|
Abstract: |
Submitted: 15 March 2016; Revised: 22 March 2016; Accepted: 24 March 2016; Published online first: 4 April 2016
Over the last decades we came to realize that high molecular weight drugs of biological origin, such as therapeutic antibodies, are of a highly complex structure, which determines their activity as well as safety in terms of immunogenicity. The manufacturing of such therapeutics requires highly complex and meticulously controlled up- and downstream processes. Still, the final drug product whose properties are determined by the process is characterized by a certain degree of ‘microheterogeneity’, i.e. the presence of several isomers of the active pharmaceutical ingredient (API).
‘Follow-on’ protein therapeutics will not be manufactured by the same process proprietary to the originator. As the process itself determines the properties, e.g. microheterogeneity, of the final drug product, an originator protein therapeutic and its follow-on product may not be considered to be identical. Hence, the generic principle applied for drugs of low molecular weight, whose properties and composition are well defined and can be reproduced by generic drug producers, cannot be applied to complex drugs such as therapeutic proteins. A regulatory strategy leading to marketing authorization of follow-on biologicals in Europe and by the US Food and Drug Administration (FDA) – the ‘biosimilar’ pathway – has thus been established and refined. This approach includes elements such as clinical testing of the follow-on against the originator product in clinical trials, as well as post-marketing surveillance [1].
Another group of complex drugs of non-biological origin (NBCDs – non-biological complex drugs) share aspects of complex structure, potential immunogenicity and impossibility of full characterization by physicochemical methods alone with biological complex drugs. Examples for these complex drugs include glatiramoids (Copaxone®), liposomal formulations (Doxil®) and nanoparticles such as iron-carbohydrate particles (Venofer®). These drugs are considered to belong to a new class of ‘nanomedicines’ [2]. Their size and attributes at the molecular scale confer these systems certain properties to interact with their biological environment.
Nanoscale drug delivery systems have been under investigation for several decades, yet only very few have actually matured to clinical application. While analytical techniques describing the physicochemical properties of these systems are being constantly refined, we had to understand that these systems need a multi-pronged analytical approach to describe their physicochemical properties. However, little is known on the relation between these properties and the clinical outcome, such that these systems always need clinical assessment to ensure efficacy and safety. The situation is rendered even more complex by the appearance of intended copies of NBCDs. Some of those, e.g. iron sucrose products (‘iron sucrose similars, ISSs’), and ‘generic’ Doxil (approved by FDA in 2013 in view of drug shortages in the US) have entered the market under the generics paradigm, partially due to the absence of a more suitable regulatory evaluation process. Therefore, an effort is needed to discover these correlations between nano-properties and biological activity, develop suitable analytical techniques and define specifications, establish clinical protocols and, last but not least, integrate this knowledge in a science-based regulatory approach to nanomedicines [3].
The principle of nanomedicines being complex drugs is well established within the scientific community. Regulatory authorities are also catching up, as shown by recent activities in the field. FDA is currently sponsoring a study comparing two iron carbohydrate products (Nulecit®, Ferrlecit®) for their similarity, involving physicochemical characterization, in vitro and clinical studies [4]. The European Medicines Agency (EMA) has published a reflection paper on the subject of ‘data requirements for iron-based nano-colloidal products’ in February 2015, which lists a range of characterization methods for these products.
In the context of ensuring the quality and safety of medicines, pharmacopoeias, as standard references for pharmaceutical drug specifications in the form of monographs, play a pivotal role. Derived from the Greek expressions φ ά ρ μ α κ ο ν (pharmakon) and π ο ι ΐ α (making), pharmacopoeias appeared as early as 50 AD (De Materia Media). In Great Britain, the national British Pharmacopoeia (BP) was published for the first time in 1864, the United States Pharmacopeia (USP) already in 1820. In addition to such national pharmacopoeias, international forms do exist as well and may replace national ones. The foundation for the European Pharmacopoeia (Ph. Eur.) was laid in 1964 by a convention of the Council of Europe, and the first volume of the International Pharmacopoeia (Ph. Int.) was published by the World Health Organization (WHO) in 1951.
Having the common goal to ensure access to good quality medicines, the organizational forms of these pharmacopoeias and the procedures followed to introduce new monographs differ largely. This manuscript gives an overview on such issues, and analyses the level of awareness for NBCDs at the European and US pharmacopoeias.
1. The European Pharmacopoeia (Ph. Eur.)
The European Directorate for the Quality of Medicines and Healthcare (EDQM) is a directorate of the Council of Europe (CoE), whose function was first defined by the ‘Convention on the elaboration of a European Pharmacopoeia’ [5] signed in 1964. These activities had the goal to define common specifications for medicinal substances and their pharmaceutical preparations, as well as to promote free movement of medicines in the European Union (EU) and the countries of the European Economic Area (EEA). In 1975, a directive by the EC makes compliance with the European Pharmacopoeia Monographs mandatory when requesting marketing authorization for medicines for human use. In 1994, the CoE signed the Ph. Eur. Convention, and the European Network for Official Medicines Control Laboratories (OMCL) was created [6], and its activities organized by EDQM from 1996 onwards. In 2001, EC Directive 2001/83/EC [7] maintains the mandatory character of Ph. Eur. monographs when requesting marketing authorization, superseding all previous directives.
With the task of protecting public health by applying one common compulsory standard in its Member States, the Ph. Eur. is the official pharmacopoeia and legally binding in 37 Member States and the EU. It is complemented by national pharmacopoeias for specific monographs of interest to only one Member State. Ph. Eur. today contains legally binding quality standards for active substances (organic, inorganic), excipients, substances of biological origin and biotechnology, herbal drugs, essential oils and fats, preparations, radiopharmaceuticals, vaccines, sera (human, veterinary), blood derivatives, and homeopathic preparations. These monographs are supplemented by general monographs on dosage forms as well as general texts on quality issues and standard analytical methods. In total, nearly 2,200 monographs currently exist. The first finished product monograph containing a chemically defined active substance (sitagliptin tablets used in type 2 diabetes) was adopted by the Ph. Eur. commission only as recent as March 2015. A second finished product, rosuvastatin tablets, was also added for monograph elaboration to the work programme, taking into account products from multiple manufacturers. In 2016, the EDQM will publish the ninth Edition of the European Pharmacopoeia, which will contain nearly 3,000 monographs and general texts and will become legally binding by 1 January 2017 [8].
The Ph. Eur. Commission (COM) consists of one delegation per each of the 37 members, a delegation from the EU consisting of a representative from Directorate General Health & Consumer Affairs and the EMA, and observers from 22 countries and WHO. Delegates come from health ministries, health authorities, pharmacopoeias, universities, or industry and are appointed by the national authorities on the basis of their expertise. There are currently 20 permanent Groups of Experts and 52 ad-hoc Working Parties dedicated to revising texts that are to be adopted by the COM which also decides on the composition of these groups.
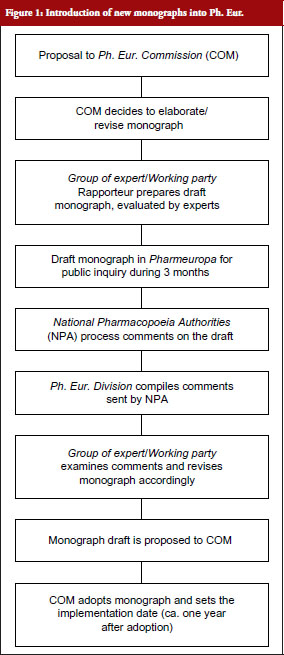
The procedure by which new texts are introduced into Ph. Eur. is depicted in Figure 1.
On submission of a proposal, typically by a national authority of a Member State, the COM decides to have the new monograph elaborated by a group of experts or working party. A draft elaborated by this group is submitted to Pharmeuropa [9], the free online publication of the EDQM for review and comments by interested parties for the duration of three months. The National Pharmacopoeia Authorities (NPA) of the Member States process the received comments on the draft monograph, which are then compiled and resubmitted to the group of experts or working party. On adaptation of the draft according to the comments received, the monograph is proposed to COM, which adopts it and sets the date of implementation into Ph. Eur., where the monograph is published about six months later.
At EDQM, the Non-Biological Complexes (NBC) Working Party was created in 2011 following an initiative of SwissMedic, the Swiss agency responsible for the authorization and supervision of therapeutic products (medicinal products and medical devices). The COM decided to add to its work programme the elaboration of a monograph on Iron Sucrose Concentrated Solution as a first example for NBCDs. Iron sucrose (IS) and its follow-on products, so-called iron sucrose similars (ISSs) products consist of a polynuclear iron core of iron(III)-oxyhydroxide, which is stabilized by a complex coating of sucrose. IS products have been used in parenteral replacement therapy of anaemia, e.g. in chronic kidney disease (CKD) patients on dialysis to stimulate the generation of erythrocytes. ISSs in Europe received marketing authorization through the generic drug pathway, suggesting interchangeability. Clinical reports such as reported in [10], however, have recently shown that IS and ISSs may not have the same or similar efficacy and safety profile, and thus appear not to be interchangeable. A regulatory pathway, comparable to the one applied for biosimilars, is therefore currently discussed as mentioned above.
The working party, consisting of experts from academia, manufacturers and regulatory authorities, is currently in the process of defining, establishing and validating assays for the characterization of iron sucrose concentrated solution to be included in the monograph. The methods considered to be included comprise assays to assess particle size by size exclusion chromatography and differential laser light scattering. The latter will also contribute to the development of a general method on size measurement of nanoparticles currently under discussion. In addition, assays for the measurement of labile iron released from the particles are being developed, as this is recognized as an important parameter for the safety of IS products.
In drafting the monograph, the working party takes into account methods described in existing monographs on Iron Sucrose Injection in the BP and USP. The working party has also participated in commenting on the draft of the EMA reflection paper on the subject of ‘data requirements for iron-based nano-colloidal products’.
2. The United States Pharmacopeial Convention (USP)
The USP was founded in 1820 in attendance of delegates of medical societies from several states, creating a list of standards and a national formulary. In 1850, colleges were invited to contribute to the revision of the USP, and in 1888, the first National Formulary (NF) was introduced by the American Pharmacists Association (APhA). USP was incorporated in 1900 in its present form as a not-for-profit organization; it is thus not a governmental organization like the Ph. Eur. The USP and NF standards for strength, quality, purity, packaging and labelling are recognized as official and enforced by FDA from 1938 onwards. In 1977, the scopes of USP and NF are redefined such that standards for drug substances and dosage forms are specified in USP, whereas excipient standards are included in NF. From 1980 onwards, both USP and NF are published under one cover. The USP-NF is also translated into Chinese and Russian. While being a national organization, USP has developed an international presence with offices and laboratory facilities in Hyderabad (India), Shanghai (China) and São Paulo (Brazil). Along with these facilities, USP also runs an office in Basel (Switzerland).
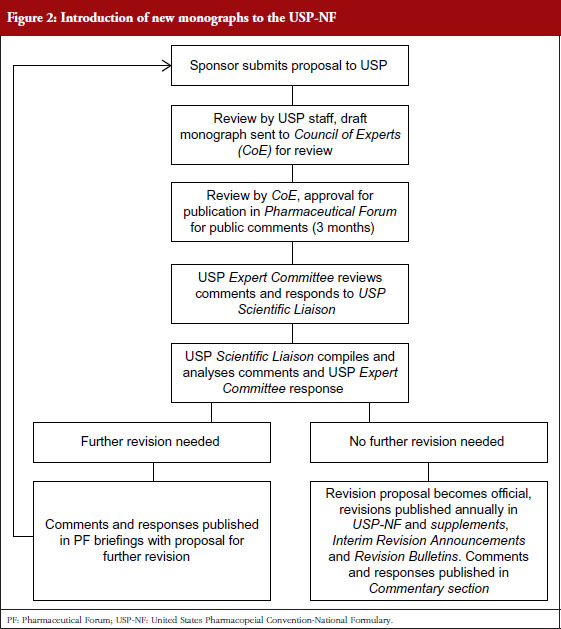
The procedure by which a USP monograph is developed is shown in Figure 2. In contrast to Ph. Eur., where the process is initiated by the submission of a proposal by a national authority, the USP process is driven by a manufacturer of an originator or generic drug sponsoring the proposal. Together with the proposal, the sponsor supplies USP with information on the drug’s specifications, relevant assays and test methods. The proposal and data submitted is reviewed by scientific staff of USP, who will also perform laboratory testing. A draft monograph is then submitted to the Council of Experts and one of its Expert Committees for scientific opinion and merits of the proposed monograph. The Council of Experts then decides to publish the draft monograph for a three-month public review at Pharmaceutical Forum (PF), the online peer-reviewed journal of USP [11].
Successively, the comments received are reviewed by the Expert Committee and integrated into the monograph draft. A USP liaison (scientific officer) compiles and analyses these comments and the draft, and submits either for further revision or for publication in USP-NF. The draft thereby becomes an official monograph, the specifications stated in the standard must be abided to when marketing the product within the US.
Besides the development and publication of monographs for prescription and over-the-counter (OTC) drugs (a relatively new activity at USP), the preparation of Reference Standards is a major activity and source of income for USP. The more than 3,500 USP Reference Standards are used in the examination of the identity, strength, quality and purity of drugs, biologicals and excipients, thus contributing to the quality of drug manufacturing. The USP Council of Experts is closely interacting with experts from industry and regulatory agencies. Until 2015, USP has made available a compendium of public standards (Medicines Compendium, MC) for approved medicines on a global basis. The MC monographs were freely available online, providing performance tests for critical quality attributes and acceptance criteria, a source-independent Reference Procedure and one or more Acceptable Procedures. The website was discontinued and shall be replaced in the future by a ‘collaborative programme’ [12].
Concerning activities in the area of NBCDs, and in addition to a published monograph on Iron Sucrose Injection as mentioned above, USP has engaged in installing an Expert Panel on glatiramers with a call for candidates published on 15 December 2015. Glatiramers (or glatiramoids) are complex drugs consisting of a large number of synthetic polypeptides of four amino acids (glutamic acid, alanine, lysine and tyrosine). Based on studies at the Weizmann Institute in the 1960s and early 1970s, which were aimed at developing experimental autoimmune encephalitis (EAE) in guinea pigs as a model for multiple sclerosis (MS), glatiramoids were shown to protect against EAE. Successively, the first glatiramoid drug for relapsing forms of MS (Copaxone®) was developed and marketed by Teva Pharmaceuticals (Teva).
As is the case for biological complex drugs, the inherent complexity of glatiramoids requires a very well controlled manufacturing process (‘the process is the product’). In fact, by slight alteration of the manufacturing process, Teva had produced a glatiramoid (TV-5010) that was similar in terms of amino acid ratio and physical properties [13] to Copaxone®. While good safety and tolerability in patients treated with TV-5010 was shown, long-term toxicity studies in rats and monkeys revealed the occurrence of fibrosis in rats and eosinophilia in monkeys. Studies comparing gene expression profiles of Copaxone® to purported ‘generic’ glatiramoids, e.g. Glatimer® (India), Probioglat® (Mexico) and Escadra® (Argentina) revealed essential differences between the originator and the follow-on drugs [14,15]. It was therefore suggested by Teva that follow-on glatiramoids should not receive marketing authorization by the generic drug pathway. However, as stated in a Denial Letter [16] to a Citizen’s Petition Letter by Teva, FDA insisted that sameness between Copaxone® and a generic glatiramoid, Momenta/Sandoz’s Glatopa®, can be demonstrated by showing equivalence in the fundamental reaction scheme, the drug’s physicochemical properties including composition, structural signatures for polymerization and depolymerization and results in a relevant biological assay. On 16 April 2015, FDA approved Glatopa® [17] as a first generic glatiramoid for the treatment of relapsing MS. Based on these discussions, FDA may issue the draft of a guidance document on glatiramoids in the future.
USP is now in the process of establishing an expert panel to discuss the draft of a monograph on glatiramer acetate. The panel will most likely include experts from academia, regulatory authorities and manufacturers. Major issues to be addressed may include the definition of critical quality attributes for glatiramoids, as well as whether bioassays should be included in the monograph to show activity and safety.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) was launched in 1990 at Brussels headquarters of EFPIA (European Federation of Pharmaceutical Industries and Associations) in the presence of representatives from regulatory agencies of Europe, Japan and the US, and from industry. The mission of ICH is to achieve greater harmonization in regulatory requirements for pharmaceutical product registration [18]. Such harmonization would result in the avoidance of the repetition of studies in the research and development of new human medicines. During the last 25 years, ICH has established harmonized technical requirements for the quality, safety and efficacy of new medicines in EU, Japan and the US.
ICH is governed by its Steering Committee (SC), which is composed of representatives from the European Commission, EFPIA, the Japanese Ministry of Health, Labour and Welfare (MHLW), the Japan Pharmaceutical Manufacturers Association (JPMA), FDA, Pharmaceutical Research and Manufacturers of America (PhRMA), Swissmedic, and Health Canada. WHO is an Observer to the SC, and the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) participates as a non-voting member of the SC. The involvement of EDQM in ICH activities is given through the SC membership of the EC.
The ICH SC has several activities. It has established a structure to develop the Medical Dictionary for Regulatory Activities (MedDRA) to facilitate sharing of regulatory information internationally for human medicines (pharmaceuticals, biologicals, vaccines and drug-device combination products). MedDRA is available for use in registration, documentation and safety monitoring of medical products both before and after marketing authorization.
SC has appointed Working Groups to review the differences in requirements between EU, Japan and the US and develop the scientific consensus required to reconcile those differences. The four types of ICH working groups include Expert Working Groups (EWG) charged with developing a harmonized guideline, Implementation Working Group that develops Q & As to facilitate implementation of existing guidelines, Informal Working Groups having the objective of developing/finalizing a Concept Paper, as well as developing a business plan for harmonization activities, and Discussion Groups dedicated to discuss specific scientific considerations or views. ICH convenes twice a year with the location of meetings rotating among Europe, Japan and the US. The five-step harmonization process is initiated by endorsement of a concept paper and business plan by the SC [19].
Several ICH guidelines are suggested for stability testing of nanomedicines [20], e.g. guideline for stability testing of non-targeted nanomedicines (Q1A (R2)) [21], new dosage forms (Q1C) [22] and biotechnology-based drug products (Q5C) [23] for targeted nanomedicines including a biological targeting moiety such as an antibody. Stability studies of liposomal formulations following ICH guidelines have been reported in literature [24]. A range of methods for testing nanotoxicology has recently been reviewed [25]. It has been suggested that the ICH-S6 guideline [26] may be applied for non-targeted nanoparticles, whereas the ICH-S8 guideline [27] may be applied for the assessment of toxicity of targeted nanoparticles conjugated to biological targeting moieties.
The Pharmacopoeial Discussion Group (PDG) [28] was formed in 1989 with representatives of EDQM, MHLW representing the Japanese Pharmacopoeia (JP), and the USP. WHO has PDG observer status since May 2001. PDG generally meets twice a year. Main activities of PDG are retrospective harmonization of general chapters and excipients monographs of the major pharmacopoeias in the three regions, serving also the mission of ICH. In some cases full harmonization of an entire monograph may not be possible. In such case, only harmonizable elements of the monograph are harmonized (harmonization by attribute). Such monograph for a drug harmonized by attribute, interchangeability is compiled only with respect to the harmonized elements, whereas for non-harmonized attributes, compliance with the individual pharmacopoeial requirements in each region is necessary. From 2003 to 2010, the ICH SC nominated an EWG with members from the three pharmacopoeias to discuss several general test chapters in the respective pharmacopoeias in connection with the ICH Q6 A [29] guideline.
The Q6A guideline has the purpose to help select common test procedures and acceptance criteria for new drug substances of chemical origin and new drug products not previously registered in the EU, Japan and the US. The guideline does not apply to the clinical test phase, but only to the marketing approval process of new drug products and combinations thereof. The guideline specifically mentions drugs such as low molecular weight peptides and (semi)synthetic antibiotics. Not covered, however, are radiopharmaceuticals and oligonucleotide-based drugs.
Increasing awareness of the intricacies of NBCDs in the science and regulatory communities was bound to have an effect on drafting of pharmacopoeial content in the form of monographs and general guidelines. Already existing content should be reviewed and revised according to the changing understanding of these complex drugs. International harmonization of pharmacopoeial contents to maintain drug quality, efficacy and safety in the three major areas will be possible through the networks (ICH, PDG) in place today.
Competing interests: The author declares that he is a member of the steering committee of the NBCD Working Group, hosted at the non-profit organization Lygature – formerly known as Top Institute Pharma, Leiden, The Netherlands (www.lygature.org/nbcd).
Provenance and peer review: Commissioned; externally peer reviewed.
References
1. European Medicines Agency. Guidelines on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev1. 22 February 2006 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
2. Duncan R, Gaspar R. Nanomedicines under the microscope. Mol Pharm. 2011;8(6):2101-41.
3. Borchard G, Flühmann B, Mühlebach S. Nanoparticle iron medicinal products – requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studies. Regul Toxicol Pharmacol. 2012;64(2):324-8.
4. U.S. Food and Drug Administration. Evaluate the therapeutic equivalence of generic sodium ferric gluconate iron complex Nulecit™ and its RLD Ferrlecit®. 16 Jul 2013 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: https://www.fbo.gov/spg/HHS/FDA/DCASC/FDA-SOL-1120929/listing.html
5. Council of Europe. Chart of signatures and ratifications of Treaty 050 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.coe.int/en/web/conventions/full-list/-/conventions/treaty/050
6. European Directorate for the Quality of Medicines & Healthcare. General European OMCL Network (GEON) [homepage on the Internet]. [cited 2016 Mar 22]. Available from: https://www.edqm.eu/en/General-european-OMCL-network-46.html
7. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. Eur-Lex. L-311, 28.11.2001, pp. 67-128.
8. European Directorate for the Quality of Medicines & Healthcare. Join the Ph. Eur. network! CALL FOR EXPERTS: Appointment of experts to prepare for the 10th Edition. 17 February 2016 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: https://www.edqm.eu/en/news/join-ph-eur-network-call-experts-appointment-experts-prepare-10th-edition
9. European Directorate for the Quality of Medicines & Healthcare. Pharmeuropa, Pharmeuropa Bio & Scientific Notes [homepage on the Internet]. [cited 2016 Mar 22]. Available from: https://www.edqm.eu/en/pharmeuropa-bio-and-scientific-notes-584.html
10. Rottembourg J, Kadri A, Leonard E, Dansaert A, Lafuma A. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10): 3262-7.
11. U.S. Pharmacopeial Convention. Pharmacopeial Forum (PF) [homepage on the Internet]. [cited 2016 Mar 22]. Available from: www.usp.org/usp-nf/pharmacopeial-forum
12. U.S. Pharmacopeial Convention. Medicines Compendium [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.usp.org/global/medicines-compendium
13. Ramot Y, Rosenstock M, Klinger E, Bursztyn D, Nyska A, Shinar DM. Comparative long-term preclinical safety evaluation of two glatiramoid compounds (glatiramer Acetate, Copaxone(R), and TV-5010, protiramer) in rats and monkeys. Toxicol Pathol. 2012;40(1):40-54.
14. Conner J. Glatiramer acetate and therapeutic peptide vaccines for multiple sclerosis. J Autoimmun Cell Responses. 2014.
15. Towfic F, Funt JM, Fowler KD, Bakshi S, Blaugrund E, Artyomov MN, et al. Comparing the biological impact of glatiramer acetate with the biological impact of a generic. PLOS One. 2014;9(1):e83757.
16. U.S. Food and Drug administration Docket No. FDA-2015-P-1050 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.fda.gov/RegulatoryInformation/Dockets/default.htm
17. U.S. Food and Drug administration. FDA approves first generic Copaxone to treat multiple sclerosis. 16 April 2015 [homepage on the Internet]. [cited 2016 Mar 22]. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm443143.htm
18. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use [homepage on the Internet]. [cited 2016 Mar 22]. Available from: www.ich.org
19. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Formal ICH procedure [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ich.org/about/process-of-harmonisation/formalproc.html#step-1
20. Muthu MS, Feng SS. Pharmaceutical stability aspects of nanomedicines. Nanomedicine (Lond). 2009;4(8):857-60.
21. European Medicines Agency. ICH Topic Q 1 A (R2). Stability testing of new drug substances and products. CPMP/ICH/2736/99. August 2003 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002651.pdf
22. European Medicines Agency. ICH Topic Q 1 C. Stability testing for new dosage forms. CPMP/ICH/280/95. January 1998 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC50
23. European Medicines Agency. ICH Q 5 C. Quality of biotechnological products: stability testing of biotechnological/biological products. CPMP/ICH/138/95. July 1996 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002803.pdf
24. Vorauer-Uhl K, Wagner A, Katinger H. Long term stability of rh-Cu/Zn-superoxide dismutase (SOD)-liposomes prepared by the cross-flow injection technique following International Conference on Harmonisation (ICH) guidelines. Eur J Pharm Biopharm. 2002;54:83-7.
25. Dobrovolskaia MA, Germolec DR, Weaver JI. Evaluation of nanoparticle immunotoxicity. Nature Nanotechnol. 2009;4:411-4.
26. ICH guideline S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. EMA/CHMP/ICH/731268/1998 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
27. European Medicines Agency. ICH Topic S 8. Immunotoxicity studies for human pharmaceuticals. EMEA/CHMP/167235/2004. May 2006 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002851.pdf
28. European Directorate for the Quality of Medicines & Healthcare. Working procedures of the Pharmacopoeial Discussion Group (PDG) [homepage on the Internet]. [cited 2016 Mar 22]. Available from: https://www.edqm.eu/medias/fichiers/Working_Procedures_of_the_PDG.pdf
29. European Medicines Agency. ICH Topic Q 6 A. Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances. CPMP/ICH/367/96. May 2000 [homepage on the Internet]. [cited 2016 Mar 22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002823.pdf
|
Author: Professor Gerrit Borchard, PharmD, PhD, Professor, Biopharmaceutical Sciences, School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, 30, quai Ernest-Ansermet, CH-1211 Geneva 4, Switzerland |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/complexity-in-the-making-non-biological-complex-drugs-nbcds-and-the-pharmacopoeias.html
Author byline as per print journal: Jon SB de Vlieger, PhD; Professor Stefan Mühlebach, PhD; Vinod P Shah, PhD; Scott E McNeil, PhD; Professor Gerrit Borchard, PharmD, PhD; Vera Weinstein, PhD; Beat Flühmann, PhD; Sesha Neervannan, PhD; Professor Daan JA Crommelin, PhD
|
Abstract: |
Submitted: 16 September 2015; Revised: 15 October 2015; Accepted: 19 October 2015; Published online first: 2 November 2015
The concept of non-biological complex drug (NBCD) products has been presented and discussed on several occasions in the GaBI Journal [1–4]. The growing interest in this topic in academic, industrial and regulatory circles led to the establishment of an Editorial Section on Non-Biological Complex Drugs in both the GaBI Journal and GaBI Online starting in the third quarter of 2015.
In 2011, the first publication on NBCDs appeared [5] as a result of a workshop held in Leiden, The Netherlands, in 2009. For the first time this class of drug products was identified and recognized. These products are more complex than small, low molecular drugs and as complex or even more complex than biologicals; sharing many of the characteristics of the latter category but not being derived from living sources. As a consequence, the authors argued that for the equivalence testing of NBCD follow-on products a regu latory pathway should be developed that was similar to the pathways developed by the European Medicines Agency (EMA) and later the US Food and Drug Administration (FDA) for approving biosimilars. This proposal was backed up by evidence provided by a (still growing) number of published studies on NBCD follow-on versions that were authorized using a ‘standard’ but inadequate generic assessment protocol.
A working group hosted by the Dutch Top Institute (TI) Pharma in Leiden was set up to raise awareness of the challenges NBCD products present worldwide, to stimulate the publication of scientific reports and discussions and to provide a rigorous, sciencebased, regulatory policy for NBCD products [6]. These efforts led to a number of publications and a book exclusively dedicated to the NBCD concept and its regulation. These publications provide a definition for NBCD products, discuss different classes of NBCDs, propose an overarching regulatory philosophy for evaluating NBCD follow-on versions and, finally, outline what issues are still unresolved [7–9].
This paper to the GaBI Journal further explains:
A definition of NBCD products that was published earlier by the NBCD working group reads: ‘A Non-Biological Complex Drug is a medicinal product, not being a biological medicine, where the active substance is not a homo-molecular structure, but consists of different (closely related and often nanoparticulate) structures that can’t be isolated and fully quantitated, characterized and/or described by stateof-the-art (physico)chemical analytical means and where the clinical meaning of the observed differences is not known. The composition, quality and in vivo performance of NBCDs are highly dependent on manufacturing processes of both the active ingredient as well as in most cases the formulation’ [7].
Present product ‘families’ and beyond
At present the following NBCD product groups or ‘families’ have been identified and discussed in the literature: liposomes, polymeric micelles, glatiramoids, iron-carbohydrate complexes, albuminanticancer drug nanoparticles and nanocrystals. The rapidly growing group of nanomedicines will add many NBCDs to this list [9–10]. Interestingly, there are also medicinal products such as the low molecular weight heparins (LMWHs) showing similar complexity but falling under different regulatory policies, i.e. by EMA LMWHs are seen as biologicals and by FDA as non-biologicals.
Over the last few years a number of studies on these NBCD product families have been published. This list is growing and expanding beyond only parenteral drugs. Recently, the present science base, including the: (1) chemistry and structure; (2) manufacturing; (3) (physico)chemical characterization; (4) pharmacology; and (5) regulatory status, of these product groups was reviewed in a book [9]. The availability of these data in the public domain should contribute to sciencebased discussions and could serve as a model to be followed for consideration of other NBCD product families. In addition, questions regarding interchangeability and substitutability of NBCD follow-on versions have important implications for the handling of such medicinal products by healthcare professionals. Requests are being made by these healthcare professionals for education on the topic of NBCDs and for further, tailor-made, reliable information for patients.
Other candidate NBCD product families that are waiting to be identified include emulsions (parenteral or ocular), dry powder inhalers and oral bioactive polymers such as complex phosphate binders.
The regulatory landscape
In earlier publications in GaBI Online the differences between the characteristics of small, low molecular weight molecules and biologicals were listed, see Table 1 [11]. If the items in the ‘biological drugs’ column (in italics) that relate to the biological source of the product and immunogenicity are removed, what remains demonstrates that there is a striking resemblance between the characteristics of biologicals and NBCD products.
Because of this similarity in product characteristics, the NBCD Working Group has proposed on several occasions that regulators should follow the same regulatory pathway for NBCD follow-on products as for biosimilars. This proposal is schematically shown in Figure 1 where ‘Totality of evidence’ is the key phrase when assessing therapeutic equivalence of NBCD innovator and follow-on products.
Neither FDA nor EMA uses special NBCD regulatory schemes. These agencies use existing pathways for the introduction of innovative and follow-on NBCD products. FDA uses the 505(b)(1)/505(b)(2) and 505(j) pathways for innovator products and follow-on versions, respectively. However, both FDA and EMA are paying increasing attention to regulatory issues related to NBCD families [12–13].
Generally speaking, there are two clearly distinct regulatory documents for these NBCD product families. On the one hand, FDA and/or EMA published ‘draft guidance’ and/or ‘reflection papers’ on new products such as liposomes [14], polymeric micelles [15], and surface coatings [16]. On the other hand, both agencies issued documents on the development of follow-on versions of NBCD products such as EMA documents on iron-based nano-colloidal products [17–19] and on existing products [20], and FDA on iron complexes [21], ciclosporin emulsions [22] and on liposome follow-on products [23–24]. Interestingly, FDA has awarded funding to characterize and clinically compare originator and follow-on versions after approving these products [25–26]. It is clear that the present arsenal of regulatory documents from these agencies will be expanded in the years to come. Hopefully, it is not too late to reach global agreement through the World Health Organization (WHO) or perhaps the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) initiatives. On the pharmacopoeial side, interest is growing in dealing with NBCD families. For example, a European Directorate for the Quality of Medicines and HealthCare (EDQM) working group is developing a monograph on ‘iron sucrose concentrated solution’ as an example for non-biological complexes and the United States Pharmacopeia (USP) is currently evaluating similar actions [27]. The British Pharmacopoeia (BP) has published a new monograph on iron sucrose injections, which is basically in line with the USP existing monograph.
One interesting feature of the European legal landscape is that certain NBCD product families are not approved through the centralized procedure (EMA). Instead, approval follows either the purely national procedures or the mutual recognition procedure under the aegis of national competent authorities. This is true for iron sucrose products and for oral bioactive polymer phosphate binders [28]. Considering the complexity of these products and the problems encountered in certain countries with generic versions of NBCD products (see below), approval thro ugh the centralized procedure and the CHMP team would be preferred.
For all NBCD product families where follow-on versions are on the market, a growing number of studies have become available in the public domain demonstrating examples of follow-on products that were approved by (national) competent authorities that differed structurally and/or in clinical practice from the originator products [9]. Often these differences were clinically relevant. Examples include publications by Rottembourg et al. [29]; Martin-Malo et al. [30]; Stein et al. [31]; Lee et al. [32] and Agüera et al. [33] for iron sucrose similars; and Weinstein et al. [34] and Towfic et al. [35] for glatiramer acetate follow-on products. These (clinical) examples raise questions to the regulatory science community in the countries mentioned in these publications about how appropriate the current systems are in ensuring equivalence in NBCD product quality, efficacy and safety. Is the current approach rigorous enough?
These examples provide lessons that should be communicated throughout the scientific community as well as to medical/pharmaceutical practitioners. More over, in the years to come, study of these examples can also help competent authorities to establish appropriate, science-based approval procedures for these complex drug products.
Expanding: The number of NBCD product families will continue to grow. It is time to pay attention to these (new) families and discuss their specific characteristics and their implications for the regulatory process at an early stage. Much information is often already available but there is a need to go through the archives and analyse these data so that scientific deficiencies are brought to the forefront.
Outstanding issues: for a list of outstanding issues concerning NBCD products one can refer to the list drawn up for biologicals. For example, labelling, comparability and attribute drift, NBCD-questionables (cf. bio-questionables [36]), extrapolation [37], interchangeability, substitution, and last but not least: a single global approach (WHO in the lead?) [38].
Facts please: For a fact-based debate on NBCD products we need to stimulate publications in the public domain. This will strengthen the science base for decisionmaking. Transparency of the regulatory process is another essential element for such a discussion. We hope that all parties (academic, industry and regulatory) involved in this debate will continue, and even step up their efforts to provide this science base.
Goal: We feel that the GaBI Journal and the GaBI Online platform offer excellent opportunities to stimulate awareness around the critical issues related to both new, innovative NBCD products and the introduction of follow-on versions. The growing science base for NBCD product legislation, e.g. in Europe and the US, and hopefully in other countries in the future, needs a non-biased publication outlet. With the GaBI publication platform we have the ambition to become the central and preferred publication hotspot for this complex topic.
Jon SB de Vlieger, PhD
Dutch Top Institute Pharma, PO Box 142, NL-2300 AC Leiden, The Netherlands
Professor Stefan Mühlebach, PhD
Vifor Pharma Ltd, 61 Flughofstrasse, PO Box, CH-8152 Glattbrugg, Switzerland; Department of Pharmaceutical Sciences, Pharmacenter, University of Basel, 50 Klingelbergstrasse, CH-4056 Basel, Switzerland
Vinod P Shah, PhD
NBCD Steering Committee member, Pharmaceutical Consultant, 11309 Dunleith Place, North Potomac, MD 20878, USA
Scott E McNeil, PhD
Director, Nanotechnology Characterization Laboratory, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research Inc, PO Box B, Frederick, MD 21702-1201, USA
Professor Gerrit Borchard, PharmD, PhD
Professor, Biopharmaceutical Sciences, School of Pharmaceutical Sciences, University of Geneva, University of Lausanne 30, Quai Ernest-Ansermet, CH-1211 Geneva 4, Switzerland
Vera Weinstein, PhD
Teva Pharmaceutical Industries Ltd, Discovery and Product Development, Global Research and Development, Netanya, Israel
Beat Flühmann, PhD
Vifor Fresenius Medical Care Renal Pharma Ltd, 37 Rechenstrasse, PO Box, CH-9001 St Gallen, Switzerland
Sesha Neervannan, PhD
Senior Vice President, Pharmaceutical Development Brands R & D, Allergan Plc, RD2-3A, 2525 Dupont Drive, Irvine, CA 92612, USA
Emeritus Professor Daan JA Crommelin, PhD
Department of Pharmaceutical Sciences, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, The Netherlands
Competing interest: All authors are members of the steering committee of the Non-Biological Complex Drug (NBCD) Working Group, hosted at the Dutch Top Institute Pharma (TI Pharma), Leiden, The Netherlands (http://www.tipharma.com/NBCD).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Mühlebach S, et al. The authorization of nonbiological complex drugs (NBCDs) follow-on versions: specific regulatory and interchangeability rules ahead? Generics and Biosimilars Initiative Journal (GaBI) Journal. 2013;2(4):204-7. doi:10.5639/gabij.2013.0204.054
2. Borchard G. Complex molecules – current developments. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):54-5. doi:10.5639/gabij.2014.0302.016
3. Walson PD, Mühlebach S, Flühmann B. First Asia-Pacific educational workshop on non-biological complex drugs (NBCDs), Kuala Lumpur, Malaysia, 8 October 2013. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):30-3. doi:10.5639/gabij.2014.0301.010
4. Nicholas MJ. Clinical development, immunogenicity, and interchangeability of follow-on complex drugs. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):71-8. doi:10.5639/gabij.2014.0302.020
5. Schellekens H, Klinger E, Mühlenbach S, Brin JF, Storm G, Crommelin DJ. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
6. TI Pharma. Non Biological Complex Drugs Working Group [homepage on the Internet]. 2015 [cited 2015 Oct 15]. Available from: http://www.tipharma.com/nbcd
7. Crommelin DJ, de Vlieger JS, Weinstein V, Mühlebach S, Shah VP, Schellekens H. Different pharmaceutical products need similar terminology. AAPS J. 2014;16(1):11-4.
8. Schellekens H, et al. How to regulate non-biological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2014;16(1):15-21.
9. Crommelin DJA, de Vlieger, JSB. Non-biological complex drugs. The science and the regulatory landscape. Advances in the Pharmaceutical Sciences series. NY: AAPS/Springer; 2015.
10. Mühlebach S, Borchard G, Yildiz S. Regulatory challenges and approaches to characterize nanomedicines and their follow-on similars. Nanomedicine (Lond) 2015; 10(4):659-74.
11. GaBI Online – Generics and Biosimilars Initiative. Small molecule versus biological drugs [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Oct 15]. Available from: www.gabionline.net/Biosimilars/Research/Small-molecule-versus-biological-drugs
12. Ehmann F, et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine (Lond). 2013;8(5): 849-56.
13. Spicher K, et al. Differences in tissue distribution of iron from various clinically used intravenous iron complexes in fetal avian heart and liver. Regul Toxicol Pharmacol. 2015;73(1):65-72.
14. U.S. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Guidance for industry. Liposome drug products chemistry, manufacturing, and controls; human pharmacokinetics and bioavailability; and labeling documentation. Draft guidance. 2002 [homepage on the Internet]. 2002 Jul 29 [cited 2015 Oct 15]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070570.pdf
15. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products. 17 January 2013 [homepage on the Internet]. 2013 Jan 23 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/02/WC500138390.pdf
16. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Reflection paper on surface coatings: general issues for consideration regarding parenteral administration of coated nanomedicine products. 22 May 2013 [homepage on the Internet]. 2013 Jul 19 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/08/WC500147874.pdf
17. European Medicines Agency. Committee for Human Medicinal Products (CHMP). Reflection paper on non-clinical studies for generic nanoparticle iron medicinal product applications. 17 March 2011 [homepage on the Internet]. 2011 Apr 1 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/04/WC500105048.pdf
18. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Overview of comments received on reflection paper on the data requirements for intravenous iron-based nano-colloidal products developed with reference to an innovator medicinal product. 26 March 2015 [homepage on the Internet]. 2015 Mar 26 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/ document_library/ Overview_of_comments/2015/03/WC500184921.pdf
19. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Reflection paper on the data requirements for intravenous iron-based nano-colloidal products developed with reference to an innovator medicinal product. 26 March 2015 [homepage on the Internet]. 2015 Mar 26 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/03/WC500184922.pdf
20. European Medicines Agency. Assessment report for: iron containing intravenous (IV) medicinal products. 13 September 2013 [homepage on the Internet]. 2013 Sep 27 [cited 2015 Oct 15]. Available from: www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/IV_iron_31/WC500150771.pdf
21. U.S. Food and Drug Administration. Draft guidance on iron sucrose [homepage on the Internet]. 2013 Nov 1 [cited 2015 Oct 15]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm297630.pdf
22. U.S. Food and Drug Administration. Draft guidance of cyclosporine [homepage on the Internet]. 2013 Jun 20 [cited 2015 Oct 15]. Available from http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358114.pdf
23. U.S. Food and Drug Administration. Draft guidance on doxorubicin hydrochloride [homepage on the Internet]. 2014 Dec 12 [cited 2015 Oct 15]. Available from http://www.fda.gov/downloads/Drugs/…/Guidances/UCM199635.pdf
24. European Medicines Agency. Committee for Human Medicinal Products (CHMP). Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal products. 21 February 2013 [homepage on the Internet]. 2013 Mar 13 [cited 2015 Oct 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf
25. Federal Business Opportunities. Therapeutic equivalence of generic iron complex product [homepage on the Internet]. [cited 2015 Oct 15]. Available from: https://www.fbo.gov/index?s=opportunity&mode=form&id=592788989854da145c8e7b6d103c898d&tab=core&tabmode=list&
26. National Institutes of Health. Michel SL, Kane MA, Polli JE. Evaluation of iron species in healthy subjects treated with generic and reference sodium ferric gluconate [homepage on the Internet]. [cited 2015 Oct 15]. Available from: http://grantome.com/grant/NIH/U01-FD005266-01
27. U.S. Pharmacopeial Convention. Monographs in need of modernization (Last updated 26 November 2014) [database on the Internet]. [cited 2015 Oct 15]. Available from: http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/2014-11-26_monographs_needing_modernization.xlsx
28. CBG-MEB. [Openbaar verslag 797e Collegevergadering verslag]. Dutch [homepage on the Internet]. [cited 2015 Oct 15]. Available from: http://www.cbgmeb.nl/documenten/vergaderstukken/2014/01/09/collegevergadering-797-verslag
29. Rottembourg J, Kadri A, Leonard E, Dansaert A, Lafuma A. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10):3262-7.
30. Martin-Malo A, Merino A, Carracedo J, et al. Effects of intravenous iron on mononuclear cells during the haemodialysis session. Nephrol Dial Transplant. 2012;27(6):2465-71.
31. Stein J, Dignass A, Chow KU. Clinical case reports raise doubts about the therapeutic equivalence of an iron sucrose similar preparation compared with iron sucrose originator. Curr Med Res Opin. 2012;28(2):241-3.
32. Lee ES, Park BR, Kim JS, Choi GY, Lee JJ, Lee IS. Comparison of adverse event profile of intravenous iron sucrose and iron sucrose similar in postpartum and gynecologic operative patients. Curr Med Res Opin. 2013;29(2):141-7.
33. Agüera ML, et al. Efficiency of original versus generic intravenous iron formulations in patients on haemodialysis. Plos One. 2015;10(8):e0135967.
34. Weinstein V, Nicholas JM, Schwartz R, Grossman I, Zeskind B. Glatiramoids. In: Crommelin DJA, de Vlieger J, editors. Non-biological complex drugs. Advances in the pharmaceutical sciences series. NY: AAPS/Springer; 2015: p. 107-49.
35. Towfic F, et al. Comparing the biological impact of glatiramer acetate with the biological impact of a generic. PLoS One. 2014;9(1): e83757.
36. Halim LA, et al. How bio-questionable are the different recombinant human erythropoietin copy products in Thailand? Pharm Res. 2014;31(5):1210-18.
37. Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191-6.
38. Crommelin DJA, et al. The similarity question for biologicals and non-biological complex drugs. Eur J Pharm Sci. 2015;76:10-7.
|
Author for correspondence: Jon SB de Vlieger, PhD; Dutch Top Institute Pharma, PO Box 142, NL-2300 AC Leiden, The Netherlands |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/non-biological-complex-drugs-nbcds-and-their-follow-on-versions-time-for-an-editorial-section.html
Copyright ©2025 GaBI Journal unless otherwise noted.