional molecular moieties in a disease-specific manner, which can be characterized by sensitive assays. If the same mechanism of action (MoA) or the same receptors are involved for the function of the biological/biosimilar product in different diseases, then extrapolation is straightforward. However, in some cases additional non-clinical or clinical data may be required, e.g. different active site, different receptor, different safety profile.
She noted that the overarching guideline on biosimilars states: if biosimilarity has been demonstrated in one indication, extrapolation to other indications of the reference product could be acceptable with appropriate scientific justification. Likewise, the general guideline on biosimilars containing biotechnology-derived proteins as the active substance, non-clinical and clinical issues, also says that extrapolation could be acceptable, but that in case of unclear relevance of the safety and efficacy data from one indication for another one, additional data will be required. In all cases extrapolation is only considered in light of the totality of evidence.
Professor Laslop presented a number of cases where extrapolation had occurred and justified the reasons for extrapolation in each example [9, 10].
In conclusion, extrapolation is essential for the success of biosimilars and must be done on the basis of the totality of data available. It is not done automatically and requires scientific justification. This is a common process in drug development and is not exclusive to biosimilars. Extrapolation is expected if comparability of the product, with respect to a reference, has been demonstrated on all levels. In addition, post-approval information for extrapolated indications is helpful and, to date, has shown the success of extrapolation.
Immunogenicity studies for assessing biosimilar products
Dr Robin Thorpe, Co-chair of the workshop, delivered a presentation on immunogenicity studies. This was similar to that given at the GaBI MENA 2018 meeting [6].
In conclusion, it was noted that immunogenicity issues occur all along the life cycle of a product and particularly when:
And that assessment requires:
Two further presentations were given by representatives from Amgen. ‘Biologicals and biosimilars – the complexity of structure and function’ was delivered by Dr Jennifer Liu, Director of Analytical Sciences, and ‘Considerations for product specific pharmacovigilance of multisource biologicals’ was delivered by Dr Thomas Felix, R & D Policy Director.
After the presentations, there was the opportunity for discussion about the topics covered. The key discussion points are summarized below.
The regulatory pathway in Colombia
The current status of biosimilars regulation in Colombia/The regulatory pathway in Colombia
Following the two presentations given on ‘Biosimilar’s regulation in Colombia: one year on’, there were some queries that required further clarification.
Dr Arley Gómez López expressed concern about the fact that Colombia does not have certain infrastructure in place for adequate drug analysis. Mr Mejía Mejía responded that the current regulation is in place to generate incentives and allow for the development of the infrastructure that is required. He emphasized that it is important to understand that the process is gradual. Having clarity regarding the standards, which has now been achieved, will allow companies to start to develop the capabilities in order to accomplish exactly the types of studies that are needed. Overall, Colombia’s Ministry of Health intends to give clarity regarding the norms so that they can be developed from the conditions. He also agreed that it is necessary to develop public and/or private initiatives to facilitate the development of the infrastructure.
Comparisons between the Colombian and other regulatory agencies
When asked about improved access to medicines in Colombia through commercial mechanisms, Mr Mejía Mejía stated that, at present, only hepatitis C medicines are bought through centralized purchasing. By focusing on the regulation of biotechnological medicines, it is hoped that access to medicines will be improved through the availability of greater therapeutic options which promote competition and reduce costs. Now that we have regulation in place, products are going through the approval process. When products are approved by INVIMA, they can start being prescribed by doctors and be covered by the health system. Eventually, in the case where the government and the ministry allow it, they can also be subject to centralized purchase or negotiations.
According to data in the presentations given, there seemed to be a lot of biosimilar applications in Colombia compared to the EU. Dr García Cortes advised that, when it comes to expensive medicinal products, it is good that there are a lot of biosimilar applications as this will promote competition and decrease prices. INVIMA’s Ms Alejandra Mayra Gomez Leal added that many biosimilars have been approved by the European Medicines Agency (EMA) and not the US Food and Drug Administration (FDA) and that Colombia is open to applications and whether or not they have been approved in other places may only be used as a reference.
There was a concluding comment by Andrea Carolina Reyes Roja, pharmaceutical chemist working at Misión Salud, a Colombian NGO. She stated that Misión Salud has followed the regulation process for biotechnological medicines in Colombia for more than 10 years. She congratulated the government for the emphasis it has put on the utilization of the abbreviated pathway as a public policy. Other health agencies, such as EMA, FDA and the World Health Organization (WHO), have a ‘questions and answers’ document published in October of 2018 which reaffirms the possibility of reducing clinical studies when supported by the vast knowledge of the molecules in analytical studies of high complexity and limited to PK/PD studies. The clarity that the Colombian government has provided on this issue will increase access and reduce the costs of medicines.
Following the presentation given by Professor Borg, Dr García Cortes wanted to know if Europe is thinking about removing the bridging studies. She noted that in Colombia they have found cases where there are significant differences between the product in the US and the European counterpart. In such cases, it is difficult to evaluate the comparability, so should bridging studies be removed? And in these circumstances should it be the US or EU product? And if only one reference product is taken, whether it be the European or the US. In response, Professor Borg noted that in the EU the reference product needs to be sourced in the EU to comply with comparability requirements. In addition, Andrea Carolina Reyes Roja stated that for the quality comparison, Europe requests the comparison to the European reference product and does not accept head-to-head comparison to a US-derived reference. She pointed out that this is different from the clinical development programme, where under certain conditions bridging exercises can be accepted. For example, a clinical study of the US reference could be used if it is also demonstrated that both the US and the EU reference product are sufficiently similar on the quality level.
Stepwise approach – PK/PD trials, comparability studies
When to use PK and PD studies and data
Ms Alejandra Mayra Gomez Leal was interested to know more about mAb studies and PK data collection. For the majority of mAb studies as presented by Dr Wang, the primary evaluation was for the efficacy aspects, and as a secondary objective, they evaluate the PK. Dr Wang explained that normally the first PK measurements will be done after the first dose of medication, and this essentially gives a single-dose PK. However, depending on how many samples can be collected, the study must be very well designed to collect multiple samples at different time points so that a PK profile can be created. In addition, and in general, PK data are collected at multiple dosing time points, so it is possible to have single-dose and multi-dose PK data.
Associate Professor Claudia Patricia Vaca González noted in her conclusion, that Dr Wang said that when there is a good indicator of PDs it can be more desirable to have a PK/PD study than a clinical efficacy study. She wondered how generalized this statement was, considering there is a growing interest from the agencies and countries to have competition and competitive products on the market. This approach could improve the speed of introduction of these products into the market. Dr Wang responded by noting that this statement is not possible to generalize to all biologicals. He reiterated that for products with well-known, well-established PD surrogates, you can generally use comparative PK/PD studies. However, agencies may also ask for a longer-term safety study which may not be powered to demonstrate equivalence and the results of which cannot be inferred from the comparative PK/PD study.
The science and rational of clinical trials
Rejection after clinical trials
There was a query regarding the number of biosimilars that were rejected in Canada, following clinical trials. Dr Wang stated that, when it comes to biosimilars and clinical trials, it is not purely a science issue, but also a regulatory issue. A biosimilar should be as good, or as bad, as the reference. If it is better, it becomes a judgement call. How much better is it? And is that going to cause safety concerns or not? In biologicals, it is often the case that you will have better efficacy but a worse safety profile. So, a proper benefit-risk assessment must be made and within the considerations of the country’s regulations. Overall, the situation is quite complicated, it is not the case that products go through all steps prior to clinical trials, without hitch, and then get rejected on the basis of trial results.
The importance/discussion of pharmacovigilance, naming issue
Improving and maintaining pharmaco-vigilance
Associate Professor Claudia Patricia Vaca González made some comments on the naming of biologicals. She said that WHO allowed countries the freedom to establish differentiation from the common international names of products. This came after lengthy discussions about both the inconvenience of having differentiation in nomenclature which causes confusion between patients and doctors; and following concerns expressed by different countries that this name differentiation could be used to reduce competition when generic or biosimilar medicines enter the market. In addition, she noted that nomenclature is very important when it comes to pharmacovigilance and tracing of the active principle. It is important to have information related to the product batch and the ability to trace a product through its logistic chain. She stated that health systems in all countries must improve upon traceability to improve pharmacovigilance.
Dr Felix responded that the concept of active ingredient traceability is very important. This is how small molecules are traced around the world. However, active ingredient traceability and batch level traceability are not two things that necessarily go together. In most countries, it is not possible to find the manufacturer of a product with only the non-proprietary name product batch number.
Professor Laslop agreed that it is important to have separate pharmacovigilance reporting for each individual biosimilar product. In Europe, pharmacovigilance data on the non-proprietary name would not be collected alone. The European implementation pathway instead requires prescription according to brand name. That is, in Europe, biosimilars are never prescribed according to non-proprietary name but rather by specific brand name together with the batch number, and this is how a product can be traced.
Dr Felix added that Europe has not implemented a specific naming approach and some countries outside Europe see this as a greenlight to do the same. However, they do not take into account certain aspects of European legislation in place to improve traceability which include brand level prescribing, good pharmacovigilance guidance and education of healthcare professionals.
With respect to this discussion, Professor Borg noted that new regulation is coming into force in the EU with respect to the falsified medicines directive. All batch numbers will be recorded where dispensed. Ideally in the future, all will be linked centrally at EMA with the help of IT software. Dr Felix explained that the directive is a European legislation which is in place to prevent the ability of counterfeit medications entering into the European system. As such, there is very high accuracy in the traceability of biological products and all products that enter Europe. In the future, this has potential to be a powerful tool and to improve patients’ safety, particularly if data input into the system occurs at the dispensing level.
A final comment was made by a member of the audience regarding pharmacovigilance and residual uncertainties. He expressed concern that these medicines were being released to the market despite the existence of uncertainties. In response, Dr Felix stated that, for all medicines, regulatory approval pathways are built on the best available evidence at a given point in time. Even for an innovative product, there is still some residual uncertainty that might exist, and the study of products must continue after they are approved. This is the case, particularly when they are used in patient populations that were excluded in clinical trials or inpatient populations that were not studied originally. In all circumstances, the collection of aggregate information is required to show that the benefit continues to outweigh risks. This is going to be especially important in the next wave of innovative medications that are being developed for patients with rare diseases which are based on clinical trial data sets that are much smaller. These will be promising in terms of their clinical benefit but due to their nature, there will be all the more reason for post-approval traceability of these products and continued measurement of the benefit-risks related to them.
Immunogenicity
After Dr Thorpe’s presentation on immunogenicity, Professor José Orozco asked if it is possible for a biosimilar to be less immunogenic. Dr Thorpe explained that there are instances where immunogenicity, at least appears, to be lower for a biosimilar. Often this is due to assay artefact problems and the real problem is that when assays are carried out the antigen must be selected with extreme care. The most logical way of doing this is to measure the antibodies against the product that the patients have received. The patients who have received a biosimilar, the new biosimilar, get screened against the biosimilar and the ones who have received the innovator get screened against that. However, this is labour intensive. As such, it is common to use just one antigen – and all the patients get screened against one antigen. This is allowed in the EU if the antigen is the biosimilar itself, as here you increase the possibility of detecting antibodies against a biosimilar which gives you a possible, slightly higher apparent immunogenicity of the biosimilar. In other circumstances, immunogenicity can be reduced in a biosimilar. This can be due to assay effects or may be because of high purity of the biosimilar (compared to older reference product made with less advanced technology). When a biosimilar has less immunogenicity, this seems good, however, this should ideally be justified in the assessment in terms of purity or assay issues.
There was a panel discussion chaired by Professor Laslop and co-chaired by Dr Gramajo.
The Regulatory panel discussion was introduced by Professor Laslop. The panel included Arley Gómez López, Research Director from the University Research Foundation ‘de la Salud’; Juan Fernando Juez Castillo, INVIMA; and Judy Hasleidy Martínez Martínez, INVIMA. Professor Laslop opened discussions and noted that the workshop presentations had thus far brought to light the two main pillars for assessment of biosimilars: the quality comparison (which includes non-clinical data) and the comprehensive clinical comparison.
Reducing the biosimilar assessment process
Mr Castillo noted that it is evident that we still have a lot to learn when it comes to biosimilars and their assessment. There is a great deal of complexity when it comes to biosimilars and their production. Therefore, it is very important to have a very strict process for analyses in Colombia. For now, he believes that shortening any analysis could result in a fall into reductionism, which is to be avoided. However, it may be necessary to have public and/or private concessions as neither the universities nor the government alone currently have the capacity to realize these types of complex analyses adequately.
When it comes to reducing the comparability exercise and the clinical studies for biosimilars, Mr Castillo also noted that this should only be done on a case-by-case basis. It is not possible to generalize and say that a certain category of products should follow a certain abbreviated ‘third pathway’ with less information required.
To emphasize this he noted that some mAb patents are expiring and, despite extensive characterization, there are still many uncertainties about the molecules. However, this is not the case for molecules like insulins which are well understood.
Dr Martínez Martínez noted that in Colombia the concept of a third pathway for certain drug products with reduced trials has been discussed. She explained that, Colombia’s Decree 1782 aims to ensure molecules are precisely characterized, have robust pharmacovigilance and good safety information. These molecules undergo clinical trials until enough information is known to prove their safety (either in Colombia or in other countries). In some cases, smaller biological molecules, such as insulins, heparins and filgrastim, can be well characterized and do not require as many clinical trials. Such molecules could be considered in the third pathway. However, to date, not many products have followed this third pathway. Overall, INVIMA is working with comparability to try and reduce the clinical and preclinical trials needed for some products but this should never impact on product safety. At present, INVIMA has not worked on the specific criteria for the different pathways. Laboratories can present the information through the pathway they chose based on the information and justifications that they selected throughout the trial. In general, if a characterization does not seem sufficient to the regulator, then more information is requested.
Professor Laslop added that this third pathway approach is similar to those adopted in Europe, Canada and the US. Here, there is potential to waive full clinical trials for certain less complex biosimilar molecules. However, if Colombia wants to adopt a similar approach to those nations, the guidelines and definitions need to be very clear. This is particularly important in terms of molecule complexity and what is, and is not, to be included in each of the different approval pathways.
In addition, Ms García Cortes noted that she did not support the idea of having a list of molecules in a third pathway. Biologicals should be assessed on a case-by-case basis, running through the three steps. These steps investigate the complexity of the molecule, the complexity of its MoA and the complexity of its production. Changes in production method such as a change in vial, can lead to massive adverse effects in the product that are not due to the molecule itself. As such, creating a list of molecules to be included in a third pathway does not seem plausible as their effectiveness is also dependent of the complexity of their action mechanism and production method.
A member of the audience noted that a third pathway should exist to give clarity in cases where it is not necessary to get to the end of the clinical trials and safety and efficacy studies. In such cases, PK/PD studies are carried out and the medicines are widely known and have robust pharmacovigilance. However, it is imperative not to compromise safety and INVIMA performs exhaustive evaluations to ensure this.
Another audience member also noted that research ethics should be part of the regulation because it ensures that patients’ rights are observed.
The applicability of the assessment pyramid
When a biosimilar undergoes evaluation for assessment of comparability, it should generally follow the stepwise approach outlined in the assessment pyramid. However, in many cases, not all stages of the pyramid are reached and undertaken prior to product approval. As such, it was proposed that this pyramid should be changed. Professor Laslop stated that she would not be in favour of removing or altering the pyramid as it is still the default pathway for biosimilar development, biosimilar assessment and, ultimately, biosimilar approval. In Europe, the possibility for abridged clinical assessments is exactly defined in product specific guidelines.
Dr Gramajo noted that the situation is the same in Canada. All assessments are done on a case-by-case basis. Those developing biosimilars should consult with agencies at an early stage to determine what information is needed and which trials are necessary. As such, the overall pyramid is still relevant.
Clinical trials
Dr Thorpe stated that classically, to prove clinical efficacy, a classical efficacy trial should be carried out. This is the standard and remains valid. However, there can be occasions when there is a better way of establishing clinical efficacy than performing a classical clinical trial. For example, with G-CSF (filgrastim) it is possible to determine PD and PK measurements more accurately than you can measure clinical responses. Clinical efficacy trials can still be carried out, but to the best of his knowledge, when these were carried out in Europe with filgrastim, the outcome was the same as shown by PK/PD studies. However, this does not mean that clinical trials do not need to be carried out with other products. The omission of clinical trials always needs to be justified.
According to Mr Castillo, if one arrives at the PK/PD step and there is little uncertainty, then this is when there seems little sense in continuing to clinical trials. They are unlikely to provide additional information. However, if there are many uncertainties following PK/PD studies it is unethical to continue to clinical trials. He believes that too many clinical trials are requested in the EU and North America. Many of these trials are huge, requiring many patients over a prolonged period of time and sample sizes required could exceed those needed for innovator products. Mr Castillo again affirmed that such decisions should be made on a case-by-case basis. He also highlighted that in Colombia, clinics are struggling to find enough patients with which to carry out large clinical trials. This is due to how the healthcare system is constructed. However, he hopes that this is changing so that more continuous studies can be done to assess long-term efficacy and trace adverse effects.
WHO reliance assessment
Mr Castillo noted that biosimilarity has not yet been officially defined in Colombia. This is an issue when choosing the exact pathway to follow for similarity assessment. It can also lead to redundant studies being done that have already been carried out in other countries.
In response, Professor Borg highlighted that WHO has a reliance assessment that helps regulators outside the EU assess biological products. As part of this, WHO has a formal agreement with EMA that allows EMA’s CHMP assessment report to be shared so that product assessment can be targeted on areas which are relevant to the country. As such, they do not need to waste time or money on reassessment and revaluation. This aims to allow for faster access to medicines for patients and improved healthcare systems in the non-EU countries. He suggested that Colombia could take advantage of this WHO reliance assessment.
Biosimilar education of the medical and patient community/Improving education about biosimilars
Professor García Cortes noted that the implementation of Decree 1782 is a big challenge for Colombia. Medical doctor education is a key part of ensuring its success. Colombia has done a lot of work to ensure correct evaluation of biosimilars from a qualitative, safety and efficacy point of view. It is now important that doctors have clarity about and trust in the medicines approved. As such, INVIMA’s next goal is to ensure that doctors and healthcare personnel are well educated when it comes to biosimilars. They should understand what they are and should trust the information available. She believes that, through this, INVIMA can create confident prescribers and good therapeutic results.
Mr Castillo stressed the importance of having a medical community that has knowledge and trust in the medicinal products it prescribes. INVIMA is working hard to ensure products receive a comprehensive evaluation and that there are channels of communication open to prevent any issues from causing problems or harm. With these mechanisms, and any other means, INVIMA will reduce the impact on the public health of Colombians.
Professor Laslop concluded that education of the medical community and the patient community is extremely important. In Europe, it became quickly apparent that it is not always efficient to simply publish something on a website. Instead, proactively approaching all stakeholders (the pharmaceutical manufacturers, or the patients, or the medical community) to educate, explain and communicate, has had far greater impact.
Dr Néstor Álvarez Lara noted that, when it comes to the communication of information, it is important to pay close attention to pharmaceutical marketing in Colombia. Advertising must be kept in check to ensure it does not negatively affect government spending or patient access to medicines.
Regulatory panel discussion conclusion and future actions
Dr Gramajo stated that the subjects discussed are very controversial, and there are a lot of things to consider which require more communication, more information, more collaboration between different agencies. Colombia is unique in that it is going to determine its own regulations and elect medical authorities in accordance to public opinion at a national and an economic level.
Discussion groups were provided with data on two semi-fictional trastuzumab biosimilar candidates. They were provided with physicochemical characteristics, selected glycan and biological attributes, and the results of a phase I study for Candidates 1 and 2.
Following the same format as previous GaBI meetings, each discussion group was asked whether the data for the candidates qualified for biosimilarity with a reference product from a quality (CMC) perspective. If not, they were asked what steps they would recommend fixing this. Discussion groups were also asked how ‘residual uncertainty’ could be addressed in preclinical or clinical studies. They were then asked, given that Candidates 1 and 2 had both the CDR (complementarity-determining region) and Fc (fragment crystallizable) region involved in their MoA for some of the indications, whether they would recommend extrapolation to all indications.
There were six discussion groups that evaluated the case studies. Overall, they all concluded that Candidate 1 is a biosimilar, from a quality perspective. However, there are differences between the candidate and the reference and some elements of residual uncertainty which require further investigation. If results of these investigations are as expected, then extrapolation is acceptable. When considering Candidate 2, all groups concluded that this is not a biosimilar, from a quality perspective. The differences between it and the reference cannot be solved and residual uncertainty is too large to warrant further study or clinical trial. Extrapolation is not acceptable.
Group 1 was moderated by Professor Fabio Ancizar Aristizábal Gutiérrez, and co-moderated by Judy Hasleidy Martínez Martínez. Based on the data presented for Candidate 1 this group suggested that further physicochemical or in vitro tests should be carried out to remediate differences. In addition, potency studies should be carried out. To address residual uncertainty, preclinical models could be used in vitro or in a parallel animal model. Here, clinical evaluation may be required for PK/PD data. Group 1 stated that Candidate 1 did qualify for extrapolation if the results obtained from the required physicochemical studies and in vitro studies resolve the uncertainties related to the quality attributes mentioned (fucosylation – antibody-dependent cellular cytotoxicity (ADCC); glycosylation – complement-dependent cytotoxicity (CDC) given that the therapeutic indication studied was relevant. Regarding Candidate 2, there are differences in some quality attributes, a larger sample of batches must be available to reduce uncertainty. Clinical studies do not allow the differences to be resolved, so do not recommend as biosimilar.
Professor Gutiérrez noted that having open communication channels and strong teamwork are very important when assessing biosimilars. This ensures everyone analyses and understands critical variables rather than criticizes information. Analysis methodology is important to optimise time use and to reduce questions following analysis. These case studies showed that the manner in which information is exchanged between different levels of experience in technical areas is critical. In addition, another important aspect is quality. The case studies put forward two quality parameters in the form of clinical trials. If preliminary quality results are obtained that raise doubt about the efficacy in the clinical trial, these can be used to confirm biosimilarity of a candidate product. In addition, Group 1 felt that the extrapolation of uses should only be allowed with better information regarding MoAs for each indication to know if they are complimentary. This is what was shown in the clinical trial provided.
Group 2 was moderated by Associate Professor Claudia Patricia Vaca González and co-moderated by Joseph Sebastián Cepeda Santamaría. The group confirmed similar outcomes to Group 1. They discussed that it is very important to compare the primary structures, yet in these cases there was no data with respect to that. It was also noted that there was no comparability data or immunogenicity data. For Candidate 2, they observed that some parameters recorded were outside the ranges expected to directly impact the immunogenicity and as such, the exercise did not enable the evaluation of immunogenicity.
Group 3 was moderated by Arley Gómez López and co-moderated by Gloria Cecilia Peñuela Sánchez. During their discussions, Group 3 noted that for Candidate 1, from a quality perspective, there are differences in the acid charge profile and the profile of deamidation which would affect the potency of the product. To remedy the differences, they should be evaluated in terms of their impact on the potency with clinical studies. They suggested that the residual uncertainty should be evaluated with the use of a small clinical study which is sufficiently sensitive in early stages of the disease. If this can be carried out, then extrapolation is possible. However, the group also noted that extrapolation cannot be done because the bioequivalence study was carried out on a single dose in healthy subjects and a more complete PK study would be required. When evaluating Candidate 2, Group 3 noted that it does not meet criteria to be a biosimilar from a quality perspective. The differences cannot be solved, and a clinical study and extrapolation should not be carried out.
Group 4 was moderated by Dr Néstor álvarez Lara and co-moderated by Giovanny de Jesús Otálvaro Rojas. This group’s opinion of Candidate 1 was aligned with that of Groups 1–3. In discussion about Candidate 2, it was noted that the clinical study design was not of good quality and the efficacy of the candidate was low.
Group 5 was moderated by Mr Castillo and co-moderated by Deyanira Duque Ortíz. With regards to Candidate 1, Group 5 noted that they had some concerns about the differences in the acid charge profile and ADCC binding test which could directly impact the PK and lead to inferior results. They suggested that information is needed to support the differences in acid charge profile. With regards to residual uncertainty, they noted that they would need the secondary evaluation results of overall survival (OS) and progression-free survival (PFS). In addition, they wanted clarification as to why the PCR evaluation variable was the primary evaluation variable. Overall, Group 5 said the candidate could be extrapolated but they would want to address the specific implications for gastric cancer.
Other observations the group made included:
Mr Castillo highlighted some brief adjustments in examinations that influenced the part of the bond strength, the power. The differences in glycosylation also affected these factors. In the PK table, with reference to the higher value, which is the expected value, the comparator result was lower than the expected value but not by much whilst the reference value was within or even higher. From an efficacy level, it caught the attention that even with these differences, the biosimilar presented a slightly higher efficacy compared to the reference, about 0.2% higher than the reference.
Group 6 was moderated by Johanna Andrea Garcia Cortes, and co-moderated by Ivan David Fonseca García. With regards to Candidate 1, the group noted that charge profile and deamidation were observed in the fucosylation particles. It was explained that the former affects ADCC and the latter, potency. However, when reviewing the data of ADCC, CDC and binding tests, they all fall within the required parameters. In addition, the group found it striking that there was nothing about comparison of comparative primary structure or comparative immunogenicity. For Candidate 2, the PK data endpoints do not fall in the correct regions and thus it is not a biosimilar.
In addition to the points made by the group moderators, Dr Wang noted that interestingly in the case of Candidate 1, the PK data is very similar to the reference. If this case had a true PD surrogate you might be able to determine a difference, but here you have no PD data so clinical studies are needed. However, looking at this case, you will need a sensitive study population as the PK values for the two products are comparable; if you had a non-sensitive study population you may reach a wrong conclusion. At each step, comparison must be sensitive to detect potential differences.
Ms Sánchez noted that the case studies were useful as it allowed workshop participants to have a complete discussion about both quality evaluation and clinical studies and the different parameters that can affect quality. Quality is very important when it comes to a products efficacy and safety and it allows for faster conclusion about whether a product is a biosimilar.
Professor Borg concluded that there was an issue with the clinical studies. The studies for the two candidates have two different designs and this could have been further discussed during the workshop. In addition, the primary endpoints warranted further discussion. For one candidate this was the overall response rate (ORR), which he believes is not the best endpoint for a study. He noted that the results clearly show that, even with an insensitive endpoint, there are huge differences in the efficacy of the product. This clearly demonstrates an inferior product. With this in mind, he added that the take home message is to consider the totality of evidence when in decision-making.
The 3rd Colombian educational workshop was successful in bringing together those involved with biological/biosimilar regulation in Colombia with experts from Canada, Europe and the US. It highlighted the progress that has been made in Colombia in terms of biosimilars regulation since the 2018 implementation of Decree 1794 and the future steps that are needed to improve biosimilars uptake and access to medicines. The workshop also highlighted many important issues surrounding biosimilars regulation and regulatory assessment and helped to clarify many regulatory concepts and concerns. The attendees shared ideas with the speakers and received clarification on issues of interest and concern.
The Generics and Biosimilars Initiative (GaBI) wishes to thank Professor Andrea Laslop and Dr Robin Thorpe, Chair and Co-chair of this workshop; the moderators and co-moderators in implementing the parallel discussions and clarifying information of the parallel discussion when finalizing the meeting report; as well as Mr Francisco Javier Sierra Esteban of INVIMA for his strong support through the offering of advice and information during the preparation of the workshop.The authors would like to acknowledge the help of the workshop speaker faculty and all participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators and co-moderators in facilitating meaningful discussion during the parallel discussions and case study working sessions, presenting the discussion findings at the meeting, and contributing in the finalization of this meeting report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Speakers
Professor John-Joseph Borg, PhD, Malta
Yolanda Elias Gramajo, MD, Canada
Thomas Felix, MD, USA
Johanna Andrea García Cortes, MSc, Colombia
Professor Andrea Laslop, MD, Austria
Jennifer Liu, PhD, USA
Aurelio Enrique Mejía Mejía, MSc, Colombia
Robin Thorpe, PhD, FRCPath, UK
Jian Wang, MD, PhD, Canada
Moderators
Néstor Álvarez Lara, PharmD
Professor Fabio Ancizar Aristizábal Gutiérrez, PhD
Joseph Sebastián Cepeda Santamaría
Deyanira Duque Ortíz, MSc
Ivan David Fonseca García
Johanna Andrea Garcia Cortes, MSc
Arley Gómez López, MD, PhD
Juan Fernando Juez Castillo
Judy Hasleidy Martínez Martínez
Giovanny de Jesús Otálvaro Rojas, PharmD
Gloria Cecilia Peñuela Sánchez, PharmD
Associate Professor Claudia Patricia Vaca González, MSc
Speakers and moderators had provided feedback on the regulatory panel discussion and the case study group discussion, respectively; read and commented the revised content of the manuscript, and approved the final report for publication.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Professor John-Joseph Borg, PhD, Malta
Yolanda Elias Gramajo, MD, Canada
Professor Andrea Laslop, MD, Austria
Robin Thorpe, PhD, FRCPath, UK
Jian Wang, MD, PhD, Canada
References
1. Generics and Biosimilars Initiative. First INVIMA Educational Workshop on Assessment of Similar Biotherapeutic Products 2016; 14 June 2016; Bogotá, Colombia. Available from: www.gabi-journal.net/about-gabi/educational-workshops/first-invima-educational-workshop-on-assessment-of-similar-biotherapeutic-products-2016
2. Generics and Biosimilars Initiative. Second Colombian Scientific Meeting on Quality Assessment of Biosimilars/Similar Biotherapeutic Products 2017; 15 August 2017; Bogotá, Colombia. Available from: www.gabi-journal.net/second-colombian-scientific-meeting-on-quality-assessment-of-biosimilarssimilar-biotherapeutic-products-2017
3. Gray E, Matejtschuk P, Thorpe R. Quality assessment of biosimilars in Colombia – reducing knowledge gaps. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):79-83. doi:10.5639/gabij.2018.0702.017
4. Generics and Biosimilars Initiative. First Latin American educational workshop on similar biotherapeutic products, Mexico; 20 January 2015; Mexico City, Mexico. Available from: www.gabi-journal.net/first-latin-american-educational-workshop-on-similar-biotherapeutic-products-mexico-city-mexico-20-january-2015.html
5. Walson PD, Thorpe R. First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars, Dubai, United Arab Emirates, 1 September 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):173–7. doi:10.5639/gabij.2015.0404.039
6. Laslop A, Wang J, Thorpe R. 2nd MENA Stakeholder Meeting on Biosimilars 2018 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2019;8(2):76-87. doi:10.5639/gabij.2019.0802.009
7. 3rd Colombian educational workshop on regulatory assessment of biosimilars 2019. 30 April 2019, Bogotá, Colombia. Available from: www.gabi-journal.net/about-gabi/educational-workshops/3rd-colombian-educational-workshop-on-regulatory-assessment-of-biosimilars-2019
8. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 22 May 2014 [homepage on the Internet]. [cited 2020 Jun 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf
9. European Medicines Agency. Guideline on similar biological medicinal products. 23 October 2014 [homepage on the Internet]. [cited 2020 Jun 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
10. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 18 December 2014 [homepage on the Internet]. [cited 2020 Jun 29]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/3rd-colombian-educational-workshop-on-regulatory-assessment-of-biosimilars-2019-report.html
|
Abstract: |
Submitted: 16 January 2015; Revised: 16 February 2015; Accepted: 20 February 2015; Published online first: 6 March 2015
In the past decades, biologicals have had a profound impact on the overall health and quality of life of patients with complex diseases such as rheumatoid arthritis, diabetes and cancers. Unlike pharmaceuticals, biologicals are produced from living organisms, e.g. human, animal, using biotechnology. The continued advances in medical research and technology have driven novel scientific platform innovations resulting in the development of newer biologicals that have expanded the treatment options for patients. For example, antibody-drug conjugates, and antisense RNA interference-, cell- and gene-based therapies are at various stages of clinical development and some have received marketing authorization. Meanwhile, the great success of many biologicals, e.g. infliximab, rituximab, along with their recent or pending expiry of patent protection have opened the door to a distinct class of biologicals ‘biosimilars’ (subsequent entry biologics [SEBs] in Canada) with one growth hormone biosimilar [1, 2] and one monoclonal antibody (mAb) biosimilar [3] authorized, so far, in Canada. This new class of biologicals poses novel regulatory and scientific challenges due to its complexities. Nonetheless, in many countries, biosimilars benefit patients and the healthcare system, due to cost factors and the opportunity, therefore, to treat a larger number of patients who might otherwise not have access to such useful products. Thus, the objectives of this review intend to highlight the Canadian regulatory review process for biosimilars, and to discuss the regulatory and scientific issues associated with the clinical assessment of biosimilars, based on the current thinking in Health Canada (HC), guidance documents and the information available for each product.
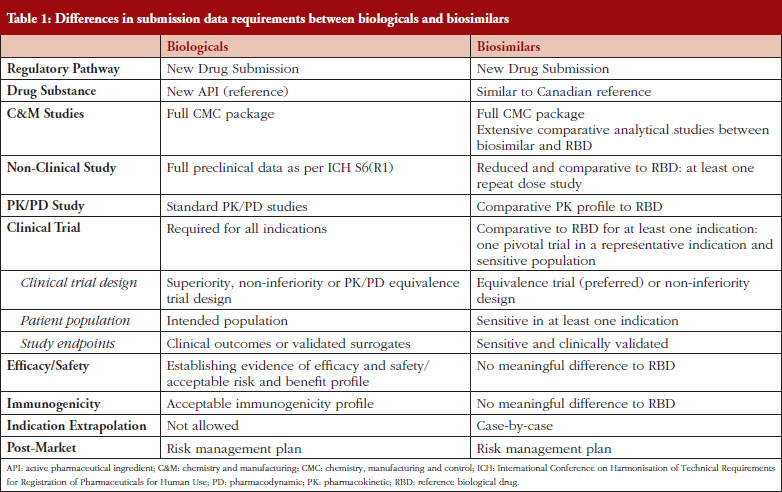
In Canada, biologicals are regulated under Schedule D of the Food and Drugs Act and Division 4 of the Food and Drug Regulations. Because biologicals exhibit a number of properties that distinguish them from pharmaceuticals, the regulatory requirements for biological submissions differ from those for pharmaceuticals [4]. Guidance documents issued by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and adopted by HC for biologicals are generally applicable to biosimilars [5–12]. Many jurisdictions as well as the World Health Organization (WHO) have published specific guidance documents regarding the data requirements for the marketing authorization of biosimilars [13–18]. In Canada, biosimilars fall under the same provision as those for new drugs: Division 8 of the Food and Drug Regulations [13]. The biosimilar regulatory framework is based on the scientific and regulatory principles within the existing regulatory framework for biologicals. A biosimilar, like a new biological, must be filed as a new drug submission, see Table 1. The premise underlying a biosimilar submission is to demonstrate similarity to a previously authorized biological (reference biological drug [RBD]) marketed in Canada and relies, in part, on prior information regarding the efficacy and safety of the RBD. The demonstration of similarity is primarily deduced from side-by-side quality studies. The biosimilar development programme not only requires a full chemistry and manufacturing (C&M) data package as is expected for a standard new biological, but also an extensive structural and functional characterization between the biosimilar and the RBD chosen. These studies should be carried out using multiple orthogonal analytical methods, e.g. physicochemical and biological analyses, with high accuracy, sensitivity and specificity. The establishment of similarity at the analytical/functional level would form the basis for a reduced non-clinical and clinical package for biosimilars, see Table 1. Any differences in quality attributes should have no adverse impact upon safety or efficacy. The type and extent of the non-clinical and clinical data are dependent on the level of ‘residual uncertainty’ that remains based on the results of the quality studies. A fingerprint like analysis algorithm to compare the quality attributes may be used to leverage a more selective approach to subsequent clinical studies [16]. Overall, the establishment of biosimilarity is based on the totality-of-evidence. The authorization of a biosimilar does not imply that the RBD and the biosimilar are considered pharmaceutically and therapeutically equivalent from the regulatory perspective in Canada, since the drug substances of the biosimilar and the RBD are not identical [13]. Any declaration of therapeutic equivalence is not within the purview of the federal regulator, but is within the authority of each Province in Canada, as health care is within the authority of the provincial health authorities [4]. Once a biosimilar is authorized, it is regarded as a stand-alone biological: manufacturers do not have to compare it with the original reference product for post-market changes.
The purpose of the clinical programme for a biosimilar is to resolve any residual uncertainties related to the similarity between the biosimilar and the RBD, and generally includes comparative pharmacokinetic (PK)/pharmacodynamic (PD), clinical safety/efficacy and immunogenicity studies. In principle, because a biosimilar is a biological, the clinical issues associated with biologicals also apply to biosimilars. However, since biosimilars usually follow a shortened clinical pathway and the clinical trials aim to exclude clinically meaningful differences in PK/PD, efficacy and safety, rather than establishing de novo risk/benefit, biosimilars are facing specific challenges and entail special considerations that are discussed below.
Choice of reference biological drug
One of the challenges associated with a global biosimilar development programme is the selection of the RBD. Clinical comparative studies should employ a suitable RBD. The Canadian biosimilar guidance document states that the RBD should be authorized for sale and marketed in Canada [13]. If multiple versions of an RBD are available on the market, it is preferable that the one licensed in Canada be used in the comparative studies. A non-Canadian RBD could be accepted if a rationale demonstrating its suitability as proxy for the version authorized in Canada is provided. The non-Canadian RBD should be marketed by the same innovator company or corporate entity that is approved to market the medicinal ingredient in the same dosage form in Canada [13]. Due to a global marketing strategy and in order to allow for a single development process, sponsors are using multiple versions of an RBD, e.g. American, Canadian and European versions made by the same manufacturer at the same or different manufacturing site(s), in clinical studies. To demonstrate that different versions of the RBD are virtually the same, sponsors would usually conduct three-way bridging studies including PK/PD studies between the different RBD versions and the biosimilar. In the selection of an RBD, it is important to note that the following products would not be considered suitable RBDs: i) different protein, e.g. granulocyte-macrophage colony-stimulating factor biological for granulocyte colony-stimulating factor biosimilar; ii) different protein modifications, e.g. non-pegylated biological for a pegylated biosimilar; iii) different amino acid sequences compared with the proposed biosimilar; or iv) protein made in a different expression system, e.g. animal-derived biological for a plant-derived biosimilar. HC considers that the absence of relevant animal-specific post-translational modifications, and/or post-translational modifications unique to plant expression systems, and their impact on immunogenicity, would make the development of biosimilars using plant-derived systems challenging. This would also increase uncertainties for decision-making due to limited regulatory experience and lack of sufficient information in the public domain [19].
Comparative PK/PD studies
PK/PD studies represent an essential part of the biosimilar clinical programme as they support biosimilarity (PK/PD comparability) between the biosimilar and the RBD. A similar PK/PD profile between the two products could alleviate some residual uncertainty and thus guide the extent of subsequent comparative clinical studies. PK/PD studies could help to monitor immunogenicity in clinical trials, e.g. via altered PK data, and could provide scientific evidence for extrapolation of indications. PK/PD data could also be used to compare different routes of administration or different strengths and formulations of a biosimilar. For example, if a biosimilar is proposed to be administered subcutaneously and intravenously, but only one route of administration is used in clinical studies, a bridging PK/PD study should be conducted to demonstrate that the two routes of administration are bioequivalent. Further, in a situation where a new strength or formulation is proposed for an intended subcutaneous (SC) or intramuscular (IM) route of administration, a bridging PK/PD study should be performed to show that the bioavailability between the different strengths or formulations, by the intended route of administration, is the same. If the intravenous (IV) route is proposed and used in the clinical studies, additional PK/PD data to compare the different strengths or formulations may not be required as the bioavailability is 100%. In cases where no studies are conducted via the SC route, it is unlikely that an indication using the SC route will be granted by extrapolation.
One key consideration for PK/PD studies is the use of a relevant patient population given that patient status, receptor internalization rate and expression of target receptor (density and subtypes) may affect the disposition and clearance of the biosimilar. In some cases, the use of healthy volunteers could be inappropriate as their PK/PD parameters may not be reflective of those observed in patients, due to differences in immune status. Also, target-mediated effects on PK cannot be fully assessed and a clinically relevant dose may induce a ceiling effect on healthy volunteers. Nonetheless, healthy volunteers may be pursued if justified, e.g. no efficacy and safety concerns. The most sensitive study design associated with comparative PK studies to detect potential differences between the biosimilar and the RBD is the single dose crossover design. However, this design could be limited by the properties of the biological such as a long half-life or by the formation of anti-drug antibody (ADA) that could impact the PK/PD profile. As well, patient population may require a continued dosing for ethical reasons. Alternatively, parallel or steady-state design could be considered. The criteria for comparative bioavailability as established in Canada for small molecules, should be generally followed [13]. However, they may not always apply to biologicals [20]. The criteria for PK comparability also differ between jurisdictions [21–23]. For instance, according to the HC guidance document for PK/PD studies, the 90% confidence interval (CI) of the relative mean area under the concentration (AUC) versus time curve to the time of the last quantifiable concentration (AUCT), as well as of the relative mean maximum concentration (Cmax) of the test (biosimilar) to the RBD should be within 80–125% inclusively [21]. At the same time, the US Food and Drug Administration (FDA) recommends applicants to provide the geometric means, arithmetic means, geometric mean ratios and 90% CI for AUC0-t, AUC0-inf, and Cmax [22]. For steady-state studies, not only the 90% CI of the relative mean area under the concentration versus time curve at steady-state over the dosing interval (AUCtau) and the ratio of the test to the reference (Cmax) at steady-state should be within 80–125% inclusively, but also the relative mean minimum concentration (Cmin) at steady-state of the test to the reference should not be less than 80% [21]. HC may accept potency correction if the measured drug content of the biosimilar and the RBD differ by 5% or more from each other. In such cases, the use of potency correction should be justified and predefined, and the applicable bioequivalence standards should be met on both potency-corrected and uncorrected data [21]. PD parameters could be investigated in the context of combined PK/PD studies or part of clinical trials. The required CI for PD parameters is usually set at 95%. The PD endpoints used should be considered as surrogate markers and be clinically validated. Otherwise, PD data would not provide strong support for biosimilarity. Note that for most biologicals, no suitable PD surrogates exist.
Equivalence versus non-inferiority design for efficacy/safety clinical studies
In line with the principle of demonstrating similarity, HC recommends that equivalence clinical trials for biosimilars be designed to show comparability to their respective RBDs. Based on the ICH definition, an equivalence trial is a trial designed to show that two interventions do not differ in either direction by more than a pre-specified insignificant margin [8]. A pre-specified and clinically acceptable equivalence margin (two-sided test) that is adequate to detect clinically meaningful differences between the biosimilar and the RBD should be selected. This equivalence margin should be based on available historical data for the RBD and should be smaller than the differences observed in superiority trials for the RBD [8–9]. Biosimilar guidelines prepared by various jurisdictions or WHO do not provide standard equivalence margins for most biologicals used to detect clinically meaningful differences in the targeted diseases. The equivalence margins should be considered on a case-by-case basis, and regulators may recommend a different margin than that proposed by sponsors. Therefore, sponsors of biosimilar products should consult with regulators prior to the initiation of pivotal trials to ensure that the selected equivalence margins are acceptable. In justified cases, a non-inferiority trial design could be acceptable such as a situation where the response rate for an RBD is very high, e.g. 90%, and superiority is unlikely to occur [24]. However, the use of non-inferiority trial requires superiority to be tested and if demonstrated, the product can no longer be considered as a biosimilar (at least in Canada). The demonstration of non-inferiority could also limit the extrapolation to other indications.
Sensitive patient population and study endpoints
Biosimilar clinical trials should be carried out in a ‘sensitive’ and homogeneous patient population. A ‘sensitive’ population is defined as one in whom potential differences between the biosimilar and the RBD are likely to be detected. For instance, metastatic breast cancer may not represent a sensitive population for trastuzumab biosimilar clinical trials because of the heterogeneous nature of metastatic disease, and the greater risk of immune impairment and of the development of secondary cancers [25]. Patients who have not received previous treatment, e.g. first-line therapy, are preferred compared with those who have been treated with different lines of therapy. Patients who previously received several or different lines of therapy are more heterogeneous and may be expected to respond differently, this may mask the detection of differences between the SEB and RBD. A patient population that received a drug as a monotherapy would also be considered to be a more sensitive population compared with one that was administered a concomitant medication, e.g. immunosuppressant, or had been treated with combination therapies. SEB sponsors are encouraged to consult HC with regard to the selection of a sensitive population prior to the initiation of a clinical trial.
Likewise, clinically relevant and sensitive study endpoints should be selected. These endpoints could be different from those traditionally used for the RBDs as they may not be considered as the most sensitive endpoints to detect differences in efficacy. If surrogate endpoints are used, they should correlate with clinical outcomes. The European Medicines Agency (EMA) states that for oncology trials, objective response rate (ORR) would be a more sensitive endpoint compared to progression-free survival (PFS) or overall survival (OS) since ORR would be less affected by patient- and disease-related factors [14]. It is also suggested that continuous outcomes, e.g. change in DAS28 over time, would be more sensitive than dichotomous outcomes, e.g. ACR20, in RA for determining clinical comparability [20]. However, regulatory bodies may not always agree upon the choice of study endpoints. Considering the case of RA, some may require ACR20, e.g. FDA [26], while others such as HC may prefer DAS28. For a biosimilar sponsor with a global development programme that is guided or required by various regulators to fulfil local regulatory or clinical practice requirements, it may be possible to pre-specify different primary study endpoints with the statistical power in the same trial to comply with various regulatory requirements. HC generally considers the commonly used surrogate markers or those defined for FDA as acceptable endpoints for clinical comparability studies. HC would not accept endpoints from an unauthorized indication in Canada for the purpose of establishing efficacy and safety comparability in pivotal clinical trials, as from a regulatory perspective this is to compare an unauthorized indication to other unknowns [27].
Immunogenicity and safety
Immunogenicity is an important aspect of biosimilar safety. The ability of biologicals to induce an immune response via the production of ADAs may result in unpredicted and unforeseen consequences that range from none to serious effects on PK, efficacy and/or safety [28]. For example, patients treated with recombinant erythropoietin developed pure red cell aplasia due to formation of neutralizing anti-epoetin antibodies (Abs) that cross-reacted with endogenous erythropoietin [29]. In a biosimilar clinical programme, immunogenicity should be evaluated in at least one clinical trial using the most sensitive population in which immune responses and adverse reactions are likely to occur. Immunogenicity testing should demonstrate that immunogenicity of the biosimilar is not increased compared to that of the RBD and that there is no change in terms of ADA concentration, titre and type between the two products. Manufacturing variations may lead to minor differences in structure, post-translational modifications, or impurities in a way that could shift the immunogenicity profile of a biosimilar and potentially affect safety [30]. Notably, glycosylation is of particular concern for mAb biosimilars since glycosylation on the Ab Fc domain could affect the activation of effector functions [31]. Immunogenicity testing should employ state-of-the-art assays that are capable of detecting difference in ADA response. Ideally, two assays should be used to validate the methodology, one using the biosimilar and the other using the RBD as the capture ligand. If only one assay is used, the biosimilar should be incorporated as the capture ligand. Neutralizing or cross-reacting Abs are the most concerning, and thus, any significant effect of these Abs on PK, efficacy and safety between the two products should be assessed.
Immunogenicity and safety, e.g. the nature, severity and frequency of adverse events, should be compared between the biosimilar and the RBD in clinical trials that enrolled a sufficient number of patients (> 100 patients) for an acceptable period of time (at least one year). Safety assessment should take into account the safety profile of the RBD throughout its life cycle to ensure that there is no new safety signal for the biosimilar. While safety data including immunogenicity are collected during the pre-market stage, it is unlikely that they are able to detect meaningful, less common (rare adverse events) or longer-term adverse drug reactions. For example, long-term safety concerns such as tumour progression or haematological malignancies have been associated with epoetin and filgrastim biosimilars, respectively [32]. Albeit analytical methods to detect ADAs are available, they remain limited and may not predict all biological properties. Therefore, a rigorous safety monitoring is required in the post-marketing setting.
Extrapolation of indications
Extrapolation to other indications for which the biosimilar has not been tested in clinical trials is appealing to many biosimilar manufacturers, but it represents a paramount concern. In Canada, an extensive and compelling comparability C&M in conjunction with reduced PK/PD and clinical studies may potentially permit extrapolation of the clinical data from the sensitive population to other indications for which the Canadian RBD is authorized at the time of filing [13]. For instance, HC granted all Canadian indications approved for the reference product to the first biosimilar somatropin, i.e. treatment of adult and children with growth hormone deficiency [1], while non-Canadian indications for the RBD were not granted, i.e. Prader-Willi syndrome, small for gestational age, Turner syndrome, and idiopathic short stature [33]. The decision to extrapolate could also be challenged by residual quality attributes and differences in the mechanism of action, pathophysiological mechanism of the disease, route of administration, posology, PK/PD profiles, concomitant therapies, clinical endpoints, study populations and/or safety/immunogenicity profiles between indications [4, 34]. Differences in clinical experience compared with the RBD may also preclude extrapolation [4]. HC’s view is that uncertainties related to the biosimilar which could result in potentially meaningful clinical effects need to be addressed prior to marketing authorization. HC, similar to many other regulatory agencies, endorses the concept of extrapolation, but each indication extrapolation has to be scientifically justified and will be considered on a case-by-case basis. Although decision-making is driven by scientific considerations, legal or public health frameworks and clinical practice vary in different countries. Indications that are still under patent or data protection in Canada would not be granted a Notice of Compliance until such protections expire or an allegation can justify immediate market entry that is either accepted by the innovator or upheld by the court. Consequently, as for any other therapeutic product, regulatory bodies may reach different decisions based on the same data package. For example, HC authorized the first mAb biosimilar infliximab for only a subset of indications and uses while other regulatory agencies granted many uses [3, 35–38].
As for any new biological product, risk management plan should be in place for biosimilars. The post-marketing pharmacovigilance programme should be tailored to each biosimilar to address specific product-related issues. In some cases, a more extensive pharmacovigilance may be needed. Recently, the government of Canada has introduced Bill C-17 also known as Vanessa’s law which gives HC the authority to take full measures to strengthen safety oversight of drugs throughout their life cycle [39]. Hence, Bill C-17 fosters a robust Canadian regulatory system that will further contribute to the safety of biosimilars for Canadians.
The advent of biotechnology has enabled the development of biologicals that have revolutionized the prevention and treatment of multiple diseases. With the patent expiry of many biologicals, the arena of biosimilars is emerging, providing patients with wider access to biologicals. Unlike new biologicals, the clinical pathway of biosimilars is usually shortened and based on the demonstration of comparability with the innovator product (RBD). As such, the clinical development of biosimilars is associated with specific regulatory and scientific issues. Biosimilar clinical trials should be thoroughly assessed and scrutinized. Key concerns related to the choice of the RBD, trial design, safety and immunogenicity discussed in this review should form the basis of the clinical assessment. The comparative PK/PD and clinical studies should also be conducted in clinically relevant settings that are the most sensitive to detect potential differences between the two products, especially the use of sensitive population and study endpoints. A rigorous post-market safety and pharmacovigilance plan must be established to ensure the long-term safety of patients. Extrapolation of indications to conditions that have not been studied remains a challenge for biosimilars. Although the concept of extrapolation appears to be widely accepted, indications granted by each jurisdiction may vary for the same product. As the knowledge and experience with biosimilars increase, regulatory agencies may be able to overcome the scientific and regulatory hurdles and harmonize their decision-making at a global level.
Competing interest: None of the authors declare any competing interests.
Provenance and peer review: Commissioned; externally peer reviewed.
Ally Pen1, PhD
Agnes V Klein2, MD, DPH
Jian Wang1, MD, PhD
1Clinical Evaluation Division – Haematology/Oncology, Centre for the Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Products and Food Branch, Health Canada, Ottawa, Ontario, K1A 0K9, Canada
2Director, Centre for the Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Products and Food Branch, Health Canada, Ottawa, Ontario, K1A 0K9, Canada
References
1. Health Canada. Summary Basis of Decision (SBD) for Omnitrope. 14 September 2009 [homepage on the Internet]. 2009 Sep 23 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/sbd-smd/phase1-decision/drug-med/sbd_smd_2009_omnitrope_113380-eng.pdf
2. Klein AV. The first subsequent entry biologic authorized for market in Canada: the story of Ominitrope, a recombinant human growth hormone. Biologicals. 2011;39(5):278-81.
3. Health Canada. Drugs and Health Products. Summary Basis of Decision (SBD) for Inflectra. 11 March 2014 [homepage on the Internet]. 2015 Feb 16 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php
4. Klein AV, Wang J, Bedford P. Subsequent entry biologics (biosimilars) in Canada: approaches to interchangeability and the extrapolation of indications and uses. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):150-4. doi:10.5639/gabij.2014.0303.033
5. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Structure and content of clinical study reports E3. 30 November 1995 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E3/E3_Guideline.pdf
6. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Ethnic factors in the acceptability of foreign clinical data E5(R1). 5 February 1998 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E5_R1/Step4/E5_R1_Guideline.pdf
7. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Guideline for good clinical practice E6(R1). 10 June 1996 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf
8. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Statistical principles for clinical trials E9. 5 February 1998 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf
9. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Choice of control group and related issues in clinical trials statistical principles for clinical trials E10. 20 July 2000 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E10/Step4/E10_Guideline.pdf
10. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Quality of biotechnological products: analysis of the expression construct in cells used for production of r-DNA derived protein products Q5B. 30 November 1995 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5B/Step4/Q5B_Guideline.pdf
11. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Specifications: test procedures and acceptance criteria for biotechnological/biological products Q6B. 10 March 1999 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf
12. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Preclinical safety evaluation of biotechnology-derived pharmaceuticals S6(R1). 12 June 2011 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf
13. Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs). 5 March 2010 [homepage on the Internet]. [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guid es/seb-pbu/seb-pbu_2010-eng.php
14. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. 30 May 2012 [homepage on the Internet]. 2012 Jun 11 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
15. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005.22 February 2006 [homepage on the Internet]. 2006 Mar 8 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
16. U.S. Food and Drug Administration. Draft guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. May 2014 [homepage on the Internet]. 2014 May 9 [cited 2015 Feb 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidandes/UCM397017.pdf
17. World Health Organization. Guidelines on the quality, safety, and efficacy of biotherapeutic protein products prepared by recombinant DNA technology [homepage on the Internet]. 2014 Aug 20 [cited 2015 Feb 16]. Available from: http://www.who.int/biologicals/biotherapeutics/TRS_987_Annex4.pdf?ua=1
18. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 19-23 October 2009 [homepage on the Internet]. 2010 Jun 4 [cited 2015 Feb 16]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
19. Health Canada. Guidance document: Plant molecular farming (pmf) applications: plant-derived biologic drugs for human use. 8 May 2014 [homepage on the Internet]. 2014 May 7 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/pmf-mcv-eng.pdf
20. Kay J, Feagan BG, Guirguis MS, et al. Health Canada/BIOTECanada Summit on regulatory and clinical topics related to subsequent entry biologics (biosimilars), Ottawa, Canada, 14 May 2012. Biologicals. 2012;40(6):517-27.
21. Health Canada. Guidance document: comparative bioavailability standards: formulations used for systemic effects. 22 May 2012 [homepage on the Internet]. 2012 Mar 21[cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-eng.pdf
22. U.S. Food and Drug Administration. Guidance for Industry. Bioavailability and bioequivalence studies for orally administered drug products-general considerations. March 2003 [homepage on the Internet]. 2003 Feb 11 [cited 2015 Feb 16]. Available from: http://www.fda.gov/downloads/Drugs/Guidances/ucm070124.pdf
23. European Medicines Agency. Guideline on the investigation of bioequivalence. 20 January 2010 [homepage on the Internet]. 2010 Mar 10 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
24. Zhang N, Chi E. Statistical considerations for the development of biosimilar products. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):21-5. doi:10.5639/gabij.2014.0301.007
25. Cortes J, Curigliano G, Dieras V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat. 2014;144(2):233-39.
26. U.S. Food and Drug Administration. Draft Guidance for Industry. Rheumatoid arthritis: developing drug products for treatment. May 2013 [homepage on the Internet]. 2013 May 13 [2015 Feb 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM354468.pdf
27. Justice Laws Website. Food and Drug Regulations [homepage on the Internet]. 2015 Feb 18 [cited 2015 Feb 16]. Available from: http://laws-lois.justice.gc.ca/PDF/C.R.C.,_c._870.pdf
28. De Groot AS, Scott DW. Immunogenicity of protein therapeutic. Trends Immunol. 2007;28(11):482-90.
29. Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469-75.
30. Brinks V. Immunogenicity of biosimilar monoclonal antibodies. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):188-93. doi:10.5639/gabij.2013.0204.052
31. Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci. 2012;1253:170-80.
32. Bennett CL, Chen B, Hermanson T, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol. 2014;15(13):e594-e605.
33. European Medicines Agency. European public assessment report for Omnitrope – product information [homepage on the Internet]. 2008 Apr 14 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000607/WC500043695.pdf
34. Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation. J Clin Pharmacol. 2014 June 26. doi:10.1002/jcph.339. [Epub ahead of print]
35. European Medicines Agency. European public assessment report for remsima (infliximab) – product information [homepage on the Internet]. 2015 Jan 15 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf
36. European Medicines Agency. European public assessment report for inflectra (infliximab) – product information [homepage on the Internet]. 2014 Dec 10 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf
37. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Japan [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Feb 16]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Japan
38. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in South Korea [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Feb 16]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-South-Korea
39. Health Canada. Protecting Canadians from Unsafe Drugs Act (Vanessa’s Law) Amendments to the Food and Drugs Act (Bill C-17). 6 November 2014 [homepage on the Internet]. 2015 Feb 16 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/legislation/unsafedrugs-droguesdangereuses-eng.php
|
Author for correspondence: Jian Wang, MD, PhD, Chief, Clinical Evaluation Division – Haematology/Oncology, Centre for the Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Products and Food Branch, Health Canada, 200 Tunney’s Pasture Driveway, Ottawa, Ontario, K1A 0K9, Canada |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/health-canadas-perspective-on-the-clinical-development-of-biosimilars-and-related-scientific-and-regulatory-challenges.html
|
Abstract: |
Submitted: 27 January 2014; Revised: 21 April 2014; Accepted: 22 April 2014; Published online first: 5 May 2014
Biosimilars (known as subsequent entry biologics [SEBs] in Canada) present new challenges in the regulation of biotherapeutic products. New and unexpected situations are encountered with some frequency, necessitating novel modes of problem solving. Here, we explain some of the principles used in decision making around SEBs. In this context, it is critical to discuss both interchangeability and extrapolation of indications as these are the two most controversial elements in decision making in this area. We also explain the approach taken by Canada, with some unique features related to extrapolating indications and clinical uses.
The advent of biosimilars followed the expiration of the patents for biological drugs. Interest had started to grow in the manufacturing of ‘copies’ of the active medicinal ingredients of some biologicals, as well copies of their final dosage forms. This new category of biological drug was named subsequent entry biologicals (SEBs) in Canada, and known in other countries as biosimilars. It is expected that these products will be cheaper [1] than the original products on which they are based. Bioequivalence studies have long been considered sufficient to prove the safety and effectiveness of generic drug products, but biosimilars (SEBs) are not generics, they are biological products that require a specified set of studies due to their unique properties, both physico-chemical and pathophysiological. It is important to note that, contrary to generic pharmaceuticals, the only a priori characteristic of a biosimilar is that, from the chemistry and manufacturing perspective, it must be highly similar [2–5] to the reference product chosen as a comparator (RBP: reference biological product).
The term SEB is used throughout this paper, except when there is a need to clarify that SEBs and biosimilars are terms that may be used interchangeably.
Biological products are complex and can vary with the batch produced, the site of production and multiple other factors, some intangible. As a consequence, the safety and efficacy parameters of a biological product may vary from batch to batch. At the same time, it is assumed that the pharmacodynamic (PD) effect of the biological stays consistent [3, 5–7].
SEBs are similar (or highly similar) to biological products already authorized for market or, in most instances, already marketed. The product to which an SEB is compared is called a reference biological product or drug (RBP or RBD). The quality attributes are comparable or highly similar and the PD action(s) of the product remain consistent with those of the product to which it is compared [5–8]
In general, a generic drug is compared with the first product of a class. SEBs may be compared to one of several original products in the class; SEBs cannot be used as reference products for new SEBs.
SEBs have some additional distinctive characteristics compared with generic drug products:
The premise underlying the development of SEBs is that they are biological products that enter the market subsequent to an existing biological and are highly similar (biosimilar) to a comparator. As such, information in the public domain may be used to shorten the development period for that drug/drug product, thereby reducing development costs. In Canada, SEB products fall under the same provisions as new drugs: Division 8 of the Drug Regulations [5]. It must be demonstrated that the active medicinal ingredient of the SEB is similar, from the chemistry and manufacturing perspective, to the reference product, and the final dosage form is equivalent to that of the reference product. As a result, no SEB can be authorized for market with bioequivalence as the sole study type. However the SEB can, and usually is, developed via a shortened clinical development pathway. This has still some advantages as far as costs are concerned.
Before developing an SEB, sponsors would do well to understand clearly, and be able to implement appropriately, the concepts of similarity.
There are many differences between pharmaceuticals and biologicals, varying from their method of manufacture/synthesis to their pharmacokinetic properties, their routes of administration and the manner in which their safety and efficacy are measured. A summary of these differences is provided in Table 1.
The differences outlined in Table 1 explain why decision making regarding SEBs is so complex. In fact, many of these characteristics – of pharmaceuticals versus biologicals – appear diametrically opposed. Experience in Canada has shown that sponsors/manufacturers of biosimilars use the unique features of each biological drug product in an unexpected way when choosing their RBP. There are examples where a concentration that is not marketed and has never been authorized in Canada is being used with the argument that the concentration does not matter. This is unwise because biologicals are commonly administered by the subcutaneous route, and practical experience has shown that the kinetic profile of a biological administered by the subcutaneous route is concentration-dependent. In other instances, subtle changes to a molecule have been introduced so as not to breach intellectual property laws; however, those changes can influence the therapeutic and immunological properties of a biological agent in manners that are unforeseen.
In general, there is an acceptance by regulatory agencies that SEBs cannot be identical copies of innovator products. The differences between a pharmaceutical and a biological require specific regulatory approaches, tailored to the concept of similarity. Most countries have created regulations specific to biosimilars. These regulations have taken various approaches with concepts ranging from approximations to the generics pathway, to hybrids between the pathways for pharmaceuticals and biologicals and, as in the case of Canada, a practical approach that takes into consideration the unique properties of biologicals. These properties are also applied in a flexible way, based on the science and its evolution. Regulatory and legal considerations are also brought in the equation when deciding on a particular product.
In Canada, SEBs fall under the same provisions as those of new drugs. The difference between an SEB and a new drug is that an SEB is developed by comparison with a reference product of similar structure and mechanism of action previously authorized and marketed in Canada.
In order to understand the pathway chosen in Canada, there needs to be a comparison between SEBs and generics, see Table 2.
Table 2 outlines the differences between generic drug products and SEBs, and explains some of the concerns regarding automatic substitution or interchangeability between two biological products. Concerns surround the correlates of quality, safety, immunogenicity, clinical and post-market traceability of biosimilars. It must be emphasized that two biologicals cannot be exactly the same. That is, the demonstration that two drug products are identical, which is the a priori requirement for a drug product to be a generic copy of another, cannot be proven for biological drugs. The US Food and Drug Administration (FDA), however, could declare some biosimilars as substitutable or interchangeable based on their fingerprint characteristics: in those instances no clinical studies would be required.
The approaches taken by different jurisdictions are documented in their respective guidelines dealing with biosimilars. In addition, the WHO has published a guidance document on biosimilars where the approach taken is very similar to that taken by Canada [14].
The meaning of substitutability and interchangeability may be defined in various ways. Although an SEB may be used instead of the reference biological, exchanging a reference product during a therapeutic course and switching between that reference and its SEB raises many concerns and could put the patient at risk. Substitution and interchangeability must be thought about carefully, for the following reasons:
Differences in interchangeability and substitutability between the European Medicines Agency, US FDA and Health Canada are outlined in Table 3. Under US legislation, interchangeable or interchangeability means that a biological product is biosimilar to the reference product and there is an expectation that the two products will produce the same clinical result in any given patient. When a given biological product is administered more than once to an individual, the risk of alternating or switching between the biosimilar and its reference product (in terms of safety or efficacy) is no greater than the risk of using the reference product without such alternation or switch. This, it can be noted, is a very high bar.
In the US, some states are moving, or proposing to move, to a vote on amendments to their generic-equivalent laws to include guidance on biosimilars. These amendments propose to establish that pharmacies can only substitute if the FDA licenses a biosimilar as interchangeable, and only then if the physician does not specify: ‘brand medically necessary’.
The extrapolation of indications, based on a limited number of studies, is an important factor to discuss because most savings in the development of SEBs can be made by extrapolating several of the indications of the comparator.
While the indication of SEBs in Canada must be the same as those of the reference product, some jurisdictions, such as EMA, have considered the extrapolation of uses to all those approved for a class of products.
Since pharmacokinetic/pharmacodynamic (PK/PD) factors may differ between indications and uses for any given biological therapeutic, extrapolation must be considered very carefully and should be based on a set of principles so as to ensure that the extrapolation is carried out in a consistent manner for all SEBs using the same principles for all.
As noted above, an SEB is not identical to the reference product chosen. While science and scientific and therapeutic principles play a role in both interchangeability/substitutability and extrapolation of indications, each country has a unique approach, based on national and local laws and practice issues and perceptions. In addition to regulations that govern interchangeability/substitutability of SEBs and the extrapolation of their indications, there exist administrative processes that differ in every country regarding the substitution of a prescribed product with another ‘equivalent product’.
It is also recognized that local medical practice and standards of practice contribute to the use of a drug which, in the case of a new drug such as an SEB, carries several unknowns. That is why in many, if not most, countries automatic substitution of SEBs for the reference product is not recommended and why Canada has taken a cautious approach to extrapolation of indications and uses.
It is hoped that with increased experience, some of these uncertainties and misgivings will be overcome.
Competing interests: None of the authors declare any competing interests.
Provenance and peer review: Commissioned; externally peer reviewed.
Jian Wang, MD, PhD
Chief, Clinical Evaluation Division, Centre for Evaluation of Radiopharmaceuticals and Biotherapeutic, Biologics and Genetic Therapies Directorate, Health Canada
Patrick Bedford, BSc, MA
Senior Policy Analyst, Office of Policy and International Collaboration, Biologics and Genetic Therapies Directorate, Health Canada
References
1. U.S. Senate. Biologics Price Competition and Innovation Act of 2009, Public Law 111–148 –Mar. 23, 2010 124 Stat. 703;351.
2. U.S. Senate. The Patient Protection and Affordable Care Act, Public Law 111–148–Mar. 23, 2010 124 Stat. 119.
3. U.S. Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. February 2012 [homepage on the Internet]. 2012 Feb 8 [cited 2014 Apr 21]. Available from: http://www.basinc.com/services/gen/BiosimilarsScientific.pdf
4. Health Canada. Fact sheet: subsequent entry biologics in Canada [homepage on the Internet]. 2006 Jul 26 [cited 2014 Apr 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/activit/fs-fi/fs-fi_seb-pbu_07–2006-eng.php
5. Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs). 5 March 2010 [homepage on the Internet]. 2010 Mar 8 [cited 2014 Apr 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2010-eng.pdf
6. U.S. Food and Drug Administration. Guidance for industry: quality considerations in demonstrating biosimilarity to a reference protein product. February 2012 [homepage on the Internet]. 2012 Feb 8 [cited 2014 Apr 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
7. European Medicines Agency. Guideline on similar medicinal products. CHMP/437/04. 30 October 2005 [homepage on the Internet]. 2006 Jun 15 [cited 2014 Apr 21]. Available from: https://www.tga.gov.au/pdf/euguide/chmp043704final.pdf
8. Health Canada. Questions & answers to accompany the final guidance for sponsors: information and submssion requirements for subsequent entry biologics (SEBs). 2014 [cited 2014 Apr 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/
applic-demande/guides/seb-pbu/01–2010-seb-pbu-qa-qr-eng.php
9. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005. 22 February 2006 [homepage on the Internet]. 2006 Sep 28 [cited 2014 Apr 21]. Available from: http://www.triskel.com/2%20Guideline%20biotech%20derived%20proteins.pdf
10. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues. EMEA/CHMP/BWP/49348/2005. 22 February 2006 [homepage on the Internet]. 2006 Dec 5 [cited 2014 Apr 21]. Available from: https://www.tga.gov.au/pdf/euguide/bwp4934805en.pdf
11. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. EMEA/CHMP/BMWP/14327/2006. 13 December 2007 [homepage on the Internet]. 2008 Jan 10 [cited 2014 Apr 21]. Available from: https://www.tga.gov.au/pdf/euguide/bmwp1432706en.pdf
12. European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products). EMA/837805/2011. 27 September 2012 [homepage on the Internet]. 2012 Sep 27 [cited 2014 Apr 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
13. Health Canada. Health Canada letter to Provincial/Territorial Drug Plans Directors: Interchangeability/Substitutabiity of Subseqnent Entry Biologics (SEBs), 29 July 2010.
14. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 19 to 23 October 2009 [homepage on the Internet]. 2010 Jun 4 [cited 2014 Apr 21]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
|
Author for correspondence: Agnes V Klein, MD, DPH, Director, Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Canada, AL 0700C, Room 0372, Tunney’s Pasture, Ottawa, Ontario, K1A 0K9, Canada |
Copyright © 2014 Pro Pharma Communications InternationalDisclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/subsequent-entry-biologics-biosimilars-in-canada-approaches-to-interchangeability-and-the-extrapolation-of-indications-and-uses.html
|
Introduction: The number of biosimilars, or similar biotherapeutic products, is expanding rapidly worldwide. A further 12 blockbuster biologicals are set to add to this number by 2020. Agreement about the best practices for the regulation, use, interchangeability and pharmacovigilance of biosimilars is lacking in many countries including in the Middle East and North Africa (MENA). |
Submitted: 18 February 2019; Revised: 24 July 2019; Accepted: 25 July 2019; Published online first: 6 August 2019
Biosimilars, or similar biotherapeutic products, have the potential to secure global supply of high quality, safe biological medicines while also cutting healthcare costs. To achieve this, several barriers need to be overcome. First, an internationally agreed system of regulatory approval will be necessary. Second, effective pharmacovigilance of biosimilars – via a system that can be accessed globally – is required. Third, a global understanding of and agreement on interchangeability must be worked out.
Countries within the Middle East and North Africa (MENA) region face a growing demand for the management of chronic diseases for which biosimilar treatments exist – including diabetes (for which biosimilar insulin can be used) and cancer (for which biosimilar rituximab and trastuzumab exist). According to His Excellency Dr Amin Hussain Al Amiri of the United Arab Emirates (UAE) Ministry of Health and Prevention (MOHAP), the MENA region has the highest prevalence of diabetes in the world, with more than one in 10 adults now living with the condition. According to the World Health Organization (WHO), cancer is the second leading cause of death globally after heart disease, being responsible for an estimated 9.6 million deaths in 2018 [1].
To achieve global agreement on regulatory approval, pharmacovigilance and interchangeability of biosimilars, open discussion across national borders and between different stakeholder groups is key. The opinion of one stakeholder group will often influence the opinion of another, and the opinion of one group is no more or less important than the opinion of another. A recent study looking at patient-reported outcomes (PROs) in separate trials of a biosimilar and originator insulin found no statistically significant differences in PRO between the biosimilar and the originator. The authors suggest that such outcomes should build confidence not only for patients but also for their healthcare providers in use of biosimilar insulins [2].
The number of biosimilars being used, or soon to be used, in MENA countries continues to grow as it does worldwide. Twelve biological products with global sales of more than US$67 billion will be exposed to biosimilar competition by 2020 [3], allowing patients access to affordable biological therapies for oncology, haematology, dermatology and autoimmune diseases.
Differences remain around the issues of regulatory approval, pharmacovigilance and interchangeability of biosimilars across the MENA region. The Generics and Biosimilars Initiative (GaBI) invited participants from countries in the MENA region and beyond including practising healthcare professionals, regulators and pharmaceutical manufacturers to discuss these issues. The goal of the meeting was to have all attendees, not just speakers, identify areas of consensus about how biosimilars should be regulated, used and monitored, as well as how to communicate the biosimilars concept to physicians, patients and other interested parties. The meeting also included case studies selected to highlight the importance of structure-function relationships for biosimilar monoclonal antibodies (mAbs), and how these relate to quality control and clinical performance.
On 10 October 2018, GaBI held the 2nd MENA Stakeholder Meeting on Regulatory Approval, Clinical Settings, Interchangeability and Pharmacovigilance of Biosimilars. As with previous workshops [4, 5], the objective of the meeting was to review and explain how biosimilars are currently regulated, used and monitored and to identify areas of consensus. In addition to presentations by expert speakers from Canada, Europe, the MENA region and the US, participants were instructed to discuss selected case studies on biosimilar mAbs in order to interpret the importance of structure-function relationships and understand their impact on quality control and clinical performance.
Regulatory standards and practices on biosimilars in UAE: safety and efficacy
Dr Rasha Sayed Salama, Public Health Consultant and Public Health Advisor for Public Health Policy and Licensing Sector at the MOHAP UAE, opened her presentation with the aim of UAE’s ‘National Strategy for Innovation’ to make UAE one of the most innovative nations in the world within seven years and within seven sectors, one of which is health. Quoting the country’s Vice President and Prime Minister, Sheikh Mohammed bin Rashid Al Maktoum, she noted that ‘change creates great opportunities, renews ideas and forces everyone to think in a different way’.
Biopharmaceuticals are one of the fastest-growing segments of the pharmaceutical industry and are expected to represent over 50% of the market by 2020. The pressure to reduce healthcare expenditure while increasing patient access to these treatments will drive the development of biosimilars. Biosimilars represent a paradigm shift in the way safety and efficacy are assessed, and education – involving academic research, policymakers and patients values – is clearly needed to support their introduction.
Referring to biosimilar guidelines around the world – from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human use (CHMP) Guideline on similar biological medicinal products [6, 7] to the US Food and Drug Administration’s (FDA) Abbreviated Approval Pathway for Biosimilars [8] – Dr Salama introduced the UAE requirements for registration of biosimilar products. These include data on consistency in the manufacturing process; heterogeneity assessment; therapeutic equivalence; safety and efficacy studies; demonstration of immunogenicity; and a pharmacovigilance plan where every person in the community can upload comments to the Ministry of Health directly.
In the UAE, biosimilars produced for overseas markets must follow international guidelines such as the US FDA and the European Union’s (EU) EMA. For national markets, biosimilars must follow UAE standards and guidance from the Cooperation Council for the Arab States of the Gulf (previously the Gulf Cooperation Council, GCC).
Any change in a biosimilar product’s specifications or characteristics is considered as a new product and will follow biosimilar procedures, see Figure 1.
Regulation of biosimilars in the EU – immunogenicity
Biosimilars are now firmly established in the EU as copy biologicals with a clear and effective regulatory route for approval, allowing for marketing of safe and efficacious biosimilar products. The EU was the first regulatory body to work on biosimilars, and thus has approved the most biosimilar products. Testing for unwanted immunogenicity is integral to product development, explained Dr Robin Thorpe, former Head of Biotherapeutics Group, National Institute for Biological Standards and Control, UK.
A biosimilar should be highly similar to the reference medicinal product in physicochemical and biological terms. Differences between originator and biosimilar versions (or, as Dr Thorpe prefers to call them, copies) are likely, and these need to be justified with regard to any potential impact on safety and efficacy. Importantly, differences are not a problem specific to biosimilars – this is an issue for biologicals in general. Testing for unwanted immunogenicity is integral to product development during both the clinical and post-marketing phases [9, 10].
Biological products (originators and bio-similars) can sometimes induce antibodies with different characteristics, including non-neutralizing antibodies against active (or inactive) product-related substances, binding antibodies against contaminants, neutralizing antibodies, or any combination of these. The clinical consequences of these vary, from benign to life-threatening.
Dr Thorpe cited an example where 13 out of 325 healthy platelet donors developed anti-platelet antibodies and transfusion dependence during treatment with megakaryocyte growth and development factor (MGDF). Only four out of 650 cancer patients treated with MGDF developed such antibodies. The reasons for this difference remain unclear, but are probably both product and patient related. The problem of induction of erythropoietin antibodies following treatment with certain EPO products and subsequent development of pure red cell aplasia was also mentioned [11]. Unwanted immunogenicity is impossible to predict, which is why immunogenicity studies are an essential component of clinical trials. Immunogenicity testing normally takes a tiered approach, including screening assays such as ELISAs and radioimmunoprecipitation assays, assays for confirming and assaying the specificity of antibodies, and neutralizing assays for discriminating neutralizing and non-neutralizing antibodies, see Figure 2. If the immunogenicity profiles of originator and biosimilar products are significantly different, they can be considered dissimilar.
Comparative immunogenicity studies – between originator and biosimilar products – need to be designed to demonstrate whether the immunogenicity of the products is the same or significantly different. For this, a homogenous and clinically relevant patient population should be selected, with head-to-head studies, using the same assays and sampling strategies. Post-approval assessment, usually as part of pharmacovigilance surveillance, is normally needed.
Biosimilars regulatory considerations in Saudi Arabia
Professor Aws Alshamsan discussed the regulation of biosimilars in Saudi Arabia by comparing the current system of department-centric evaluation for biosimilar approval – where each department separately approves each new therapy – with an alternative product-centric evaluation – where the departments work together in sequence to approve a therapy, see Figure 3. Professor Alshamsan says the process of department-centric approval is a disconnected, inefficient process taking 290 days, whereas product-centric approval takes much less time. Currently, only five biosimilars are approved in Saudi Arabia (Omnitrope, Remsima, Zarzio, Grastofil and Binocrit), compared with at least 50 approved in the EU [12].
Biosimilars are, on average, 37% cheaper than originator products, driving the need for a more efficient, faster, but no less safe and effective route to biosimilar approval. A product-centric evaluation would be considerably faster and more efficient, cutting approval time significantly, see Figure 3. Learnings from the product-centric system would help build internal capacity for the regulator.
Currently, changing from an innovator drug to its biosimilar, or changing between biosimilars, is acceptable in Saudi Arabia following discussion between physician and patient. Pharmacists in Saudi Arabia cannot substitute biosimilars without consultation with treating physicians. Interchangeability is approved at the regulator level; switching is approved by the local authority, prescriber and the patient; substitution is approved by the local authority and pharmacist. To date, no interchangeable biosimilars have been approved in Saudi Arabia.
Biosimilars: Canada’s approach to inter-changeability, biosimilarity, extrapolation of indications and uses – a comparison to the US FDA
Dr Jian Wang, Division Manager at the Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Canada (HC), compared the FDA’s approach to biosimilars with that taken by Canada. In the US, biosimilars follow a biosimilar specific regulatory pathway – 351(K) – whereas in Canada, biosimilars are considered as new drugs. In Canada, a stepwise approach to biosimilar approval progresses from comparative quality studies, to comparative non-clinical studies, to comparative clinical studies, see Figure 4. FDA compares biosimilars against US-licensed reference products, whereas the use of a non-Canadian version of the Canadian authorized reference product is acceptable by HC. Bridging to the national product is not required by HC, whereas FDA does require bridging to demonstrate that any foreign-sourced reference product and the corresponding domestic product are highly similar [13].
Looking at pharmacokinetic (PK) endpoint acceptance limits, FDA considers that the 90% confidence interval (CI) of the relative mean maximum concentration (Cmax), area under the serum concentration–time curve (AUCt) and area under the curve with respect to increase (AUCi) of the test to the reference should be within 80% to 125%. HC considers that the 90% CI of the relative mean AUCt and the 90% ratio Cmax of the test to the reference should be within 80% to 125%. With the pharmacodynamic (PD) endpoint, FDA considers that the 90% CI, for mean ratio (test to reference) should be within the predefined acceptance limits of 80% to 125%. HC considers that the 95% CI, for mean ratio (test to reference) should be within the predefined acceptance limits of 80% to 125%.
Dr Wang identified several potential differences in clinical trial design (in oncology) between the US and Canada, see Table 1, including differences in statistical analysis on endpoint (risk ratio for FDA; risk difference for HC), and in study endpoint (pathological complete response (pCR) for FDA, HC also accepts pCR, even though pCR-based indication has not been authorized in Canada and overall response rate (ORR) is preferred.
Interchangeability
On the issue of interchangeability, HC’s authorization of a biosimilar is not a declaration of equivalence to the reference drug, and the authority to declare two products interchangeable rests with each province and territory. At FDA, interchangeability designation and standards are mandated by law (Final guidance published in May 2019), and there are additional data requirements.
FDA has different and distinct statutory approval requirements for biosimilars compared with interchangeable (IC) products. Additional data are required for IC products: they are expected to produce the same clinical result in any given patient; and any risk in terms of safety or diminished efficacy of alternating or switching between IC and reference products should be no greater than the risk of continuing with the reference product alone. It is the applicant’s responsibility to choose their approach; and provide adequate support for their approach in addressing these additional requirements.
At the time of this meeting (10 October 2018), no interchangeable biosimilar product had been licensed by FDA [14].
No switch study
On the issue of switching, HC does not require a switch study from a reference biological drug to a biosimilar for market authorization.
When HC and relevant regulatory agencies authorize the marketing of a biosimilar, that biosimilar will have met all quality, safety and clinical standards. The final authorized indications are not extrapolated from one single comparative clinical study. The decision to authorize the requested indications is dependent on the demonstration of similarity between the biosimilar and reference biological drug based on data from comparative structural, functional, non-clinical, PK/PD and clinical studies and a detailed scientific rationale.
The approved biosimilar will be structurally and functionally (highly) similar to the reference product. Residual uncertainty from quality assessment does not cause clinically meaningful differences in efficacy, safety and/or immunogenicity. A biosimilar approved by HC may receive all or some therapeutic indications of the reference product.
Biosimilars receive indications of the reference based on the totality of evidence collected from all comparative studies, see Figure 5.
Principles and challenges related to manufacturing process development and demonstration of analytical comparability for biosimilars
The concept of analytical similarity, from the EU biosimilar perspective, was discussed by Dr Niklas Ekman, Vice Chair of the EMA’s Biosimilar Medicinal Products Working Party (BMWP). He looked at this from development of the manufacturing process for biosimilars, and from product experience, reflecting on two recent marketing authorization application assessments (Remsima/Inflectra infliximab; and Ontruzant trastuzumab).
The development programmes for biosimilars start with a comprehensive characterization of the chosen reference medicinal product, the Quality Target Product Profile (QTPP). This requires extensive analytical testing, including physicochemical and biological assays. The biosimilar manufacturer aims to achieve a deep understanding of the quality profile of the reference product, including the naturally occurring batch-to-batch variability, see Figure 6.
The characterization data also guides the development of the manufacturing process for the biosimilar. Once the process has been established, the pivotal evidence of analytical similarity is obtained through characterization studies which should, as far as possible, be conducted in a side-by-side manner. The comparison should include a sufficient number of biosimilar and reference batches using orthogonal, highly sensitive and appropriately qualified analytical methods that are able to detect all relevant differences in the quality profiles. When properly completed, the analytical similarity exercise significantly reduces the remaining uncertainty with regard to biosimilarity.
The experience of the EMA CHMP in assessing biosimilarity was highlighted by the examples of two cases, one of which addressed the impact of glycosylation differences observed for Remsima/Inflectra infliximab; the other of which focussed on challenges associated with a shift in critical quality attributes of the reference product, Herceptin.
In the first case, high similarity was shown between the biosimilar Remsima/Inflectra and the reference (Remicade) for primary, secondary and tertiary structure, in vitro tumour necrosis factor-alpha (TNF-α) neutralization, binding affinity and in vitro functional tests. However, minor differences were reported for C-terminal lysine content, aggregates, intact IgG level, charged molecular variants, glycosylation pattern, and binding to FcγRIIIa.
Largely based on the results from additional functional in vitro studies, the CHMP concluded that the differences were not clinically meaningful. Functional difference was seen only in an antibody-dependent cell-mediated cytotoxicity (ADCC) assay employing artificially high transmembrane ™ TNF-α expressing Jurkat target cells in combination with highly purified natural killer (NK) effector cells. Furthermore, there were no published reports of ADCC being induced by TNF antagonists in patients.
In the second case, analytical and functio-nal similarity were demonstrated between Ontruzant and EU Herceptin. However, in extended clinical characterization studies, comparability within the Herceptin originator could not be demonstrated using equivalence testing. As a result, the biosimilar had effectively become superior to the reference product, see Table 2.
Such differences raise important issues related to safety, but in this case the originator and biosimilar were shown to have similar safety profiles. Ontruzant was considered highly similar to Herceptin, and any differences in efficacy have since been attributed to a change in batches of the reference product.
Quality shifts of the reference product do happen, notes Dr Ekman. These can be challenging to handle for the biosimilar developer. Without reference product characterization data spanning over a long period of time, quality shifts might remain undetected. A thorough understanding of both the candidate biosimilar and the reference product characteristics is critical for successful biosimilar development, see Box 1.
Clinical and non-clinical assessment of biosimilars
Professor Andrea Laslop, Head of the scientific office at the Austrian Agency for Health and Food Safety, Austria, summarized the stepwise approach to demonstrating comparability between biosimilars and originators. After having shown biosimilarity at the physicochemical, biological and in vitro functional level, the clinical comparability exercise establishes biosimilarity in vivo.
If differences are observed at the non-clinical level, it is important to ask whether there is a plausible rationale (based on the quality characterization). It is important to ascertain whether the differences concern a major pathway or mode of action, and to consider whether further investigations using either more sensitive assays or under more physiological conditions would be useful.
All non-clinical findings need to be interpreted in the context of clinical results and vice versa – this is the principle of the totality of data.
PK similarity to the reference product is investigated in comparative phase I studies, mainly in healthy volunteers. These studies also yield comparative PD, supporting the evidence for biosimilar efficacy with limited information.
For PK/PD results outside equivalence limits, this might be because the study was not sufficiently powered, because the variability was higher than expected or because the drug concentration was higher in the test/reference batches. Variability can be difficult to anticipate if PK data on the originator product is scarce. Variability is also impacted by sample size. For these reasons, a crossover design may reduce or even eliminate variability, a further decrease in variability might also be seen by using only male subjects.
A clinical phase III trial confirming equivalence in efficacy completes the clinical comparability exercise and provides safety data pre-approval. When such an efficacy trial is performed, limited PK/PD sampling in the patient population can qualitatively confirm the results observed in healthy volunteers and at the same time allows the assessment of PK after repeat administration. In certain instances, when a validated PD surrogate marker is available, the PK/PD study may suffice to confirm similar efficacy. Considering the low sensitivity of the clinical phase III trial in detecting potential differences between the products, its importance may be questioned. Likewise, waiving of the phase III study could enable the development of biosimilar orphan drugs by mitigating feasibility constraints due to small population sizes.
If differences in efficacy are seen, it is important to ask whether the study was sufficiently powered. Similarly, if the variability was higher than expected, this could potentially be addressed by providing additional PD data, in case these data would represent established surrogate markers of efficacy. Before investigating further, it should be determined whether the observed differences are clinically relevant, as usually defined by the chosen non-inferiority margin.
If differences in safety are observed it is important to check that these were not just a chance finding. They could be the result of differences in antigenicity or in impurities, in which case a stringent risk management plan should be proposed in order to especially control the development of antibodies. However, differences in immunogenicity could also be the result of an artefact due to assay variability or due to a difference in assay sensitivity. Above all, strong post-marketing collection of safety and immunogenicity data is paramount.
Once biosimilarity has been established in one indication, extrapolation to other indications is generally accepted on the basis of sound scientific justification. Extrapolation is the most important principle for biosimilars, but it is also the most contentious. It is not a new concept – extrapolation is routinely seen with generics, for example, and in paediatric indications where trials on child subjects are often not deemed ethical. Another situation most relevant for the understanding of extrapolation for biosimilars is a change in manufacturing of an approved biological product. This leads to a new version of the active substance, which also corresponds to the definition of a biosimilar, indicating that in fact an originator biological after changes in manufacturing can be regarded as its own biosimilar. However, clinical data are typically not required to substantiate manufacturing changes and extrapolation is done on the basis of comprehensive in vitro comparison of the product before and after the manufacturing change. In any case, extrapolation should always be seen in the light of the totality of data.
An update on biosimilars and switching experience – the clinical perspective
Professor Tore Kristian Kvien, who leads the rheumatology department at Diakonhjemmet Hospital in Norway, provided an update on the Norwegian government-funded NOR-SWITCH trial [15]. The trial involved 482 patients on stable treatment with Remicade across six indications including rheumatoid arthritis, Crohn’s disease, ulcerative colitis, spondyloarthritis, psoriatic arthritis and psoriasis. Patients were randomized to continue with the reference product or switch to Remsima/Inflectra and there were no significant differences in primary (disease worsening) or secondary (including drug discontinuation and remissions rates) endpoints. An extension study showed that switching from reference product to CT-P13 was not inferior to maintained treatment with CT-P13 over 26 additional weeks [16], see Figure 7. Professor Kvien concluded that these data add to the increasing real-world evidence that switching from originator biological disease modifying antirheumatic drugs (bDMARDs) to their biosimilars is safe and efficacious.
Despite this, Professor Kvien noted persistent objections to biosimilars based on subjective complaints – the Nocebo effect. This will need to be addressed in future studies [17].
Professor Aws Alshamsan introduced the Saudi approach to interchangeability and switching by explaining that biosimilars are not automatically interchangeable. Of the 10 biosimilars that have been approved in Saudi, only two are deemed interchangeable. Biosimilarity alone is not enough for substitution or switching – this requires another level of scrutiny.
This approach is quite different to that of Tunisia, as explained by Dr Sonia Sebai Ep Ben Amor. In Tunisia, medicines are procured by pharmacy centralized purchase (PCP). When the biosimilar of trastuzumab Hertraz was approved by the Biosimilar Specialized Committee, the Minister of Health’s tender committee and the national insurance company chose to purchase the biosimilar for economic reason, which meant Herceptin was no longer available. There was no choice. Since then, a second biosimilar has been chosen and patients have been switched to this. Patients who prefer to get the originator have to pay for it themselves. Ongoing pharmacovigilance is the only way to record any differences in safety and efficacy between biosimilars and originator.
Tunisia’s Biosimilar Specialised Committee makes decisions on interchangeability on a case-by-case basis. The committee includes representatives of regulatory authorities, national control laboratory (dossier assessors), pharmaceutical inspection and the purchasing department as well as expert clinicians from different specialities who use biosimilars. There are several brands of insulin available in Tunisia, from Sanofi, Novo Nordisk and Julphar (Juslin). Two Tunisian insulins – human insulin and insulin glargin – are currently going through registration.
No monoclonal antibody biosimilars are currently marketed in Egypt, but two are under discussion, according to Ms Doaa Mohamed Abdelrady Mohamed. Decisions regarding interchangeability will be made by the Ministry of Health, and the patient will not be given a choice, said Ms Mohamed.
Dr Niklas Ekman summarized Finland’s, and more broadly the EU’s, approach. He highlighted issues around terminology. In the EU, interchangeability describes a property of two products, that are expected to gain the same result in the patient. Interchangeability can be seen in two ways: either as switching by the prescriber; or as substitution at the pharmacy level, without the knowledge of the prescriber. In Finland, all biosimilars that have been approved in the EU are interchangeable, which means that the physician can change between the two. Finland has not yet taken a position on substitution at the pharmacy level, but from a scientific point of view, the challenges of substitution should be similar to those of switching. There are, however, still practical questions associated with pharmacy-level substitution that need to be solved. EMA approves biosimilars, but Member States can decide whether to approve interchangeability. Most Member States agree that EMA-approved biosimilars are interchangeable when the switch is overseen by the prescriber.
Ms Dunia Ama AlBastaki, who works in drug regulation in Kuwait, asked whether approved biosimilars were themselves interchangeable. Dr Ibrahim A Aljafalli, former Vice President of the Drug Sector in Saudi FDA, replied that biosimilars approved by EMA were considered inter-changeable.
In response to a question from Ms Suna Mohammad Ibraheem Habahbeh, Professor Alshamsan explained that, in order to approve switching, a clinical trial that involves switching must be run. This must happen before the biosimilar is approved.
Professor Ahmed Elmelegy of Egypt asked about the extent of biosimilar price differences in Saudi and Tunisia, Dr Sebai Ep Ben Amor of Tunisia said that there was a 50% difference between Herceptin and its biosimilar – an enormous reduction. Professor Alshamsan said that there was currently no price difference between Remicade and Remsima, but for Neupogen and Zarxio the biosimilar was a fifth of the price. He expected a new pricing policy would be introduced soon. Professor Abdalla Mohamed Elsayed Abotaleb of Egypt added that Egypt has a pricing policy for biosimilars based on a concept called second brand innovator. There is a 30% variation from innovator to second brand pricing for Hervive and Retuksera.
Regulatory standards
Mazen Kurdi, Professor of Pharmacology at the Lebanese University, argued that most regulatory bodies in the MENA region are made up of pharmacists, with a few physicians. Based on the information shared at this meeting, Professor Kurdi said it was clear that regulatory bodies need scientists. The science is not always understood by regulatory bodies, and this is particularly a problem for locally-produced drugs (rather than those imported from the US and Europe, which have been subject to more informed regulation).
Dr Aljafalli, responded that Professor Kurdi’s generalization was not fair to mature regulatory bodies in the region. Many regulatory bodies in the region have experts other than pharmacists. In the Saudi FDA, for example, there are chemists, biochemists, microbiologists, analytical chemists, biostatisticians, pharmacists, physicians and experts in veterinary medicine. Dr Aljafalli, a pharmacist himself, said many regulatory authorities in the region were similarly diverse.
Dr Rasha Sayed Salama, UAE, said biosimilars produced in the UAE are approved by the country’s Public Health Policy and Licensing Sector, which has a Drug Department responsible for approval. This includes pharmacists, academics and members of the pharmaceutical sector. For applications from the EU and the US, EMA and FDA guidelines are followed (with some revisions).
Immunogenicity
Dr Robin Thorpe, UK, said that the latest immunogenicity guidance from EMA was issued at the end of 2017 but came into effect in 2018. There is currently no better guidance and it can only improve as fast as technology improves. Asked if improvements in post-approval assessment were also needed, Dr Thorpe replied that they were and warned that, particularly in developing nations, harmful side effects could be going undetected.
Dr Ekman, Finland, joined Dr Thorpe in explaining that immunogenicity is always tested in clinical studies. It is difficult to predict immunogenicity, but it is important to take into account features, for example, aggregated products, that might have the potential to be immunogenic. Dr Thorpe added that rheumatology patients are more ‘immune active’ and so need to be assessed as such.
Interchangeability and switching
Professor Alshamsan, King Saud University, Saudi Arabia, said that whereas guidance on interchangeability was the regulator’s responsibility, switchability is the decision of the prescriber. If two biosimilars are proven interchangeable, they need a switching study, he said. The regulator cannot force the prescriber to switch between biosimilars, even if they are interchangeable.
Biosimilars in Saudi Arabia
Professor Alshamsan said that two trastuzumab biosimilars are currently going through the registration process. Alongside these, there are registration plans for biosimilar rituximab and bevacizumab.
Biosimilar indications
Dr Jian Wang, Health Canada, explained that the number of indications a biosimilar has varies depending on the country. A biosimilar might have 10 indications in the US, but only eight in Canada. It cannot claim that it has 10 in Canada if the Canadian reference product only has eight.
Batch variability
Dr Ekman explained that EMA approves a product rather than batches. The approval process ensures consistent manufacturing, limiting batch-to-batch variation. Both Drs Ekman and Thorpe explained that the original approval is designed to take into account immunogenicity, usually including between six to 12 months of comparative immunogenicity data. The original approval should provide assurance, but pharmacovigilance is needed – as it is for all drugs.
Pharmacovigilance
Professor Andrea Laslop of the Austrian Agency for Health and Food Safety, responded to questions on pharmacovigilance in Europe, and explained how data are collected and assessed via periodic safety update reports (PSURs). Initially, these are based on a six-monthly interval and then once yearly and then, once the product has been on the market for over three years, not collected unless there is an issue with efficacy or safety.
Clinical trials
Although more extensive than a normal, quick and small PK trial, Professor Laslop said that crossover trials (where subjects receive each treatment in succession – acting as their own controls), are a worthwhile investment. Professor Laslop added that relatively large sample sizes – including PK studies with 180 to 190 healthy volunteers – were helpful when addressing high variability and increasing dropout rates.
Totality of evidence
A presentation on the totality of evidence illustrated the critical role of structure–function in biosimilar development using the example of monoclonal antibodies. The relationship between chemistry, manufacturing and controls (CMC) and clinical studies was examined. All biologicals, including biosimilars, have complex structures and multiple quality attributes. The relationship between product quality attributes and biological function is complex, as is the relationship between product quality attributes and process.
Clinical perspective on biosimilars use: clinicians, pharmacists, regulators
European and US approaches to pharmacovigilance of multisource biologicals were presented prior to a stakeholder panel discussion. When multiple sources of biologicals are available it is challenging to accurately trace and attribute adverse events (AEs) to the correct product. Misattribution of AEs can damage reference product safety databases and prevent biosimilar manufacturers from accurately measuring the benefit-risk of their products.
In Europe, via legislation and the release of good pharmacovigilance practice guidance, brand names and lot numbers are required to be recorded in patient medical records and AE reports. The intent is that all Member States will comply, and that accurate traceability will be achieved by efforts to improve reporting processes by users.
In the US, FDA has finalized a naming policy that adds a four-letter suffix to a common root non-proprietary name for all biologicals and biosimilars that are newly approved [18]. The intent is that a distinguishable naming convention will allow for automatic use on packaging and in systems during prescribing, dispensing, documentation in patient records, and when an AE is reported.
An effective system of pharmacovigilance for biosimilars from many different sources is key to fostering confidence in the biosimilar marketplace and to empowering manufacturers to take full accountability of their product profiles.
Biosimilar naming
Professor Aws Alshamsan questioned the value of the four-letter suffix. If a patient was receiving Neupogen, for instance, and then a week later was switched to Zarxio, how would an immunogenicity incident be reported? Would it be attributed to Zarxio or to Neupogen because the immune response took its time to build up, perhaps it was a delayed type of hypersensitivity. Professor Alshamsan wondered if the four-letter suffix would mislead the regulator to interpret the data incorrectly.
Dr Thomas Felix, Amgen, explained that a single report of an AE is not sufficient evidence to link an AE to a particular product. Each individual report is filed, and healthcare professionals are alerted when a signal or a cluster of reports becomes apparent in the records. A single report might go to the wrong place, but when an investigation is underway, the hyphenated suffix will help. Without the suffix you might only have the non-proprietary name. If you have a long history of different products, maybe cycling through three or four different biosimilars, you would notice over time how a particular product was linked to a particular event.
Dr Robin Thorpe, UK, added that Japan developed their biosimilar naming system with the prefix BS1, BS2, BS3 and so on before WHO developed its biological qualifier (BQ) system. FDA embarked on biosimilar naming much later and tried to base their system on some of the principles of the WHO BQ system, but this has created problems that Dr Thorpe does not see getting resolved.
Pharmacovigilance
Dr Mohamed A Omair from the rheumatology department at King Saud University, Saudi Arabia, agreed that pharmacovigilance is essential but also difficult to find time for alongside a busy physician’s workload. He suggested that hospital pharmacists should take the lead, particularly where there is a clinical pharmacy service.
Dr Omair cited a small study that found only 25% of Saudi rheumatologists thought biosimilars might have a role in patient management. A subsequent study during the Arab League Against Rheumatism meeting, which included Arab rheumatologists from across the Middle East region, asked physicians about their level of knowledge. Rheumatologists were asked whether they thought the evidence for biosimilars was adequate, whether they would use biosimilars, and how many years’ experience they would need before prescribing biosimilars to most of their patients. Thirty per cent of those questioned responded that there was insufficient evidence in favour of biosimilars, and they would not use them. About 30%−40% responded that they were unsure. Dr Omair and colleagues conclude that there is a clear knowledge gap among physicians. Education is needed, and this must focus on healthcare providers rather than decision makers and officials.
Switching
Dr Adeeba Al-Herz, President of the Kuwait Association of Rheumatology, based at the Amiri Hospital; Faisal and Medical One Specialized Polyclinic in Kuwait, raised the issue of difficulty in switching between known, effective biological originators and unknown biosimilars. Despite literature in support of a biosimilar, a physician is more familiar with the efficacy and safety of the originators given the fact that they have been using them for many years. In addition, physicians still need more data regarding their long-term safety. Patients will also be worried of taking unknown drugs, regardless of published evidence. Dr Jian Wang, from Health Canada, asked how much evidence would be needed to convince patients. Dr Al-Herz said that the evidence was sufficient, but evidence alone is not enough to convince patients (or even physicians) on a practical level. Dr Wang echoed Dr Niklas Ekman’s earlier presentation showing that there had been no examples – in the past 12 years – of an approved biosimilar behaving differently to its originator. Dr Ekman cautioned that this was true for the EU; but may be different in other regions. Dr Wang understood that patients would question why they were being switched when there was no problem with the originator: trust must be built between regulators, physicians and patients. Dr Wang suggested differences between originators and biosimilars might have been overemphasized, overlooking the high similarity between them.
Professor Tore Kristian Kvien emphasized the role of communication, and the importance in improving compliance. In Norway, the pressure to switch to a biosimilar is particularly great because of the cost differences, which are felt directly by hospital budgets. This press ure is not so marked in MENA countries.
Patient choice
Dr Ravi Mohan Pedapenki of the Ministry of Health in Bahrain, at the Salmaniya Medical Complex, Medical Oncology, was reminded of a patient in India treated successfully with an originator biological treatment for HER2+ (human epidermal growth factor receptor 2 positive) breast cancer (trastuzumab). When the patient’s insurance stopped covering the cost of treatment, the patient refused to switch to the cheaper biosimilar but could not afford the originator. Her symptoms returned. Finally, she had to be treated with the biosimilar, which was as successful as the originator had been. In this way both the physician and the patient were convinced of the efficacy of the biosimilar. Being provided with evidence by regulators is not enough, physicians and patients need to see evidence at first hand.
There was wide agreement from physicians across the MENA region that the economic argument in favour of biosimilars is off-putting. Switching from a known successful treatment to an unknown treatment because it is cheaper does not instil confidence, particularly if neither the patient nor the physician is under tight financial constraints.
In response to a question about how many times patients could be switched between biosimilars, Professor Tore Kristian Kvien, Norway, replied that a multi-switch trial had been designed and was waiting to receive funding. Professor Kvien says multi-switch trials are needed. It is not sensible to switch if disease symptoms are not being controlled. In this case it would be better to choose a therapy with a different mode of action.
Patient choice is key in Kuwait and Dr Yasser Mustafa Ali Ghadanfar from Kuwait predicts this will present a barrier to switching. Professor Kvien responded that patients in Norway were willing to switch to the biosimilar when they were told how much money would be saved, and how this could be used to offer more innovative therapies. Dr Wang agreed that the financial argument was the only way to convince patients – that improved treatments will be made available with the savings. Patients who still prefer not to switch have to pay for the difference for their own therapy.
Asked how much money was saved by switching to biosimilars in Norway, Professor Kvien said that in 2014 Remsima, biosimilar infliximab, was 39% less expensive than Remicade. In 2015, it was 69% less expensive. Pricing information is now publicly confidential, so Professor Kvien was unable to provide more recent figures.
Dr Wang, said savings were variable. The listed savings are between 15% to 50%.
Dr Mohammed A Omair, Saudi Arabia, mentioned a patient programme being designed to explain what biosimilars are and how they are used worldwide. This should improve patient acceptance of biosimilars and reduce the nocebo effect (caused by unwarranted negative expectations).
Discussion groups were provided with data on two semi-fictional trastuzumab biosimilar candidates. They were provided with physicochemical characteristics, selected glycan and biological attributes, and the results of a phase I study for Candidates 1 and 2. Similar case study was carried out at previous GaBI meetings.
Each discussion group was asked whether the data for the candidates qualified for biosimilarity with a reference product from a quality (CMC) perspective. If not, they were asked what steps they would recommend fixing this. Discussion groups were also asked how ‘residual uncertainty’ could be addressed in preclinical or clinical studies. They were then asked, given that Candidates 1 and 2 had both the CDR (complementarity-determining region) and Fc (fragment crystallizable) region involved in their mechanism of action (MoA) for some of the indications, whether they would recommend extrapolation to all indications.
Summary discussion of case study of therapeutic protein monoclonal antibody – Candidates 1 and 2
Professor Ahmed Elmelegy from Egypt led discussion Group 1, concluding that they would approve Candidate 1 and not approve Candidate 2. Candidate 1 was deemed a biosimilar because its physicochemical characteristics – the MoA, the binding capacity and binding results – were similar. His group found no residual uncertainty and so did not require additional preclinical or clinical studies. The group agreed about extrapolation in all cases in which HER2 is expressed – breast cancer, metastatic breast cancer and gastric cancer. Candidate 2 was not biosimilar because the quality attributes were different. Importantly, the MoA, the binding to the HER2, was inferior. The clinical study results were also inferior. His team suggested the drug company would have to go back to the beginning with Candidate 2.
Professor Ahmed Aljedai of Saudi Arabia led Group 2; and agreed with the conclusions of the first group. However, Professor Aljedai’s group would have liked to see additional studies because there were some differences with regards to deamidation which might affect potency. The group would ask for a comparative non-clinical study and would recommend PD studies for this purpose. The group would address issues of residual uncertainty with these preclinical studies and with clinical studies. The group agreed that the study design was adequate for the given indication but they would require further studies before extrapolation to other indications.
Professor Laslop asked if Professor Aljedai’s group would recommend PD studies in patients. Professor Aljedai said they would start with animals and move to patients. They chose this way of making sure differences in deamidation were not affecting potency, otherwise they might be wasting time and money. Professor Laslop said that no PD studies would be recommended in Europe, particularly as animal studies would not give relevant information for human subjects.
Dr Ravi Mohan Pedapenki, Bahrain, said that he would treat all HER2+ cancer patients with Herceptin, regardless of the cancer type because the mechanism remains the same. Dr Jian Wang, who was in Professor Aljedai’s discussion Group 2, was concerned that the concept of biosimilarity was being overlooked by the group. Biosimilarity was accepted, but extrapolation was not recommended. Professor Aljedai asked who would extrapolate based on the MoA only or the totality of evidence including MoA as well as the physicochemical properties and others, without the need for clinical trials. If you take the example of rituximab, which has been studied in lymphoma patients, said Professor Aljedai, would you extrapolate to rheumatoid arthritis?
Dr Pedapenki said he would not, because he only extrapolated in cancers. Professor Laslop said that they do extrapolate in Europe. In Europe, they agree to the extrapolation for rituximab from rheumatoid arthritis to oncology and the other way around. Rituximab depletes CD-20 positive B cells, said Professor Laslop. That is the same PD effect, in each of the diseases where the originator has been approved, and this is why extrapolation works. She added that regulators would ask for an additional PK trial in the other population to check for differences in the PK profile. It could be different because in the oncology setting there is target mediated clearance which is not relevant for the immunological disease.
Group 2 was co-moderated by Professor Abdalla Mohamed Elsayed Abotaleb, Egypt, who explained that his group agreed with Group 1 that Candidate 2 did not qualify as biosimilar due to differences in physicochemical characteristics that might impact potency and immunogenicity. As before, the group said that this candidate would need to be started from scratch.
Group 3, moderated by Professor Mazen Kurdi, Lebanon, reported that his discussion group would ask for extra clinical data before extrapolating with Candidate 1 ‘even if it was clear that we should not’. He and his group concluded that this was needed because the candidate was a mAb and showed differences in deamidation. Dr Niklas Ekman commented that this would depend on where the deamidation was, adding that a clinical study might not reveal features relating to the deamidation. Deamidation differences will not always affect function.
Group 4, moderated by Dr Adeeba Al-Herz, Kuwait, concluded any differences in deamidation could be remediated by optimizing the manufacturing process, for example, by improving deamidation, which might increase potency. Her group would have preferred more data on ADCC (antibody-dependent cell-mediated cytotoxicity and CDC (complement dependent cytotoxicity) to close in on any differences in immunogenicity. Nevertheless, the group would recommend extrapolation to all indications for Candidate 1 because if it is effective in one indication it can be extrapolated to all, particularly since the CDC and ADCC are similar. Dr Al-Herz added, however, that since potency (deamidation) is affected by structure, more data would be needed for all indications. The group concluded that Candidate 2 would not qualify as a biosimilar due to marked differences and the physicochemical characteristics that might impact potency and immunogenicity.
Group 5, moderated by Professor Ali K Abu-Alfa, Head of the Division of Nephrology and Hypertension at the American University of Beirut, Lebanon, would be happy with extrapolation for Candidate 1 following a period of learning more about the drug. As before, Candidate 2 was not judged a biosimilar.
Dr Ekman asked Professor Laslop about the endpoints that should be used to determine biosimilarity for the two candidates. She replied that the ideal endpoint in early breast cancer would be pathologically complete response. This is very sensitive because it can be measured more accurately than all of the other endpoints.
Overall survival, especially in a setting with patients with different sizes and numbers of metastases, is influenced by many additional factors leading to a lot of background noise in the trial. As a result, you might see no difference and conclude on similarity simply because of the fact that the whole background noise is masking any minor differences in the efficacy outcome.
Delegates at the 2nd MENA Stakeholder Meeting agreed that they had learned a great deal; but cautioned that what they had learned needed to be made more widely available. ‘In terms of clinicians, I think their knowledge in biosimilars is not as good as you may expect’, said Dr Adeeba Al-Herz from Kuwait. ‘We need to step down and go to the clinicians and try to educate them about the different aspects of biosimilars’. Dr Ibrahim A Aljufalli agreed, comparing the current position of biosimilars with the introduction of generics in the 1970s and 1980s. As regulatory authorities improve alongside improved sensitivity in the detection of differences between products, and experience in switching grows, physicians and patients will grow to accept biosimilars and the savings this will mean for healthcare budgets.
Dr Niklas Ekman concluded by encouraging meeting attendees to return to the clinicians in their countries and explain what biosimilars are and what they are not. Once this information has been made widely available, clinicians can then decide whether or not to prescribe biosimilars.
Speakers
Respected Dr Lubna AlShaali, PhD, UAE
Professor Aws Alshamsan, BPharm, RPh, PhD, Saudi Arabia
Niklas Ekman, PhD, Finland
Thomas Felix, MD, USA
Professor Tore Kristian Kvien, MD, PhD, Norway
Professor Andrea Laslop, MD, Austria
Jennifer Liu, PhD, USA
Rasha Sayed Salama, MD, PhD, UAE
Robin Thorpe, PhD, FRCPath, UK
Jian Wang, MD, PhD, Canada
Moderators
Professor Abdalla Mohamed Elsayed Abotaleb, PhD, Egypt
Professor Ali K Abu-Alfa, MD, FASN, FAHA, Lebanon
Adeeba A A H Al-Herz, MD, FRCPC, FACP, Kuwait
Professor Ahmed Aljedai, PharmD, MBA, BCPS, FCCP, FAST, Saudi Arabia
Professor Ahmed Abdelsalam Mohamed Elmelegy, MSc, PhD, Egypt
Professor Siham Hamaz, MD, Morocco
Professor Mazen Kurdi, PhD, Lebanon
Doaa Mohamed Abdelrady Mohamed, MSc, Egypt
Mohamed A Omair, MD, Saudi Arabia
Ravi Mohan Pedapenki, MD, Bahrain
Sonia Sebai Ep Ben Amor, MD, Tunisia
Speakers and moderators had provided the discussion/conclusion of the group discussion, read the report and revised the content of the summary discussion.
The Generics and Biosimilars Initiative (GaBI) wishes to thank Dr Rasha Sayed Salama from the MOHAP UAE for her support to the organization of the meeting; the moderators in clarifying the information of the case study discussion when finalizing the meeting report; as well as Professor Andrea Laslop and Dr Robin Thorpe, Chair and Co-chair of the 2018 meeting, for their strong support through the offering of advice and information during the preparation of the meeting.
The authors would like to acknowledge the help of all the workshop speaker faculty and participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators and co-moderators in facilitating meaningful discussion during the parallel case study working sessions, presenting the discussion findings at the meeting, and contributing in the finalization of this meeting report.
Lastly, the authors wish to thank Dr Bea Perks, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. World Health Organization. Cancer [homepage on the Internet]. [cited 2019 Jul 25]. Available from: https://www.who.int/cancer/en/
2. DeLozier AM, Ilag LL, Perez-Nieves M, Kaushik P, Duan R, Pollom RK, et al. Patient-reported outcome measures in phase III trials of LY2963016 insulin glargine and reference insulin glargine products: ELEMENT 1 and ELEMENT 2. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):56-62. doi:10.5639/gabij.2018.0702
3. GaBI Online – Generics and Biosimilars Initiative. US$67 billion worth of biosimilar patents expiring before 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Biosimilars/General/US-67-billion-worth-of-biosimilar-patents-expiring-before-2020
4. Aljedai AH, Alhomaidan AM, Trifirò G, Alshamsan AWs, Al-Foheidi M, Alsenaidy MA, et al. First GCC Stakeholder Meeting on Approval Process, Interchangeability/Substitution and Safety of Biosimilars 2017 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(4):158-63. doi:10.5639/gabij.2018.0704.032
5. Thorpe R, Griffiths E, Ekman N. First ASEAN educational workshop on regulation and approval of biosimilars/similar biotherapeutic products 2017 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):127-32. doi:10.5639/gabij.2018.0703.025
6. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 18 December 2014 [homepage on the Internet]. [cited 2019 Jul 25]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf
7. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Guidelines/EU-guidelines-for-biosimilars
8. GaBI Online – Generics and Biosimilars Initiative. US guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from www.gabionline.net/Guidelines/US-guidelines-for-biosimilars
9. European Medicines Agency. Immunogenicity assessment of biotechnology-derived therapeutic proteins [homepage on the Internet]. [cited 2019 Jul 25]. Available from: https://www.ema.europa.eu/immunogenicity-assessment-biotechnology-derived-therapeutic-proteins
10. European Medicines Agency. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. 24 May 2012 [homepage on the Internet]. [cited 2019 Jul 25]. Available from: https://www.ema.europa.eu/documents/scientific-guideline/guideline-immunogenicity-assessment-monoclonal-antibodies-intended-vivo-clinical-use_en.pdf
11. Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469-75.
12. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
13. Kurki P. Potential changes to the FDA approach to biosimilars have a global impact. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):53-5. doi:10.5639/gabij.2018.0702.011
14. Niazi SK. Rationalizing FDA guidance on biosimilars—expediting approvals and acceptance. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):84-91. doi:10.5639/gabij.2018.0702.018
15. Perks B. Randomized non-inferiority trial fails to find inferiority switching from infliximab originator to CT-P13 biosimilar. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):188-9. doi:10.5639/gabij.2017.0604.042
16. Goll GL, Jørgensen KK, Sexton J, Olsen IC, Bolstad N, Haavardsholm EA, et al. Long-term efficacy and safety of biosimilar infliximab (CT-P13) after switching from originator infliximab: open-label extension of the NOR-SWITCH trial. J Intern Med. 2019;285(6):653-69.
17. Tweehuysen L, van den Bemt BJF, van Ingen IL, de Jong AJL, van der Laan WH, van den Hoogen FHJ, et al. Subjective complaints as the main reason for biosimilar discontinuation after open-label transition from reference infliximab to biosimilar infliximab. Arthritis Rheumatol. 2018;70(1):60-8.
18. GaBI Online – Generics and Biosimilars Initiative. FDA update on naming biologicals [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Reports/FDA-update-on-naming-biologicals
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2019 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/2nd-mena-stakeholder-meeting-on-biosimilars-2018-report.html
Copyright ©2024 GaBI Journal unless otherwise noted.