First Latin American educational workshop on similar biotherapeutic products, Mexico City, Mexico, 20 January 2015
Published on 2015/03/25
Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):143-8.
|
Introduction: Similar Biotherapeutic Products (SBPs or biosimilars) comprise a rapidly expanding area of therapeutics worldwide. However, best practices for their regulation are lacking in many Latin American countries. |
Submitted: 6 February 2015; Revised: 4 August 2015; Accepted: 6 August 2015; Published online first: 19 August 2015
Introduction
Follow-on medications, especially small-molecule generic drug products, often provide equal efficacy with the same or fewer adverse effects than the original, patented medicinal products they are designed to mimic at lower costs to patients and healthcare systems.
The success of small-molecule generic drug products is largely the result of the fact that these are relatively easily characterized and synthesized chemicals that require only proof of similar composition and bioavailability to assume equal therapeutic and toxic effects (the generics paradigm). The efficacy and safety of biological products, i.e. biosimilars or similar biotherapeutic products (SBPs), however, are more complex to evaluate because they are difficult or even impossible to completely characterize by their physiochemical properties and even minor changes in their manufacturing processes can result in clinically meaningful differences in efficacy or safety. For these reasons SBPs are often regulated using a much more complicated similarity approach [1] but there are large differences in how SBPs are regulated in different countries including countries in Latin America [2], such as Mexico.
There are a large and growing number of follow-on biotherapeutic products either being used in, or proposed for use in Latin American countries but, as in most countries, there is also a lack of consensus as to the best practices for their evaluation, approval and post-marketing surveillance.
As part of its educational commitment, the Generics and Biosimilars Initiative (GaBI) therefore organized and ran a workshop to try to identify such best practices in order to maximize the chances that such follow-on products provide patients with non-inferior quality, safety and clinical responses.
Methods
On 20 January 2015, GaBI held the First Latin America Educational Workshop on Similar Biotherapeutic Products with a focus on regulatory, approval, pharmacovigilance and risk management in Mexico City, Mexico. The objective of the workshop was to discuss the role and importance of structure–function relationship in biologicals including biosimilars. The workshop was designed to promote active discussion amongst various stakeholders concerning the best practice methods for the evaluation, clinical testing and post-marketing surveillance of both originator and follow-on biotherapeutic products.
Speakers included experts from academia, regulatory agencies and biological manufacturer. Participants included regulators, academicians and practicing physicians from Mexico as well as other Latin American and non-Latin American countries, most of whom were involved in the day-to-day regulation, evaluation or use of biological products. Presentations were in English or Spanish with simultaneous translations into Spanish or English, respectively.
Results
The workshop began with the welcoming remarks from the COFEPRIS (Federal Commission for the Protection against Sanitary Risk) Federal Commissioner Mikel Arriola Peñalosa, JP, MPP, who outlined the efforts Mexico has made in the area of SBPs. He emphasized the legal framework upon which Mexican federal governmental pharmaceutical policies are based. These policies are designed to provide consumers access to a wide variety of innovative products, including generic drugs and SBPs, at the highest possible quality and best price, and are based on four fundamental requirements. The first and main mandate is to maintain quality and efficiency of supplies. Second, that safety, quality and efficacy must not be separated from efficiency. Third, that Mexican consumers need to have a transparent regulatory agency that can translate efficiency into economic benefits. Finally, there is a need to gradually eliminate the many barriers that exist to entrance of new pharmaceuticals.
The Mexican generic drug policy began in 2005 with a new, reformed legal framework for both innovative and generic drugs that required generics to be tested for bioequivalence, which was not required in most other Latin American countries. In 2011, Mexico focused its generics policy on the causes of 75–80% of mortality, mainly chronic degenerative diseases, and rapidly licensed a large number of generic medicines. There are currently 240 generic drugs registered in Mexico being used to treat millions of patients. Prices have decreased 60% with a per capita saving of Pesos 1,000. For the 32 active substances with a monopoly in 2011, there are now 340 health registries, which means that Mexico is a leader in generics penetration with units dispensed going up from 51% to 85% and invoicing from 30% to 54%, resulting in major cost reductions; especially for drugs used to treat cardiovascular diseases, cancer and diabetes where prices have decreased 90%.
Secondary reforms were added in 2009 to manage biological products. Simplified requirements and registries were established to deal with biologicals approved prior to this and requirements (Standard 257) were established in December 2014 for the approval of new biologicals as well as a transition framework that gave manufacturers of already marketed follow-on biological products a certain period (ending 2016) to conduct the clinical studies required to approve new SBPs. While previously approved drugs will not have to perform the same studies as new products, by 2016 all biologicals approved in Mexico will have to have been supported by clinical studies if they are to be marketed. The new process for approval of new biologicals has been implemented and 35% of biologicals have gone through this; with 26 biocomparables already approved. Plans are to increase the number and value of SBPs that Mexican citizens have access to.
The Commissioner concluded by saying that Mexican generic and biosimilar drug products have been very successful, as evidenced by the 4,500% increase in the number of drugs available (from three to 133, with 150 projected at the end of 2015) as well as cost decreases greater than in any other country while maintaining quality.
Dr Robin Thorpe from the UK, the Workshop Chair and GaBI Journal’s Deputy Editor-in-Chief, presented an overview entitled ‘Introduction to a global view of biologicals, biosimilars and non-originator (non-comparable) biologicals’. He clarified the differences between a ‘follow-on biological’ and a true biosimilar. Both are biological medicinal products that contain a version of the active substance in an already authorized original biological medicinal product (reference medicinal product), but to be a true ‘biosimilar’, the follow-on product must demonstrate similarity to the reference medicinal product in terms of quality characteristics, safety and efficacy based on a comprehensive comparability exercise. Biosimilars are firmly established in the European Union (EU) with a clear and effective regulatory route for approval. Although some other countries have adopted a similar regulatory stance for biosimilars, the situation regarding the procedure used to evaluate ‘biosimilars’ is not always clear. In some countries, including some in Latin America, products that are approved as ‘biosimilars’ are not evaluated using the comparability approach formulated in the World Health Organization (WHO) guidelines and so would not be regarded as biosimilars in either the EU or by WHO. Such non-innovator products should not be called ‘biosimilars’.
Dr Thorpe then considered ‘Reference products and International Standards/Reference reagents: appropriate and inappropriate uses in biosimilar product development’ where he clarified the difference between these often confused terms. Both reference products and WHO International Standards/Reference Reagents have distinct and important roles to play in the development and characterization of SBPs. It is important to understand that these roles are distinct and that their uses are not interchangeable.
The reference product is key and fundamental in the development of SBPs. It is the ‘comparator’ for all the comparability studies, i.e. for quality, non-clinical and clinical assessment. It is ideally a product that has been approved and marketed in the relevant country or geographical area, which has a long established history of good efficacy and safety.
The use of a reference product for all comparability studies is essential for the development of biosimilars. In contrast, WHO International Standards/Reference Reagents are primary standards intended for calibrating procedures, characterization and validation of potency assays.
Dr Gustavo Grampp summarized ‘The role of analytical and structure–function studies in the assessment of biosimilarity’. He explained why state-of-the-art structural and functional preclinical assays are essential for the stepwise evaluation of candidate biosimilar products. These methods can achieve significant precision and sensitivity, although the ability to compare certain aspects of structure and function remains challenging in some cases. As increasingly sensitive methods are likely to identify some physicochemical differences between products, a significant challenge for a biosimilar sponsor is to study the potential impact of observed differences on safety and efficacy via structure–function studies. The results of such studies can help inform the scope of additional non-clinical and clinical studies. However, non-clinical structure–function studies are not without limitations and sponsors and regulators should properly weight these data within the overall assessment of similarity.
Dr Meenu Wadhwa gave a presentation on ‘Immunogenicity testing for biotherapeutic products’. As she explained, all biological therapeutics have the potential to induce an immune response in recipients of these products. Elicitation of an immune response can result in variable clinical impacts ranging from benign to severe adverse effects, a diminution in clinical efficacy or in some cases hypersensitivity or allergic reactions. Unwanted immunogenicity is an important component of regulatory submissions for product approval. Immunogenicity assessment requires carefully planned prospective studies in a suitable indication, a well-considered strategy and a panel of appropriately validated (or ‘fit for purpose’) assays for antibody detection and characterization in clinical samples. She presented an overview of the bioanalytical methods in use, see Table 1, for assessment of the immunogenicity of biotherapeutics and available guidance and briefly discussed how biosimilarity should be addressed in the context of immunogenicity.

Dr Vladamir Hanes reviewed the totality of information required for the global, regulatory approval of SBPs. The definition of what is required to be approved as a biosimilar in the US is well known. There are two distinct elements that must be met to fulfil the requirement. The product has to be highly similar chemically, and there must be no clinically meaningful differences versus the reference product. Hence, the law is clear that products must be highly similar in both preclinical and clinical studies. Both requirements must be met and not just one or the other. This requires a stepwise assessment of the Totality of Evidence for a proposed biosimilar. The first step is the assessment of quality, which includes the analytical and functional evaluation of the biosimilar candidate. It must have identical amino acid sequence and potency as the originator; however, minor differences in, for example, glycan pattern and in post-translation modifications cannot be avoided and since these can impact the product’s performance or activity, any potential differences must be assessed. The second step is the pharmacological evaluation of the product. Pharmacokinetic (PK) evaluation in healthy volunteers is generally considered the most sensitive model to test the product’s PK similarity. Demonstration of equivalent PK, when combined with proof of highly similar structure and function, removes major causes of uncertainty and establishes whether the same dose of the biosimilar as the reference product can be used in clinical trials. The third sequential step involves a clinical confirmation study or studies. The efficacy of a biosimilar candidate product should be confirmed in a head-to-head comparative study in a sensitive population, using sensitive endpoints, usually with an equivalence statistical approach. Finally, confirmation of an acceptable and comparable clinical safety profile is tested to identify the incidence of known risks and to assess any potential unknown risks. Such testing includes assessment of immunogenicity using state-of-the-art tests for evaluating anti-drug antibody formation. Each of these steps provides a critical contribution to the totality of evidence for the candidate product. Each step can reduce the residual uncertainty; and each step has to utilize the most sensitive state-of-art methodologies. No single step can be used to overcome a failure in another step.
To illustrate how these steps are performed, Dr Hanes presented some illustrative data from two monoclonal antibodies currently in development; both of these monoclonal antibodies bind to a receptor and have antibody-dependent cell-mediated cytotoxicity (ADCC) effector functions.
Dr Thijs J Giezen presented on ‘Safety assessment and risk management of biosimilars: a regulatory perspective’. Dr Giezen’s presentation focused on the main aspects involved in the safety assessment and pharmacovigilance of biosimilars including safety aspects related to any exaggerated pharmacology, immunogenicity and the extrapolation of indications; as well as biosimilars in clinical practice covering switching and substitution [3]. He discussed from both a regulatory and a clinical perspective the requirements for a Risk Management Plan and additional pharmacovigilance activities, risk minimization and traceability.
Ms Prapaassorn Thanaphollert presented data on, and current Thai FDA regulatory approach to ‘Unwanted immunogenicity of EPO (epoetin) products and related clinical problems (PRCA, pure red cell aplasia); the experience in Thailand’. EPO products were being approved and granted marketing authorization in Thailand for more than 20 years before biosimilar guidelines existed or were being implemented. Most of the EPO products that are widely used in Thailand to treat patients with chronic renal failure (CRF) are given by the subcutaneous (SC) rather than the intravenous (IV) route in order to allow CRF patients to self-administer them at home. The Thai FDA (Food and Drug Administration) had received an increasing number of PRCA case reports. These were brought to the attention of the Subcommittee on ADR (adverse drug reactions) and prompted the agency to mandate the inclusion of a statement warning on the labels (package inserts and box) of all marketing authorization holders (MAHs) of EPO products of the possible increased risk of PRCA after SC use. Despite this warning, the number of PRCA cases detected in Thailand is still increasing when compared to the incidences reported from other countries worldwide. The cause(s) of this problem and any potential relationship between products, shipping and storage, or drug administration practices is still under investigation but this example illustrates that there is still residual uncertainty as to the actual or relative safety of such biological products. In response to this problem the Thailand EPO registry was established in most medical school hospitals who now follow a protocol with the objective of describing the incidence of PRCA cases in Thailand over time and with respect to drug product and administration used. This protocol requires any switching of EPO products to be identified and reported. A preliminary report obtained from this registry showed that SC administration is still commonly used despite the label warning and that the increased immunogenicity resulting in PRCA continues to be reported after administration by the SC route regardless of which EPO product was used. It was pointed out that EPO products given subcutaneously might also be associated with different product handling than with IV, in-hospital use. Product handling is a potential confounder in the relationship between route of administration and PRCA incidence.
Professor Philip D Walson, Editor-in-Chief of GaBI Journal, summarized the results of a recently published review describing the current state of SBP evaluation and approval in Latin American countries [1]. Professor Walson urged participants to read the manuscript and consider implementing the steps proposed, including: (1) Enhanced training of regulatory authorities on how to evaluate biosimilars since having staff with appropriate skills and expertise and the sharing of knowledge between health authorities in the region are important; (2) Establishment of a region-wide working group, under the auspices of PAHO, comprising representatives from regulatory authorities from various Latin American countries with expertise in biosimilars; (3) Each country in Latin America should establish its own working group, comprised of people with interest in biosimilars, to assist regulatory authorities in their efforts to develop and introduce biosimilars into their respective countries; (4) A dedicated portion on the PRAIS website to promote discussion on biosimilars, topics such as ongoing studies, and problems and issues of concern; (5) Countries in Latin America must enhance their efforts to improve pharmacovigilance; and (6) Products previously approved as ‘intended copy’ biological drugs should be evaluated according to regulations specific to biosimilars.
The final didactic presentation entitled ‘The challenges of nomenclature – INN, biosimilars and biological qualifiers’ summarizing the current, still evolving status of proposed methods to name SBPs was given by Dr James S Robertson, member and Rapporteur of the WHO INN Expert Committee. The International Nonproprietary Name (INN) is an important attribute of any medicinal compound. Whilst SBP developers are eager to establish an appropriate INN for their products, the WHO INN Expert Committee cannot take into consideration whether a product is an SBP or not, as this relates to a specific regulatory process and is outside the remit of the INN Expert Committee. Nevertheless, the use of Greek letter suffixes for glycosylated SBP has been debated for several years. A further issue arises when different regulatory jurisdictions develop their own distinct nomenclature system for SBPs. In order to establish a common global system, the WHO INN Programme has proposed the development of a Biological Qualifier (BQ) system which is proposed to apply to all biologicals, including SBPs. The BQ would not be part of the INN but would be distinct for each biological although its use would be voluntary. The draft format of the scheme being considered was described and discussed.
Parallel case study working sessions
The two fictional cases of a follow-on biological therapeutic, one a native protein product and the other a monoclonal antibody were explained to the participants. The data presented in the case studies were fictional. However, the cases were designed based on real scientific observations and were considered suitable to highlight important aspects in a biosimilarity evaluation. The quality attributes presented in the case studies covered only a subset of all quality attributes required to be assessed for biosimilarity. Participants were provided simplified graphical data that compared the preclinical testing performance of the follow-on products with that of the originator products and were then divided into four discussion groups. Groups 1 and 2 discussed how they would evaluate the data from the follow-on native protein product, and Groups 3 and 4 discussed the monoclonal antibody data.
Each group had a non-faculty moderator from Mexico who guided the discussions and collated the opinions for presentation to all participants in an open session that followed. Two of the sessions had simultaneous translations and two were conducted in Spanish only.
While there was some general agreement, there were individual and group differences of opinion as to how the two products should be handled, see Tables 2 and 3.
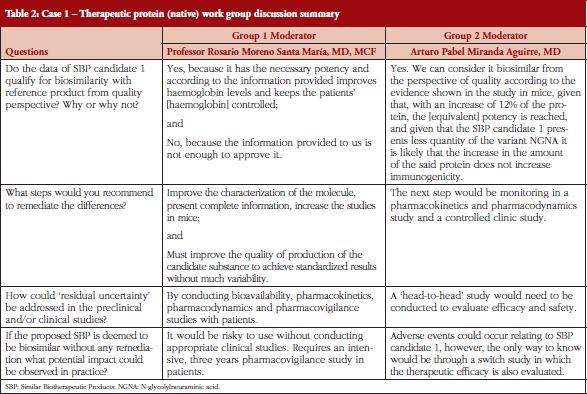
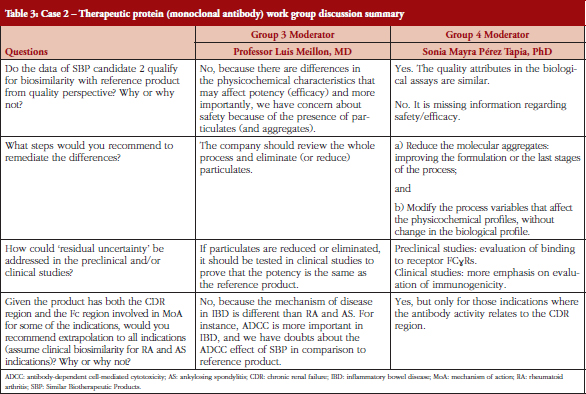
The lack of consensus as well as the questions and concerns raised suggested that there is a need for clear regulatory pathways to be developed, basic scientific and clinical data as well as training and education of regulators who implement these pathways are needed. It was also suggested that making applicable documents, e.g. from WHO, European Medicines Agency (EMA) and US Food and Drug Administration (FDA), more available and publishing the proceedings of this workshop might lead to greater international regulatory cooperation and more effective approval, use and monitoring of these products.
Discussion summary of the four work groups
Dr Thorpe briefly summarized the reports from the groups. The conclusion from Group 1 with the EPO study was that the product should not be approved as a biosimilar yet. Although the material does have appropriate potency and leads to increases in haemoglobin the group thought that more information was necessary. The information provided was incomplete, and more studies were needed with larger numbers of mice. If these were satisfactory it will then be necessary to conduct clinical studies in humans. It was also considered that the quality of the product may need improving.
Group 1 really took a regulatory view, whereas Group 2 took a more pragmatic view. The latter group thought that the quality evidence presented suggested that the product could be considered to be a biosimilar, but this required head-to-head PD and pharmacovigilance studies for confirmation of this.
Group 3 thought that the mAb product does not qualify as biosimilar mainly for two reasons; potential potency problems and even more importantly safety problems due to of the presence of particles and aggregates. The latter reflects the potential of the products to be immunogenic. The group also thought the manufacturing process needed modifying to remove or at least reduce aggregates and particles and this would be necessary before clinical trials could be conducted. Clinical studies will be needed to address potency issues and also probably to evaluate immunogenicity. It was considered that extrapolation is not possible because ADCC is important for inflammatory bowel disease indications, and this is lower in this product.
Group 4 was split between whether the mAb should be considered as a biosimilar or not. Some thought the data presented was deficient in various ways. As well as requiring more quality data they also needed more studies on Fc characterization, because the Fc is clearly important for this monoclonal in some of the indications. Concerning extrapolation, it was considered that the product can be used in all indications which have the same mode of action but not others.
Conclusion
A large and growing number of follow-on biotherapeutic products are either being evaluated by regulators for use or are already being used in Mexico and in other Latin American countries. However, approval of such products is difficult because even minor changes in production methods may have major but unpredictable impacts on their therapeutic and toxicologic profiles when used in patients. In addition, products can have varying performance characteristics in a number of currently available preclinical testing methods but it is not always clear how important these differences are in predicting clinical performance.
These facts make rational approval of these follow-on versions challenging to regulate and may explain why there was no consensus among the Mexican stakeholders who attended this workshop as to how two such biologicals are best regulated including how to best evaluate product composition, efficacy and safety used, or monitored after authorization (pharmacovigilance). In addition, there appeared to be gaps in the understanding of the properties of and the preclinical testing of these agents that need to be addressed before harmonized, uniform approaches to their approval can be developed. These gaps might be at least partially addressed by educational programmes such as the workshop described herein.
Speaker Faculty and Moderators
Thijs J Giezen, PharmD, PhD, The Netherlands
Gustavo Grampp, PhD, USA
Vladimir Hanes, MD, USA
James S Robertson, PhD, UK
Prapassorn Thanaphollert, BS, Thailand
Robin Thorpe, PhD, FRCPath, UK (Chair)
Meenu Wadhwa, PhD, UK
Professor Philip D Walson, MD, USA/Germany (Co-Chair)
Professor Luis Meillon, MD, Mexico
Arturo Pabel Miranda Aguirre, MD, Mexico
Professor Rosario Moreno Santa María, MD, MCF, Mexico
Sonia Mayra Pérez Tapia, PhD, Mexico
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank Commissioner Mikel Arriola Peñalosa, JP, MPP, Cofepris (Comisión Federal Para La Protección Contra Riesgos Sanitarios – Federal Commission for the Protection against Sanitary Risk), Department of Health, Mexico, and his colleagues (specifically Dra Daphny Trejo Núñez) for their strong support through the offering advice and information during the preparation of this educational workshop.
The authors would like to acknowledge the help of the workshop speaker faculty and participants, each of whom contributed to the success of the workshop and the content of this report as well as the support of the moderators: Professor Luis Meillon, Dr Arturo Pabel Miranda Aguirre, Professor Rosario Moreno Santa María, and Dr Sonia Mayra Pérez Tapia, in facilitating meaningful discussion during the parallel case study working sessions, and presented the discussion findings at the workshop.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Co-author
Robin Thorpe, PhD, FRCPath
Deputy Editor-in-Chief, GaBI Journal
References
1. Alvarez AA, Mysler E, Ruiz de Castilla EM, Flores-Murrieta FJ, Hughes J, Azevedo VF. Recommendations for the regulation of biosimilars and their implementation in Latin American. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):143-8. doi:10.5639/gabij.2014.0303.032
2. Pan American Health Organization. Regional platform on access and innovation for health technologies [homepage on the Internet]. [cited 2015 Aug 4]. Available from: http://prais.paho.org/rscpaho
3. Ebbers HC, Chamberlain P. Interchangeability. An insurmountable fifth hurdle? Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):88-93. doi:10.5639/gabij.2014.0302.022
|
Author for correspondence: Professor Philip D Walson, MD, Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


