Building stakeholder confidence in biosimilar medicines through evidence-based information sharing
|
Abstract:
The European Commission held a stakeholder event to discuss biosimilar medicinal products in May 2017. A session on building stakeholder confidence in biosimilar medicines provided an update of the latest available clinical experiences with biosimilar medicines, focusing on switching between biological medicines and interchangeability. Assistant Professor Gianluca Trifirò from the University of Messina, Italy, discussed these issues from the perspective of physicians in Italy.
|
Submitted: 23 October 2017; Revised: 6 November 2017; Accepted: 7 November 2017; Published online first: 20 November 2017
Introduction
As both clinical experience and real-world evidence increases, knowledge sharing and identifying best practices can support the appropriate use of biologicals, including biosimilar medicines. Gianluca Trifirò, Assistant Professor of Pharmacology at the University of Messina and a Clinical Pharmacologist in the Unit of Clinical Pharmacology of the Academic Hospital ‘G. Martino’ of Messina, Italy provided an update of the latest available clinical experiences with biosimilar medicines in Italy, focusing on switching between biological medicines and interchangeability. He provided clarity on the European Crohn’s Colitis Organisation (ECCO) position on the use of biosimilars for inflammatory bowel disease (IBD) and discussed the challenges ahead [1, 2].
EC stakeholder event
The European Commission (EC) held a stakeholder event to discuss biosimilar medicinal products in May 2017. A session on building stakeholder confidence in biosimilar medicines at this event provided an update of the latest available clinical experiences with biosimilar medicines, focusing on switching between biological medicines and interchangeability.
Professor Trifirò discussed the issue of stakeholder confidence from the perspective of physicians in Italy. He reported the results of a survey of 816 Italian physicians including their experience with regional or national drug policies and how this has affected their prescribing behaviour.
In addition, Professor Trifirò presented the conclusions of an updated position statement from ECCO on the use of biosimilars for IBD. He concluded his presentation by looking at the future challenges facing biosimilar uptake in Italy and worldwide and how these challenges should be met.
Italian physician survey
Professor Trifirò presented the results of a survey involving 816 physicians in Italy [3] and which was coordinated by Cittadinanzattiva that is a non-profit organization which is aimed at promoting civic participation and protection of citizens’ rights in Italy. The survey questionnaire was developed in a roundtable with representatives of several Italian scientific societies and patient and physicians organizations. The same roundtable participants distributed the survey questionnaire electronically using the web platform ‘esurveypro’ through a dedicated link through mailing, newsletter, Internet and participation at events and conventions.
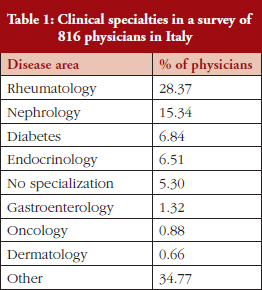
The physicians, who worked across a range of specialties, see Table 1, were asked whether the way that they prescribe medicines was influenced by regional or national drug policies.
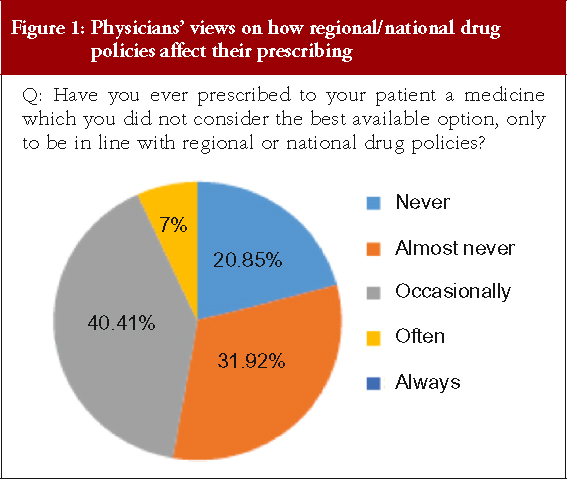
Physicians were asked whether they had ever prescribed a medicine that they did not consider was the best available option in order to conform to regional or national drug policies, see Figure 1, there was no response for ‘Always’.
ECCO position statement on the use of biosimilars for IBD
Professor Trifirò presented a position statement on the use of biosimilars for IBD released in 2017 by ECCO [1, 2]. The statement is the result of a consensus meeting held on 15 October 2016 and is based on the current regulatory guidance from the European Medicines Agency and evidence about efficacy and safety of biosimilars in IBD patients, see Box 1.
|
Box 1: ECCO 2017 position statement on the use of biosimilars for inflammatory bowel (IBD) disease [2]
1. Biosimilarity is more sensitively characterized by performing suitable in vitro assays than clinical studies.
2. Clinical studies of equivalence in the most sensitive indication can provide the basis for extrapolation. Therefore, data for the usage of biosimilars in IBD can be extrapolated from another sensitive indication.
3. When a biosimilar product is registered in the European Union, it is considered to be as efficacious as the reference product when used in accordance with the information provided in the Summary of Product Characteristics.
4. Demonstration of safety of biosimilars requires large observational studies with long-term follow-up in IBD patients. This should be supplemented by registries supported by all involved stakeholders [manufacturer, healthcare professionals and patients’ associations].
5. Adverse events and loss of response due to immunogenicity to a biological drug cannot be expected to be overcome with a biosimilar of the same molecule.
6. As for all biologicals, traceability should be based on a robust pharmacovigilance system and the manufacturing risk management plan.
7. Switching from the originator to a biosimilar in patients with IBD is acceptable. Studies of switching can provide valuable evidence for safety and efficacy. Scientific and clinical evidence is lacking regarding reverse switching, multiple switching and cross-switching among biosimilars in IBD patients.
8. Switching from originator to a biosimilar should be performed following appropriate discussion between physicians, nurses, pharmacists and patients, and according to national recommendation. The IBD nurse can play a key role in communicating the importance and equivalence of biosimilar therapy.
|
Future challenges
Several challenges for the future of bio-similars were identified. Establishing an effective system of pharmacovigilance will be key. The first challenge highlighted by Professor Trifirò will be to explore comparative long-term safety and effectiveness of first generation biosimilars thanks to data that have been cumulated over time. As experience with biosimilars grows, it will be important to evaluate clinical effects of switch between originator and biosimilars and vice versa, and between different originators.
Alongside these challenges, the secondary use of healthcare databases for post-marketing surveillance needs to be considered also for second-generation biosimilars in cancer patients.
While promoting the use of low cost biologicals, warned Professor Trifirò, prescribing biologicals wisely remains the highest priority over cost considerations. Every stakeholder (payers, healthcare professionals, patients) needs to be involved in the collection of real-world evidence about biosimilars that has to be integrated with pre-marketing evidence from randomized clinical trials [4].
Professor Trifirò concluded by noting that, given the growing number of biosimilars to be marketed in the near future across several different therapeutic areas, an international post-marketing surveillance system specifically for biosimilars must be established [5].
Acknowledgements
The author wishes to thank Dr Bea Perks, GaBI Journal Editor, in preparing this Special Report manuscript.
Competing interests: None.
Provenance and peer review: Not commissioned; internally peer reviewed.
Author
Assistant Professor Gianluca Trifirò1,2, MD, PhD
1Unit of Clinical Pharmacology, Azienda Ospedaliera Universitaria, Farmacologo Clinico, Policlinico ‘G Martino’, Dipartimento di Medicina, Clinica e Sperimentale, 1 Via Consolare Valeria, IT-98125 Messina, Italy
2Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina, 1 Via Consolare Valeria, IT-98125 Messina, Italy
References
1. Danese S, Fiorino G, Raine T, Ferrante M, Kemp K, Kierkus J, Lakatos PL, Mantzaris G, et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease-an update. J Crohns Colitis. 2017;11(1):26-34.
2. GaBI Online – Generics and Biosimilars Initiative. ECCO position statement on biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Nov 6]. Available from: www.gabionline.net/Biosimilars/General/ECCO-position-statement-on-biosimilars
3. Cittadinanzattiva. Squillace A, Amoroso C, Nardi S, Aceti T. Indagine civica sull’esperienza dei medici in tema di aderenza alle terapie, con focus su farmaci biologici e biosimilari [homepage on the Internet]. [cited 2017 Nov 6]. Available from: http://www.cittadinanzattiva.it/files/rapporti/salute/indagine-aderenza-terapie-focus-farmaci-biologici-biosimilari.pdf
4. Trifirò G, Ingrasciotta Y, Marcianò I, Genazzani A. Biosimilars in Italy: what do real-world data reveal? Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):114-9. doi:10.5639/gabij.2017.0603.023
5. van Gelder T, Lunddahl B, OM Claus B. Pharmacovigilance, traceability and building trust in biosimilar medicines. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):135-40. doi:10.5639/gabij.2017.0603.026
|
Author: Assistant Professor Gianluca Trifirò, MD, PhD, Unit of Clinical Pharmacology, Azienda Ospedaliera Universitaria, Farmacologo Clinico, Policlinico ‘G Martino’, Dipartimento di Medicina, Clinica e Sperimentale, 1 Via Consolare Valeria, IT-98125 Messina, Italy
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/building-stakeholder-confidence-in-biosimilar-medicines-through-evidence-based-information-sharing.html
Biosimilars in Italy: what do real-world data reveal?
Author byline as per print journal: Assistant Professor Gianluca Trifirò, MD, PhD; Ylenia Ingrasciotta, MSc; Ilaria Marcianò, MSc; Armando A Genazzani, DPhil, MD
|
Abstract:
This paper aims to provide an overview of the available real-world data about the pattern of use and the comparative effectiveness of biosimilars and originator biological drugs in Italy.
Different observational studies resulting from an Italian Ministry of Health funded project were described. These studies evaluated the pattern of use and the comparative effectiveness of biological drugs and biosimilars. In addition, further studies using Italian administrative databases to explore the switching patterns between biosimilars and originators and the clinical consequences of switching are described.
The included studies highlighted a remarkable heterogeneity in biosimilar uptake across Italy, and an overall increasing trend in biosimilar use. During the first year of treatment, switching between drugs of the same class was common, occurring mostly between originators.
When investigated, erythropoiesis-stimulating agent biosimilars and originators, a comparable effectiveness in terms of the haemoglobin levels gained after treatment beginning was demonstrated.
Given the expected rise in the number of biosimilars, a combination of multiple healthcare databases from several countries may represent an opportunity for post-marketing monitoring of biological drugs.
|
Submitted: 18 April 2017; Revised: 18 July 2017; Accepted: 19 July 2017; Published online first: 1 August 2017
Introduction
In recent years, national health services (NHSs) across the globe have been faced with ever increasing expenditures due to the marketing of high-cost innovative drugs. In Italy, the amount spent on pharmaceuticals by public hospitals almost doubled from Euros 2,124 million in 2011 to Euros 4,095 million in 2015 [1]. This trend is unlikely to stop as there is an increasing human life expectancy (from 1970–2010, the average life expectancy increased at an annual average rate of 0.21 for males and 0.17 years for females [2]), and the constant development of highly innovative therapies such as biological drugs.
Biological drugs contain one or more active substances, produced or extracted from biological systems or via biotechnological procedures [3, 4]. Biological drugs include hormones, growth factors, interleukins, interferons, vaccines, monoclonal antibodies and fusion proteins. They have led to a dramatic change in the management of several high burden diseases in different therapeutic areas, such as rheumatology (e.g. rheumatoid arthritis and ankylosing spondylitis), dermatology (e.g. psoriasis), gastroenterology (e.g. ulcerative colitis and Crohn’s disease), and oncology. These drugs selectively target specific proteins which play a key role in the disease progression or the tumour growth [5], thus offering substantial benefits in terms of response rate and quality of life [6].
With these therapeutic innovations, NHSs of western countries face new challenges. They must find a balance between economic sustainability and ensuring market access to innovative medicines. Research and development also needs to be rewarded financially. In order to ensure the sustainability of health systems where payers require the value of innovation to be demonstrated, strict cost containment strategies must be adopted [7].
When chemically synthesized drugs were considered, the introduction of generics represented a great opportunity for NHSs to achieve relevant savings. In the US in 2015, around 90% of all prescriptions were for generic drug products [7]. In European countries in the same year, the generics market share was more heterogeneous. It ranged from around 5% in Greece, to 70% in The Netherlands [8]. However, when biological drugs were considered, there are more hurdles to overcome to get access to markets. This is mainly due to the fact that biologicals are highly reliant on their production process, which can lead to subtle differences in the product quality. Biosimilars are biological drugs containing a version of the active substance of an already approved original biological drug (reference product, whose patent has expired) and, due to the subtle differences, are required to demonstrate their biosimilarity to the corresponding reference product, in terms of: quality characteristics, biological activity, safety and efficacy, based on a comprehensive comparability exercise [4, 9].
In general, biosimilars are considered as comparable, effective and safe therapeutic alternatives to their reference products [10]. They can provide around 20–30% purchase cost reduction in comparison to the reference product [11]. Such cost reductions can reach significantly higher percentages (69% in 2015) where a higher uptake of biosimilars occurs as demonstrated in the case of infliximab in Norway [12]. In cases such as these, tendering is usually the preferred purchasing tool of health services [13]. Overall, this means that biosimilars may represent a valid opportunity for additional cost savings.
Biosimilars in Italy: an overview of dispensing and monitoring
In 2015, the 30 most costly molecules to Italy’s hospitals included drugs that did not have biosimilars available yet, i.e. adalimumab, and drugs whose corresponding biosimilar is already on the European market, i.e. etanercept, insulin glargine, somatropin, epoetin alfa. Biosimilar uptake in Italy was relatively low when compared to countries such as Norway. Uptake was also widely variable and varied for different drugs. For example, it varied from 0.3% for biosimilar follitropin-alfa, to 31.2% for biosimilars of erythropoiesis-stimulating agents (ESAs), in terms of defined daily dose (DDD) per 1,000 inhabitants of the total of the therapeutic class. However, an increasing trend in biosimilar consumption was observed in comparison to the previous year (+49.0% for biosimilar ESA, +21.5% for biosimilar somatropin and +16.5% for biosimilar granulocyte colony-stimulating factors [G-CSFs]) [14].
In the preliminary ‘Italian report on Drug use, 2016’ (January to September) a specific indicator to monitor the appropriateness of biosimilar ESA use was developed, highlighting an increasing number of naïve patients starting ESA therapy with a biosimilar (+20.2%, compared to the same period of the previous year) [1]. This indicator reflects the recommendation from the Italian Medicines Agency (Agenzia Italiana del Farmaco, AIFA), which clearly outlines that biosimilars are the preferred therapeutic alternative to be used to ensure economic savings, especially in naïve patients (both never-treated and previously exposed patients with an adequately long wash-out period), as no scientific or regulatory reasons justify the use of a more expensive product in these patients [4]. However, the AIFA recommendations also state that, whenever a patient is successfully treated with a biological drug, and this is substituted with another drug (even a cheaper one), the corresponding responsibility is that of the individual prescribing physician [4]. In practice, this discourages substitution. Automatic substitutions of reference products and biosimilars by healthcare professionals other than the prescribing physician are not allowed.
The Italian health service is run at the regional level. This allows the adoption of different loco-regional health interventions to influence the uptake of the most cost-effective molecule (usually the biosimilar over the reference product), but no systematic monitoring of biosimilars in clinical practice has been put in place at a national or regional level. Even though some biosimilars are subjected to AIFA monitoring registries, the corresponding data have never been shared, nor has any study been conducted using such registries. However, in some Italian regions specific web-based prescription monitoring systems for biologicals have been set up but no results have been published to date.
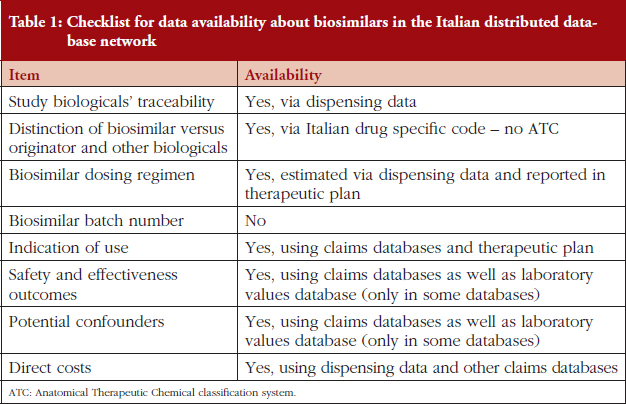
In this complex context, the drug-utilization studies may represent the first step of post-marketing assessment of biosimilars in real-world settings. The ideal data source for such assessments should allow:
a) Tracing biological drugs for which biosimilars are marketed.
b) Distinction between biosimilars and originators (both reference products and biological drugs still covered by patent), as the Anatomical Therapeutic Chemical classification system identifies the specific molecule but does not distinguish between the medicinal products and the drug-specific name is therefore necessary.
c) Plus: identification of information about biosimilar dosing regimen, batch number, indication of use, effectiveness outcome as investigated in pre-marketing randomized clinical trials (RCTs), safety outcomes as reported in risk management plans, potential confounders for effectiveness and outcome analysis, direct costs of the biosimilars for economic evaluations.
The Italian administrative healthcare databases are a valid source to explore the treatment pattern of biosimilars in different geographic areas over time, as well as the short- and long-term comparative effectiveness and safety of biosimilars versus originators in naïve users and switchers, together with the economic impact of biosimilars. Several observational studies have been conducted throughout Italy over time to explore the pattern of use of the available biosimilars and corresponding originators, as well as their comparative effectiveness [15–19].
This paper provides an overview of the available real-world data on the pattern of biosimilar use and the comparative effectiveness of the biosimilars and their corresponding originator biological drugs. The data were collected in Italy outside the constraints of traditional clinical trials.
Real-world data in Italy
The Italian Ministry of Health recently funded the 4-year project entitled ‘Assessment of short- and long-term risk-benefit profiles of biologicals through healthcare database network in Italy’ (RF-2010-2320172). This aimed to create a network of administrative healthcare databases from different Italian local health units (LHUs) and regions, to evaluate the pattern of use and the comparative safety and effectiveness of biological drugs for which biosimilars were available on the Italian market when the project was approved, i.e. ESAs, G-CSFs, somatropin. Six centres participated in the project: Caserta, Treviso and Palermo LHUs; and Tuscany, Umbria and Lazio regions, covering a total population of 14,133,687 persons (around 25% of the whole Italian population), see Figure 1. Various healthcare claims databases, linkable through unique and anonymous patients’ identifiers, were made available from the participating centres: demographic and mortality registries, emergency department visits and hospital discharge diagnosis, drug dispensing, healthcare service payment exemptions, outpatient diagnostic tests and specialists’ visits and, in some cases, also laboratory findings, see Table 1. From these sources, more than 65,000 ESA users, more than 30,000 G-CSF users, and almost 7,000 somatropin users in the years of 2009–2014 were retrieved.

In addition, two different population-based drug-utilization studies analysed the switching patterns between biosimilars and originator ESAs, using the administrative healthcare databases from the Sicilian LHU of Messina in the period January 2010 to May 2011 [20], and those from the Umbria region in the period July 2011 to December 2014 [19], respectively. One retrospective study analysed data from 11 Italian dialysis centres, from 2011 to 2014, to evaluate the effect of switching from originators to biosimilar ESAs [21].
Loco-regional healthcare interventions in Italy and biosimilar uptake
To date, two drug-utilization studies have been published in the context of the Italian Ministry for Health project (RF-2010-2320172). These were aimed at evaluating the prescribing patterns of ESAs and G-CSFs (both biosimilars and originators) in the participating Italian centres where different health policy interventions promoting biosimilar use were implemented [14–16]. Considering the uptake of biosimilars, Ingrasciotta Y et al. highlighted an increasing trend in biosimilar ESA use in all centres considered from 2009 to 2013 (overall, +33.6%) [15]. Heterogeneity in uptake was observed in the different centres, with a greater increase being observed in Treviso (up to 45.0% of biosimilar ESA users on total ESA users) than in the Caserta LHU (from 7.5% to 22.9%) during the study years. Similarly, an increase in the proportion of biosimilar G-CSF users over time was described by Marcianò I et al. Here, there were 0.2% of G-CSF users in 2009 and 66.2% in 2014, together with a notable difference between individual centres [16]. Ongoing analyses from the same project documented a similar trend for somatropin in the same study years. However, the biosimilar of this drug accounts for a much lower proportion of the total somatropin use (8.0%).
Potential reasons for such variability in biosimilar uptake may be ascribed to the different healthcare policy interventions promoting biosimilar use adopted at a loco-regional level. In some centres, naïve users must be dispensed a biosimilar; in others, clinicians are requested to justify any prescription of drugs other than the cheapest and are otherwise charged with the cost of the prescribed drug; and in others, achievement of predefined thresholds of biosimilar consumption assures economic incentives. These policies, as well as different tender procedures for the purchase of originators and biosimilars by public hospitals, the scepticism surrounding biosimilars, and the differences in patients’ access to biological drugs, may all have influenced the uptake of biosimilars in regions across Italy.
Switching patterns in Italy: biosimilars and originators
As mentioned previously, AIFA does not allow automatic substitution of a prescribed biological drug with another one, even if cheaper [4], probably because the safety of the substitution of biosimilars and originators is not supported by robust studies [22]. However, switching between different active pharmaceutical ingredients belonging to the same therapeutic class appears to be quite common in clinical practice. This generally concerns switching from one originator to another.
The analysis of the switching patterns of the Italian project (RF-2010-2320172) revealed that, during the first year of treatment, switching was frequent. This was both among different G-CSFs (20.3% of naïve G-CSF users [16]) and among different ESAs (17.0% of naïve ESA users [15]). In general, switching occurred most frequently from biosimilars to reference products, from other ESAs still covered by patent to biosimilars, and between originators in both patients affected by chronic kidney disease (CKD) (17.0%) and those with chemotherapy-induced anaemia (17.4%), see Figure 2. These results support previous data from a drug-utilization study in southern Italy [20] where switching between ESAs was also very frequent (21.8% of incident users, during a 1.5 years observation period). Among switchers, 27.0% switched more than once and switching back accounted for 20.7%. Furthermore, the most frequent switches occurred towards the reference product (32.9%). An additional real-world study on ESAs, using administrative healthcare databases from the Umbria region, documented that 12.2% of incident ESA users having at least two prescriptions of ESAs, experienced a switch during a 48-month observation period. The switching practice equally affected all ESAs and occurred most frequently between originators. The probability of switching increased with the duration of therapy, from 15% of incident users switching within the first year of treatment, up to 25% within two years [19].

Switching practices concern all biological drug users equally, not only those using biosimilars [15, 16, 19, 20]. This suggests that it is not influenced by the drug cost or by evidence provided by the comparability exercise. Furthermore, a switch between different biological drugs of the same therapeutic class is not associated with adverse events. Ebbers H et al. [23] reviewed data from clinical trials and pharmacovigilance databases, identifying almost 60 studies that evaluated the safety of switching between different ESAs or G-CSFs or somatropin (regardless of whether biosimilars or originators). Here, no safety issues were described following switching to and from different biological drugs (even though the studies were too short to evaluate long-term safety) [23].
Comparative effectiveness of biosimilar versus originator in an Italian post-marketing setting
According to AIFA, ESAs are prescribed to patients affected by chronic kidney disease (CKD) with haemoglobin (Hb) levels lower than 11 g/dL, and in the case of chemotherapy-induced anaemia with Hb levels lower than 10 g/dL. In both conditions, haemoglobinaemia ranges between 11–12 g/dL, avoiding an increase higher than 2 g/dL over a 4-week period [24].
To date, four pre-marketing clinical trials have been conducted on patients with CKD-related anaemia, and no differences in achieving the target Hb value after 12, 24, 28 or 56 weeks of treatment were found between biosimilar and reference product [25–28]. Furthermore, a meta-analysis of RCTs, comparing the efficacy and safety of ESAs in CKD patients, highlighted a lack of evidence suggesting the superiority of any ESA formulation [29]. Only one randomized study was conducted to evaluate the efficacy and safety of 12-week treatment with biosimilar epoetin alfa in chemotherapy-induced anaemia due to solid tumours [30]. On average, less than 500 patients were enrolled in each pre-marketing RCT on biosimilar ESAs.
With respect to post-marketing assessment, several observational studies have proven the effectiveness of epoetin zeta, darbepoetin alfa, and biosimilar epoetin alfa, using real-world data but they have not been compared to originators [31–33]. Only one European multicentre retrospective study compared the real-life clinical effectiveness and safety of biosimilar epoetin alfa versus darbepoetin alfa in patients with chemotherapy-induced anaemia, showing no differences [34].
In the context of the Italian project (RF-2010-2320172), a population-based study was conducted to evaluate and compare the effects of biosimilar and originator ESAs on haemoglobinaemia in CKD and cancer patients between 2009 and 2014. This used the administrative database from Treviso LHU which also included haemoglobinaemia values [17]. In this study, incident ESA users with at least one Hb measurement taken within the month prior to the first ESA dispensing, i.e. Index Date [ID], defined as the baseline Hb value, and another measurement between the second and the third month after ID (follow-up Hb value) were considered. This allowed for the identification of more than 1,000 incident users. Considering the effects of ESAs on Hb values during the follow-up period, no statistically significant differences were found for biosimilars versus reference product/other ESAs in patients being treated for either indications of use, when considering mean baseline Hb value and follow-up Hb value. Based on the difference between follow-up and baseline Hb values, i.e. delta Hb (ΔHb), ESA users were classified as non-responders (ΔHb ≤ 0 g/dL), responders (0 ‹ ΔHb ≤ 2 g/dL) and high responders (ΔHb › 2 g/dL). Around 15–20% of ESA users were non-responders in both CKD and cancer. Considering the distribution of responders and high responders in both indications of use, no differences across types of ESA users were found. The strength of ESA therapy, defined as the total number of dispensed DDD during the follow-up, divided by the days of follow-up, was investigated stratifying by type of administered ESA, i.e. reference product, biosimilar and other ESAs still covered by patent. No statistically significant difference in the strength of treatment between different types of ESAs was found in either CKD or cancer patients. The difference related to the higher doses used in chemotherapy-induced anaemia was in line with the dosing regimens described in the Summary of Product Characteristics.
In terms of responsiveness to the drug, no statistically significant difference between biosimilars (Binocrit, Abseamed, Retacrit) and reference product (Eprex)/ESAs covered by patent (Aranesp, Nespo, Neorecormon, Eporatio, Mircera) was found, despite a comparable prescribed cumulative dose of drug during the observation period. This finding is in contrast to the results reported in a recently published research letter [21] where Minutolo R et al. analysed data from 11 Italian dialysis centres between 2011–2014. Here, the goal was to evaluate the effect of switching from originator to biosimilar ESAs when treating anaemia. The authors concluded that switching from originator to biosimilar ESAs may require higher doses of biosimilar ESA to maintain Hb levels (so called ‘dosing penalty’) [21]. Several factors may explain the differences observed by Ingrasciotta [17] and Minutolo [21]: a) Ingrasciotta Y et al. investigated all naïve users of different ESAs while Minutolo R et al. analysed only those who switched from originators to biosimilars. As renal functionality declines progressively, thus requiring higher doses of all ESAs to achieve target Hb over time, it may be more appropriate to investigate all possible switches between biosimilars and reference product/other ESAs covered by patent and vice versa; b) Ingrasciotta Y et al. conducted a population-based study, thus using real-world data, while Minutolo R et al. conducted a multicentre study where patients received stable originator ESA doses, thus preventing the generalization of the results to the whole spectrum of the haemodialysis population.
Despite the AIFA recommendations surrounding the specific Hb values to start ESA treatment, 14–24% of CKD patients had a baseline Hb value higher than 11 g/dL. In addition, 22–38% of cancer patients started ESA treatment having a baseline Hb value higher than 10 g/dL, regardless of the type of ESA dispensed at ID. More worryingly, 33–54% of CKD patients and 26–39% of cancer patients reached a follow-up Hb value higher than 12 g/dL [17]. Given the widely known negative effects of high Hb values [35], these data may reflect an inappropriate use of ESA thus warranting further investigation.
Conclusions and future assessment of biosimilars in Italy
The Italian administrative healthcare databases and their distributed database network have provided real-world data about biosimilar patterns of use and the comparative efficacy of ESAs. In the future, post-marketing assessment of biosimilars will be required and this will need: a) data supporting the long-term safety and effectiveness of first generation biosimilars versus originators; b) further investigation and evaluation of the clinical effects of switching between originators and biosimilars, biosimilars and originators, and between different originators; c) the risk–benefit profile of the most recently marketed and more complex biosimilars, i.e. monoclonal antibodies.
In light of the growing number of biosimilars set to reach the market in the future across therapeutic areas, an international post-marketing monitoring system for biosimilars, based on combination of multiple healthcare databases from several countries, may provide useful data. The secondary use of healthcare administrative databases represents a great opportunity for post-marketing assessment of biosimilars and biological drugs in general.
This paper reported a heterogeneous uptake of first generation biosimilars over time and across regions of Italy. This is likely due to the specific loco-regional healthcare policy interventions, and confirms comparable effectiveness of the different types of ESAs in clinical practice, used for treatment of both cancer and CKD patients. However, a better monitoring of biosimilar prescribing is necessary to ensure that savings can be made without putting patients at risk.
Competing interests: Assistant Professor Gianluca Trifirò declares his participation to advisory boards on biosimilars, organized by Sandoz and Hospira; furthermore, he coordinates studies funded by pharmaceutical companies to his department. Ilaria Marcianò, Ylenia Ingrasciotta and Dr Armando Genazzani declare that they have no conflicts of interest.
Provenance and peer review: Commissioned; externally peer reviewed.
Authors
Assistant Professor Gianluca Trifirò1,2, MD, PhD
Ylenia Ingrasciotta2, MSc
Ilaria Marcianò1, MSc
Armando Genazzani3, DPhil, MD
1Unit of Clinical Pharmacology, Azienda Ospedaliera Universitaria, Farmacologo Clinico, Policlinico ‘G Martino’, Dipartimento di Medicina, Clinica e Sperimentale, 1 Via Consolare Valeria, IT-98125 Messina, Italy
2Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina, 1 Via Consolare Valeria, IT-98125 Messina, Italy
3Department of Pharmaceutical Sciences, University of Piemonte Orientale, 6 Via Bovio, IT-28100 Novara, Italy
References
1. Italian Medicines Agency. L’uso dei farmaci in Italia – Rapporto OsMed Gennaio-Settembre 2016 [homepage on the Internet]. [cited 2017 Jul 18]. Available from: http://www.aifa.gov.it/sites/default/files/Rapporto_OsMed_gennaio_settembre_2016.pdf
2. Timonin S, Shkolnikov VM, Jasilionis D, Grigoriev P, Jdanov DA, Leon DA. Disparities in length of life across developed countries: measuring and decomposing changes over time within and between country groups. Popul Health Metr. 2016;14:29.
3. European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products) [homepage on the Internet]. 2012 [cited 2017 Jul 18]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
4. Italian Medicines Agency. Position Paper sui Farmaci Biosimilari [homepage on the Internet]. 2013 [cited 2017 Jul 18]. Available from: http://www.agenziafarmaco.gov.it/sites/default/files/AIFA_POSITION_PAPER_FARMACI_BIOSIMILARI.pdf
5. Dos Reis C, Teixo R, Mendes F, Cruz RS. Biosimilar medicines – review. Int J Risk Saf Med. 2016;28(1):45-60.
6. Mach JP. Introduction to monoclonal antibodies. Cancer Immun. 2012;12:11.
7. Chin WW. A delicate balance – pharmaceutical innovation and access. N Engl J Med. 2015;373(19):1799-801.
8. IMS Health. The role of generic medicines in sustaining healthcare systems: a European perspective [homepage on the Internet]. 2015 [cited 2017 Jul 18]. Available from: https://www.imshealth.com/files/web/IMSH Institute/Healthcare Briefs/IIHI_Generics_Healthcare_Brief.pdf
9. European Medicines Agency. Guideline on similar biological medicinal products [homepage on the Internet]. CHMP/437/04 Rev 1. 23 October 2014 [cited 2017 Jul 18]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf
10. Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111-7.
11. Genazzani AA, Biggio G, Caputi AP, Del Tacca M, Drago F, Fantozzi R, et al. Biosimilar drugs: concerns and opportunities. BioDrugs. 2007;21(6):351-6.
12. Asbjørn M. Norway, biosimilars in different funding systems. What works? Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(2):90-2. doi:10.5639/gabij.2015.0402.018
13. Curto S, Ghislandi S, van de Vooren K, Duranti S, Garattini L. Regional tenders on biosimilars in Italy: an empirical analysis of awarded prices. Health Policy. 2014;116(2-3):182-7.
14. Italian Medicines Agency. L’uso dei farmaci in Italia – Rapporto OsMed 2015 [homepage on the Internet]. [cited 2017 Jul 18]. Available from: http://www.agenziafarmaco.gov.it/sites/default/files/Rapporto_OsMed_2015_AIFA.pdf
15. Ingrasciotta Y, Giorgianni F, Bolcato J, Chinellato A, Pirolo R, Tari DU, et al. How much are biosimilars used in clinical practice? A retrospective Italian population-based study of erythropoiesis-stimulating agents in the years 2009–2013. BioDrugs. 2015;29(4):275-84.
16. Marcianò I, Ingrasciotta Y, Giorgianni F, Bolcato J, Chinellato A, Pirolo R, et al. How did the introduction of biosimilar filgrastim influence the prescribing pattern of granulocyte colony-stimulating factors? Results from a multicentre, population-based study, from five italian centres in the years 2009–2014. BioDrugs. 2016;30(4):295-306.
17. Ingrasciotta Y, Giorgianni F, Marcianò I, Bolcato J, Pirolo R, Chinellato A, et al. Comparative effectiveness of biosimilar, reference product and other erythropoiesis-stimulating agents (ESAs) still covered by patent in chronic kidney disease and cancer patients: an Italian population-based study. Mantovani LG, editor. PLoS One. 2016 May 17;11(5):e0155805.
18. Trotta F, Belleudi V, Fusco D, Amato L, Mecozzi A, Mayer F, et al. Comparative effectiveness and safety of erythropoiesis-stimulating agents (biosimilars vs originators) in clinical practice: a population-based cohort study in Italy. BMJ Open. 2017;7(3):e011637.
19. D’Amore C, Da Cas R, Rossi M, Traversa G. Switching between epoetins: a practice in support of biosimilar use. BioDrugs. 2016;30(1):27-32.
20. Loiacono C, Sgroi C, Coppolino S, Cannata A, Ferrara R, Arcoraci V, et al. How much are biosimilars used in southern Italy?: a retrospective analysis of epoetin utilization in the local health unit of Messina in the years 2010–2011. BioDrugs. 2012;26(2):113-20.
21. Minutolo R, Borzumati M, Sposini S, Abaterusso C, Carraro G, Santoboni A, et al. Dosing penalty of erythropoiesis-stimulating agents after switching from originator to biosimilar preparations in stable hemodialysis patients. Am J Kidney Dis. 2016;68(1):170-2.
22. Pani L, Montilla S, Pimpinella G, Bertini Malgarini R. Biosimilars: the paradox of sharing the same pharmacological action without full chemical identity. Expert Opin Biol Ther. 2013;13(10):1343-6.
23. Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12(11):1473-85.
24. Italian Medicines Agency. Determinazione AIFA. Aggiornamento del Piano terapeutico AIFA per prescrizione SSN di Eritropoietine (ex Nota 12) [homepage on the Internet]. [cited 2017 Jul 18]. Available from: http://www.agenziafarmaco.gov.it/sites/default/files/2010-07-29_determina_aggiornamento_template_nota_12_g.u._75_del_31-03-2009.pdf
25. Wizemann V, Rutkowski B, Baldamus C, Scigalla P, Koytchev R, Epoetin Zeta Study Group. Comparison of the therapeutic effects of epoetin zeta to epoetin alfa in the maintenance phase of renal anaemia treatment. Curr Med Res Opin. 2008 Mar 18;24(3):625-37.
26. Krivoshiev S, Todorov V V, Manitius J, Czekalski S, Scigalla P, Koytchev R, et al. Comparison of the therapeutic effects of epoetin zeta and epoetin alpha in the correction of renal anaemia. Curr Med Res Opin. 2008;24(5):1407-15.
27. Krivoshiev S, Wizemann V, Czekalski S, Schiller A, Pljesa S, Wolf-Pflugmann M, et al. Therapeutic equivalence of epoetin zeta and alfa, administered subcutaneously, for maintenance treatment of renal anemia. Adv Ther. 2010;27(2):105-17.
28. Haag-Weber M, Vetter A, Thyroff-Friesinger U, INJ-Study Group. Therapeutic equivalence, long-term efficacy and safety of HX575 in the treatment of anemia in chronic renal failure patients receiving hemodialysis. Clin Nephrol. 2009;72(5):380-90.
29. Palmer SC, Saglimbene V, Mavridis D, Salanti G, Craig JC, Tonelli M, et al. Erythropoiesis-stimulating agents for anaemia in adults with chronic kidney disease: a network meta-analysis. Cochrane Database Syst Rev. 2014 Dec 8;(12):CD010590.
30. Weigang-Köhler K, Vetter A, Thyroff-Friesinger U. HX575, recombinant human epoetin alfa, for the treatment of chemotherapy-associated symptomatic anaemia in patients with solid tumours. Onkologie. 2009;32(4):168-74.
31. Michallet M, Luporsi E, Soubeyran P, Amar NA, Boulanger V, Carreiro M, et al. BiOsimilaRs in the management of anaemia secondary to chemotherapy in HaEmatology and Oncology: results of the ORHEO observational study. BMC Cancer. 2014;14(1):503.
32. Bustos A, Álvarez R, Aramburo PM, Carabantes F, Díaz N, Florián J, et al. Evaluation of clinical use and effectiveness of darbepoetin alfa in cancer patients with chemotherapy-induced anemia. Curr Med Res Opin. 2012;28(1):57-67.
33. Kerkhofs L, Boschetti G, Lugini A, Stanculeanu D-L, Palomo AG. Use of biosimilar epoetin to increase hemoglobin levels in patients with chemotherapy-induced anemia: real-life clinical experience. Future Oncol. 2012;8(6):751-6.
34. Rodriguez Garzotto A, Cortijo Casacajares S, Pernaut C, Ruiz Ares GJ, Otero Blas I, Heine O, et al. Erythropoiesis-stimulating agents for the treatment of chemotherapy-induced anemia: comparisons from real-world clinical experience. J Blood Med. 2014;5:43-8.
35. KDIGO Clinical practice guideline for anemia in chronic kidney disease. Kidney Int. Suppl. 2012;2(4):Aug (2).
|
Author for correspondence: Ylenia Ingrasciotta, Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina, 1 Via Consolare Valeria, IT-98125 Messina, Italy
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/biosimilars-in-italy-what-do-real-world-data-reveal.html
First GCC stakeholder meeting on approval process, interchangeability/substitution and safety of biosimilars 2017 – Report
Author byline as per print journal: Gianluca Trifirò, MD, PhD; Meteb Al-Foheidi, MD, FRCPC; Ali M Alhomaidan, PhD; Ahmed H Aljedai, PharmD, MBA, BCPS, FCCP, FAST; Musaed Abdullah Alkholief, PhD; Mohammad A Alsenaidy, MSc, PhD; Aws Alshamsan, BPharm, RPh, PhD; Tore Kristian Kvien, MD, PhD
|
Introduction: A meeting was organized by the Generics and Biosimilars Initiative (GaBI) in collaboration with Saudi Pharmaceutical Society (SPS), to discuss the regulation, approval process, interchangeability/substitution, and post-marketing surveillance of biosimilars in Gulf Cooperation Council (GCC) countries. This ‘First GCC Stakeholder Meeting on Approval Process, Interchangeability/Substitution and Safety of Biosimilars’, took place on 20 November 2017, in Riyadh, Saudi Arabia and gave relevant stakeholders an opportunity to discuss these concepts between themselves and with experts from Saudi Arabia and abroad.
Methods: The meeting brought regulators from GCC countries and academics, medical specialists, and pharmacologists and pharmacists from Saudi Arabia together with experts from Italy, Norway, Saudi Arabia and the US, to share knowledge and exchange information. There were a number of expert speaker presentations with an interactive panel discussion. Following this, the audience was presented with data for two semi-fictional similar biotherapeutic products. The participants were divided into two discussion groups where they evaluated the fictional data supplied.
Results: The presentations were successful in conveying information about the current state of biosimilar regulation, approval, interchangeability/substitution, and post-marketing surveillance of effectiveness and safety. This resulted in a useful interactive discussion from which clearly defined action points could be extracted.
Conclusion: Biosimilar medicines are becoming increasingly available and used across GCC countries. The First GCC biosimilars stakeholder meeting was successful in bringing representatives from GCC nations together with those from Europe and the US, to discuss the best routes forward for successful biosimilar approval and regulation and enabled action points to facilitate biosimilar uptake with appropriate pharmacovigilance to be outlined.
|
Submitted: 22 November 2018; Revised: 27 November 2018; Accepted: 28 November 2018; Published online first: 11 December 2018
Introduction
Appropriate methods for the approval and regulation of similar biotherapeutic products (SBPs or biosimilars) are subject to global discussion [1, 2]. To ensure that biosimilars successfully enter markets and maintain the safety and efficacy achieved by originator products, approval and regulation guidelines need to be clearly outlined. In addition, it is important to define interchangeability/substitution so that products are used in a way that does not impact on safety and efficacy.
To discuss the regulation and approval of biosimilars in the Gulf Cooperation Council (GCC) countries such as Bahrain, Kuwait, Oman and Saudi Arabia, the First GCC Stakeholder Meeting on Approval Process, Interchangeability/Substitution and Safety of Biosimilars, took place on 20 November 2017, in Riyadh, Saudi Arabia. It was organized by the Generics and Biosimilars Initiative (GaBI) in collaboration with Saudi Pharmaceutical Society (SPS). Fifty-four participants, speakers included, attended the meeting.
This first GCC biosimilars stakeholder meeting was an interactive scientific meeting on the regulation, approval and use of biosimilars with a focus on their interchangeability/substitution and safety. It brought regulators from GCC countries, and academics, medical specialists, and pharmacologists and pharmacists (clinical, hospital) from Saudi Arabia; together with experts from Italy, Norway and the US, to share knowledge and exchange information.
The meeting aimed to address the issues of physicochemical characterization, analytical comparability, interchangeability/substitution and safety of biologicals/biosimilars. The participants engaged in active discussion concerning regulatory approval process, cell line development, hospital formulary selection, switching, and set out to identify future educational needs.
Expert speaker presentations
The format of the GCC meeting was similar to that followed in previous educational workshops and scientific meetings as reported in the GaBI Journal [3–5]. There were a number of expert speaker presentations followed by Q&A and an in-depth panel discussion. The presentations are downloadable from the GaBI website [6].
The GCC meeting began with a welcome from Professor Yousif A Asiri, Vice Rector of Planning and Development and Professor of Clinical Pharmacy at King Saud University (KSU), Saudi Arabia. This was followed by an introduction by Abdulaziz Alhossan, Assistant Professor at KSU, and then a series of presentations given by expert speakers [6].
The false myths of biosimilars
Clinical Pharmacologist and Professor Gianluca Trifirò, of the University of Messina, Italy gave a presentation during which he discussed and dispelled various ‘false myths’ surrounding biosimilar products. These were outlined as follows:
– Biosimilars are not identical but only similar to the reference product, thus they should be considered as different drugs
– Pre-marketing evidence on biosimilars are much more limited than what is available for the reference product at the time the drug is marketed
– Biosimilars are less safe than reference product in routine care
– Interchangeability of biosimilars and reference product should never be considered due to serious immunogenicity risks potentially associated with switching among therapeutic proteins
In each case, Professor Trifirò provided appropriate evidence and explanation as to why these myths should not be considered a concern.
Challenges related to physicochemical characterization and analytical comparability of biologicals/biosimilars
Mohammad A Alsenaidy, Assistant Professor at KSU, described biologicals/biosimilars as structurally complex, highly specific macromolecules used therapeutically to compensate for body deficiencies, e.g. hormones and clotting factors; and to treat diseases, e.g. cytokines and monoclonal antibodies; and to prevent illnesses, e.g. polyclonal antiserum and certain vaccines. He noted that the structural complexity of proteins makes it challenging to preserve their conformational integrity, biological activity and stability throughout manufacturing, storage and distribution. Professor Alsenaidy then discussed how physiochemical characterizations, stability and degradation profiles of protein-based drugs can be carried out. These are key exercises for the development of protein-based drugs, whether originator or biosimilar. He also noted that, the ability to establish high quality analytical profiles determines the extent for additional animal and/or clinical evaluations such as pharmacokinetics (PK), pharmacodynamics (PD), and/or immunogenicity studies.
Biosimilar cell line development
Musaed Abdullah Alkholief, Assistant Professor at KSU, outlined the key factors necessary for successful biosimilar cell line development. He noted that during the upstream development of these molecules, cell line selection and cell culture processing represents the most critical step in biosimilar development and as such should be carefully evaluated. When developing a biosimilar the slightest variability between the originator cell line and the biosimilar cell line may significantly affect the characteristics of the product, consequently limiting its utilization. It is anticipated that by 2022 the global market size of cell line development will reach US$6 billion. In conclusion, Professor Alkholief noted that a better understanding of the current status and future directions of cell line development is needed to fully exploit biosimilar development processes.
Biosimilar regulations in Saudi Arabia
The Executive Director for Pharmaceutical Products Evaluation of the Saudi Food and Drug Administration (SFDA), Dr Ali M Alhomaidan, gave a talk in which he described the biosimilar approval pathway in Saudi Arabia, quality, safety and efficacy considerations surrounding biosimilars, and their pricing and interchangeability. Background on the pricing of biosimilars in Saudi Arabia and the system used for pricing these products is explained in the paper by Alhomaidan et al. [7]. Further information can be found in the summary of discussions of this meeting below.
Interchangeability for biosimilars: considerations and concerns
Aws Alshamsan, Associate Professor at KSU, discussed the concepts of interchangeability, switching and substitution of biosimilars, and how they differ across Europe, Saudi Arabia and the US. He highlighted that a number of concerns surround the regulatory authority definitions and the differences between them. In addition, he highlighted four key challenges that need to be overcome to facilitate interchangeability that is safe and efficacious. These include: the extrapolation to indications only studied for the reference product; ethical and medical–legal aspects of approval; developing strategies for adequate post-marketing pharmacovigilance; and developing effective pricing policy that distinguishes high quality products.
Formulary considerations for biosimilars for health systems
According to Professor Ahmed Al-jedai of Alfaisal University, Saudi Arabia, global spending on medicinal products is expected to reach Euros 1.3 trillion by 2020 and that, with price reductions of 20%–40% with respect to reference products, biosimilars could create an estimated cumulative savings of Euros 50–100 billion during this timeframe. He highlighted the differences in biosimilar approval approaches between Europe and the US and noted that SFDA and the GCC have adopted the European Medicines Agency’s (EMA) approach. Currently, only five biosimilars are available in the Saudi markets. He also outlined the cost analysis of the biosimilars already approved in Saudi Arabia and discussed the role that pharmacists can play in increasing biosimilar adoption.
Switching from originator product to biosimilars in rheumatology, dermatology and gastroenterology: clinical evidence
The keynote presentation was delivered by Tore Kvien, Professor of Medicine and Rheumatology at Diakonhjemmet Hospital in Norway. This presentation outlined the current need for biosimilar products that are less expensive and as such, can increase patient access to biological medicines. The details of a randomized clinical trial that compared the tumour necrosis factor (TNF) inhibitor infliximab with the less expensive biosimilar CT-P13, also known as the NOR-SWITCH study [8], were presented. This study addressed questions surrounding the efficacy, safety and immunogenicity of switching from an originator to its biosimilar for the first time and showed that switching is not inferior to remaining with the originator product, and that switching can be considered for non-medical reasons.
During a 6-month extension study, patients on the reference infliximab were switched to CT-P13 and compared to the group whose treatment with CT-P13 was maintained. Results from the switch group was not inferior to the maintenance group. The point estimates were actually generally in favour of the switch group and this extension study supported the results of the primary study published in The Lancet 2017.
Oncologist perspective – the use of biosimilar trastuzumab in breast cancer: clinical experience
Assistant Professor Meteb Al-Foheidi of King Saud Bin Abdulaziz University for Health Science in Saudi Arabia discussed the need for oncology biosimilars, noting that they could lead to cancer treatment savings of up to 30%. However, there is currently no consensus on which endpoints to use in oncology biosimilar studies and long-term endpoints are often not feasible in these studies.
Professor Al-Foheidi also described the case of biosimilar trastuzumab in breast cancer treatment and noted that there had been no issues or safety concerns regarding its use in the EU.
Summary of the discussions that followed the expert presentations
After the presentations, there was the opportunity for discussion about the topics covered. The key discussion points are summarized below.
Biosimilar variability
The presentation on ‘Biosimilar cell line development’ prompted a number of questions about biosimilar product variability. Assistant Professor Alkholief stressed that when developing a biosimilar the most critical step is choosing the initial cell line. Having the right components and conditions for growth is also very important in reducing variability, but these are all dependent on the cell line chosen to begin with.
It was suggested that a ‘cell line bank’ could be used as a reservoir of standard cell lines. However, he noted that cell lines are already available to buy and that this is not the critical issue affecting variability. Instead, pharmaceutical companies develop their own processes and set the conditions to grow the biologicals/biosimilars as required and the pharmaceutical companies are not required to share this information at any point, even after patent expiry. As such, variability is inevitable for biosimilars as cell lines and conditions will likely differ from those used to produce the reference products. Despite this, Professor Aws Alshamsan informed that the World Health Organization (WHO) is starting a programme to create a master cell line of bevacizumab so that it can be distributed to manufacturers in order to harmonize biosimilar product outcomes. Dr Ibrahim Aljuffali from the Ministry of Health Saudi Arabia advised that this was done in an attempt to reduce the cost of biosimilars and lessen the burden on national healthcare budgets. He also stated that this, and other similar cell lines, are likely to have a significant impact on the biosimilar landscape in the future.
The cell line bank approach could be rolled out to produce other biologicals after their patents have expired. Such banks would enable governmental agencies to harmonize globally by providing the starting material. However, such an initiative is the responsibility of the government regulators and not private companies.
Global harmonization of biosimilar regulatory approval procedures
Across the globe, biosimilar approval processes differ and harmonizing these processes could reduce the number of clinical studies carried out and make biosimilars more accessible. However, achieving global harmonization of regulation and approval procedures for biosimilars is likely to be complicated and lengthy.
Professor Trifirò highlighted that EMA and the US Food and Drug Administration (FDA), who have differing policies, regularly communicate regarding approval of biosimilar products. However, having different policies in different countries can reduce the credibility of biosimilars and the amount of trust practitioners and patients have in these products. For example, in the case of infliximab, which underwent studies verifying its use as a treatment option for rheumatology indications, the biosimilar drug product was also approved for treatment of inflammatory bowel disease (IBD) in Europe based on extrapolation of indication. Nevertheless, this extrapolation was not accepted by Health Canada and these contradicting policies may lead physicians to have a lack of confidence in the biosimilar product.
A lack of harmonization does not only cause a lack of trust in biosimilar products, but it also creates an increased workload for pharmaceutical manufacturers. They have to submit applications to different regulatory authorities worldwide which have different requirements and this means separate bioequivalence studies and applications are needed. This is expensive for producers in terms of both time and money.
Currently, there is no global harmonization for the regulation and approval of generic drug products. ‘These are far simpler than biologicals/biosimilars and as such, consensus is that we will not see global harmonization of biosimilars for some time. However, biosimilar regulation development is at an early stage in some countries and there are still only relatively few products approved globally, which means that regulatory processes can still be altered and developed to allow for harmonization’, said Professor Trifirò.
Biosimilar pricing
Biosimilars are designed to be lower-cost alternatives to originator products and facilitate access to biological medicines worldwide. Professor Kvien advised that in most markets, the cost reduction for the biosimilar infliximab is 30%. In Norway, when Remsima was approved it was initially 39% less expensive than the originator Remicade, in its second year it was 69% less expensive (possibly the highest cost reduction seen so far), and it is currently about 60% cheaper. This large cost reduction was largely achievable due to the Norwegian national tender system.
In Saudi Arabia, the price of each registered product is accessible through the SFDA website. Dr Alhomaidan described how biosimilar prices are set in Saudi Arabia according to three approaches and each product is priced on a case-by-case basis. The first pricing approach is based on reference pricing, where the price is set relative to its price in 30 reference countries. The second approach requires pharmacoeconomic evaluation and the price is set according to the results of this. And, the third is based on recommendations from the company/manufacturer. In the latter case, the company recommendation may be applied if it offers a product at a significantly lower price than reference product prices.
Professor Kvien added that the pricing transparency in Saudi Arabia is positive. In Europe, pricing can be concealed, but he believes that patients should be aware of the magnitude of saving that is achieved when choosing to switch to a biosimilar product.
The discussion highlighted that in some cases, a reference product manufacturer will gain biosimilar market access after originator patent expiry through production and marketing of a subsidiary’s biosimilar. In these cases, Professor Al-jedai believes that it is highly likely that some biosimilars will come from the same cell line as the reference products as several major pharmaceutical companies have started producing biosimilars of their own biological products. It would cost pharmaceutical companies and its subsidiaries a lot in terms of time and money, to start from scratch and create a new biosimilar when they know they already have a functioning and effective cell line that produces the already approved medicine. Under these circumstances, Professor Kvien believes the product is the same as the originator but re-packaged to be able to take a share of the biosimilar market. Here, the biosimilar must still undergo the same process of evaluation and approval of any other biosimilar product.
Biosimilar nomenclature
Much debate has surrounded the nomenclature of biosimilars in recent years. Across the globe, different approaches have been adopted. With respect to this, Professor Trifirò emphasized that traceability is key for any biological/biosimilar/chemical drug product. It should be possible to achieve this through product naming or identification via batch number. If products can be traced back to the batch level or at least brand name, adequate pharmacovigilance will be achieved so that the source of any adverse reactions/effects can be immediately identified.
In Europe, legislation has been passed to improve pharmacovigilance. In the event of an adverse event, the brand name must be evident (this differs for each biosimilar version of a reference product); and in the absence of a brand name, the manufacturer and non-proprietary name, plus batch level information, must be available. This is very important if a quality investigation is required.
In the US, the situation is slightly different. All biosimilar versions of a reference product currently have the same non-proprietary name which makes them indistinguishable from one another. With respect to this, it is also noted that if one company’s biosimilar product is flagged as a concern, this affects the credibility of all biosimilars with the same non-proprietary name, even if they are made by different manufacturers.
To address this problem and improve traceability of biosimilars, FDA implements a non-proprietary name policy with a four-lowercase letter suffix [9]. This aims to improve and facilitate pharmacovigilance in a multi-source environment which will prevent adverse event data collection that cannot be disaggregated. It should be possible to disaggregate all data and have product-level traceability that allows manufacturers to be held accountable and to develop product safety signalling. In addition to this, FDA’s new naming approach hopes to prevent inadvertent substitutions occurring as a result of product names being the same. This can occur at the pharmacy, rather than prescriber level, if non-proprietary names are shared by different products. At present, when two products have the same non-proprietary name, they can be perceived as being therapeutically equivalents. However, for biologicals and biosimilars, healthcare professionals need to examine the trial data to confirm that they are therapeutically similar and ensure a switch is acceptable.
Dr Aljuffali noted that the new naming guideline issued by FDA is likely to cause chaos and confusion. The idea is that approved products will be named retrospectively and as such, there will be significant impact on electronic health records, utilization of products, existing national drug codification, and finances. Dr Hajer Almudaiheem of the Ministry of Health Saudi Arabia agrees that the new US naming guideline will cause confusion. She also added that if products have different names this could create an artificial barrier to interchangeability due to the misperceptions of prescribers/practitioners.
Professor Trifirò emphasized that it is important to reach a balance in which nomenclature allows the traceability of products and also facilitates the interchangeability of drug products to ensure effective pharmacovigilance.
There are various other naming approaches being adopted around the world. Overall, this makes it difficult to assemble adverse event data in a manner that can be used and accessed internationally. A globally harmonized naming approach for biologicals/biosimilars would facilitate universal pharmacovigilance.
Patient choice, nocebo effect and education on biosimilars
In the future, it is hoped that patients will have a far greater role when it comes to decisions made about their treatment. However, ‘In Saudi Arabia, patients do not yet have this level of autonomy regarding their treatment and their physician makes most of the treatment decisions on their behalf’, said Professor Alshamsan. He stressed that this is likely a cultural issue governed by patient attitudes in GCC countries that are different to those seen in the west.
Professor Al-Foheidi reiterated that in Saudi Arabia the healthcare provider is generally in charge of treatment and switching. Here, both the practitioner and pharmacist have a say in the ultimate treatment decision. Clinicians weigh up the benefits and risks of treatment options based on the information available to them and although they may discuss this with patients, ultimately clinicians make the decision.
‘Patients should always be fully informed about their treatment options and decisions made by their clinicians. Through adequate patient education, the nocebo effect can be dramatically reduced’, said Professor Kvien.
This final comment was supported by Dr Nabila Al Lawati who noted that generally, patients are either educated or non-educated with respect to biosimilars and it is relatively easy to persuade the educated patients to take biosimilars. However, non-educated patients, particularly those who are older, often believe that the most expensive medication is the best and in such cases; it may be beneficial to leave the decision in the hands of the clinician. Professor Al-Foheidi commented that it can be more effective to make a general rather than an individual decision regarding biosimilars and patient advocacies can play an important role here.
Biological/biosimilar switching
The issue of switching between biologicals and biosimilars and the safety of multi-switching was raised. Professor Trifirò advised that the pre- and post-marketing evidence gathered so far, with respect to switching from different sources, had not identified any clinical issues. In Europe, a lot of data have been accumulated which allow for the assessment of the long-term effects of switching, however, this is difficult to investigate in a post-marketing setting. He added that ideally, studies similar to the NOR-SWITCH study [8] for each biosimilar would be carried out, but in reality performing such expensive RCT (randomized clinical trials) to explore all possible reference product/biosimilar switches is impossible. So, efforts need to be made to investigate the effects on long-term switching especially for drugs that have long-term effects, such as rituximab in real-world settings. He noted that healthcare databases including claims databases and electronic medical records as well as drug registries have the potential to help here. Understanding the long-term effects of switching and multi-switching between biosimilars is one of the key challenges that needs to be addressed and this requires an international concerted effort to develop the best methodological approach.
Professor Kvien informed about the Norwegian disease-modifying antirheumatic drugs (DMARD) registry established in 2000. The registry continues to enrol patients who are switching to a biosimilar and those starting treatment with a biosimilar. He estimates that 80% of patients using biosimilars enrol with the registry. However, more data are needed to conduct analysis of biosimilars in a way similar to that carried out in the NOR-SWITCH trial [8]. This registry will soon merge with other registries, such as the death and cancer registries, so that there is more robust data on safety.
Biosimilars in Saudi Arabia
Dr Alhomaidan confirmed that in Saudi Arabia there are guidelines on the quality requirements for biosimilars and there is a pricing system in development for biosimilars, but this is not well established yet. The safety and efficacy requirements are the same as those adopted by EMA. In both Europe and Saudi Arabia, guidelines do not aim to promote biosimilar usage, but instead promote the dissemination of the correct information about them to ensure their quality, safety and efficacy.
In Saudi Arabia, biosimilar products are evaluated during registration, with the first consignment, and then are continually assessed by taking random market samples. When it comes to manufacturer inspections, each manufacturer is inspected by a team of three inspectors before biosimilar production is initiated. He added that Saudi Arabia is in the process of setting up a centre that will enable phase I clinical trials to be carried out by an approved, licensed clinical trials team. These will subsequently be reviewed by the benefit-risk assessment team to facilitate the safe and efficacious entry of biosimilars to the market.
|
Action points highlighted in the discussions
The key findings from the discussions of the stakeholder meeting are summarized as below:
- Further research and investment into ‘master cell bank’ could enable further cost reductions in biosimilar products and reduce variability between products and batches.
- Action needs to be taken to achieve global harmonization of biosimilar approval and regulation procedures. This should be done in the near future, while currently there are only few products approved in the global market.
- Biosimilar pricing transparency is positive and should continue.
- Action should be taken to ensure that all biosimilar products globally are traceable to batch level to ensure adequate pharmacovigilance is upheld.
- Global consistency, transferability and/or harmonization of biosimilar nomenclature needs to be achieved.
- Strong governmental regulations should be in place to ensure drug products can be tracked without necessarily having to initiate new biosimilar nomenclature.
- The long-term effects of switching and multi-switching between biosimilars and/or reference products need to be addressed. This requires an international concerted effort to develop an optimal methodological approach.
- Electronic healthcare records need to be developed and implemented to facilitate pharmacovigilance and gather further data on switching.
- Biosimilar patient registries could be established and implemented to gather further data on switching.
|
Parallel case study working sessions
After the formal presentations and discussions, the audience was presented with data for two semi-fictional SBPs, both are a trastuzumab monoclonal antibody. The participants were divided into two discussion groups where they evaluated the fictional data supplied. This was carried out in a similar way to that which occurred at previous GaBI meetings [2].
Summary discussion of case study of therapeutic protein monoclonal antibody – candidate 1
Based on the information the two groups received, Dr Nabila Jawad Al Lawati and Associate Professor Khalid Alsaleh agreed that the first candidate, monoclonal antibody IgG1, did qualify as a biosimilar of the reference product. Dr Al Lawati noted that, with respect to the physiochemical attributes, the charge profile acidity and deamination were not similar to the originator, but this did not affect the products activity or quality attributes. In terms of biological attributes, all is within predefined limits, so the candidate does qualify as a biosimilar from a quality perspective. Professor Alsaleh noted that his discussion group was aware that there are some differences between the candidate and the reference product, but these were not major.
To remediate the differences between the IgG1 candidate and the biosimilar, Dr Al Lawati’s group thought that the product applicant could be asked to alter the charge profile to ensure it is more acidic. Professor Alsaleh’s group said the chemical structure of the candidate could be altered but this would then need further biosimilarity studies to be carried out on it which would negate the point in developing a biosimilar as it would cost a lot in terms of time and money.
To address the ‘residual uncertainty’ in the preclinical and/or clinical studies, Dr Al Lawati’s group suggested that the PK/PD parameters be obtained in clinical studies. Professor Alsaleh’s group said that the bio-immunogenicity could be addressed through clinical studies. Here, the group noted that there was a hard endpoint observable when using the biosimilar. This was seen in the complete initial pathological response when treatment was given, and in the follow-up studies carried out in the year after initial treatment. The overall response to the treatment was good – 46% for the candidate versus 48% for the reference product.
Dr Al Lawati’s group said that, for extrapolation to be recommended, the applicant would need to submit more information about the mechanism of action and site of action. Only with this additional information could a decision be made. On the other hand, Professor Alsaleh’s group said that the IgG1 candidate could be extrapolated but only if there was no other choice. However, if the reference product was available this would be used in preference to the biosimilar for extrapolated indications. He explained that for the reference product trastuzumab, there is a combination indication for metastatic settings which makes extrapolation complicated. He noted that, if a trastuzumab, biosimilar were to be used in such a situation, a combination study should be required, not just clinical data.
Summary discussion of case study of therapeutic protein monoclonal antibody – candidate 2
Based on the information the two groups received, Dr Al Lawati and Professor Alsaleh agreed that the second candidate did not qualify as a biosimilar of the reference product as there8 were many differences in the physiochemical properties of the candidate and the originator. Dr Al Lawati noted that there were particularly significant differences in the total protein content which is an important parameter. The charge profile and deamination also differed. In addition, there were differences in the aggregates and particulates which are important factors when considering the immunogenicity.
Dr Al Lawati said that the physiochemical data need to be a closer match to those of the reference product before further steps are taken towards candidate approval. According to Professor Alsaleh, there is a very soft endpoint to the study that should not generally be used as a primary endpoint and this needs to be amended. Based on the physiochemical data, both groups agreed that the candidate should not undergo preclinical/clinical trials at this point. As the candidate is not a suitable biosimilar, it cannot be extrapolated for any indications.
Conclusion
Biosimilar medicines are being increasingly used and available across GCC countries. The first GCC meeting was successful in bringing representatives from GCC nations together with experts from Europe, Saudi Arabia and the US, to discuss the best routes forward for successful biosimilar approval and regulation and enabled action points to facilitate biosimilar uptake with appropriate pharmacovigilance to be outlined.
Speaker Faculty and Moderators
Speakers
Assistant Professor Meteb Al-Foheidi, MD, FRCPC, Saudi Arabia
Assistant Professor Khalid A Alburikan, PharmD, BCPS, Saudi Arabia
Ali M Alhomaidan, PhD, Saudi Arabia
Assistant Professor Abdulaziz Alhossan, PharmD, MPH, BCPS, Saudi Arabia
Professor Ahmed H Al-jedai, PharmD, MBA, BCPS, FCCP, FAST, Saudi Arabia
Assistant Professor Musaed Abdullah Alkholief, PhD, Saudi Arabia
Assistant Professor Mohammad A Alsenaidy, MSc, PhD, Saudi Arabia
Professor Aws Alshamsan, BPharm, RPh, PhD, Saudi Arabia
Thomas Felix, MD, USA
John Bradley Jordan, PhD, USA
Professor Tore Kristian Kvien, MD, PhD, Norway
Professor Gianluca Trifirò, MD, PhD, Italy
Moderators
Nabila Jawad Al Lawati, PhD, Oman
Associate Professor Khalid Alsaleh, MD, Saudi Arabia
Editor’s comment
All moderators had provided the discussion/conclusion of the group discussion, read the report and revised the content of the summary discussion of the case study.
Acknowledgements
The Generics and Biosimilars Initiative (GaBI) wishes to thank Assistant Professor Abdulaziz Alhossan, for his support through the offering of advice and information during the preparation of this scientific meeting; as well as Professor Gianluca Trifirò, chair of the 2017 GCC stakeholder meeting; for his strong support in facilitating the discussion in the scientific meeting.
The authors and speakers would like to acknowledge the help of all the meeting speaker faculty and participants, each of whom contributed to the success of the meeting and the content of this report, as well as the support of the moderators: Dr Nabila Jawad Al Lawati and Associate Professor Khalid Alsaleh in facilitating meaningful discussion during the parallel case study working sessions, presenting the discussion findings at the meeting and contributing in the finalization of this Meeting Report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Competing interests: The scientific meeting was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. Gray E, Matejtschuk P, Thorpe R. Quality assessment of biosimilars in Colombia – reducing knowledge gaps. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):79-83. doi:10.5639/gabij.2018.0702.017
2. Griffiths E, Ekman N, Thorpe R. First ASEAN educational workshop on regulation and approval of biosimilars/similar biotherapeutic products 2017 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):127-32. doi:10.5639/gabij.2018.0703.025
3. Walson PD, Thorpe R. First Latin American educational workshop on similar biotherapeutic products, Mexico City, Mexico, 20 January 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):143-8. doi:10.5639/gabij.2015.0403.031
4. Walson PD, Thorpe R. First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars, Dubai, United Arab Emirates, 1 September 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):173-7. doi:10.5639/gabij.2015.0404.039
5. Walson PD, Thorpe R. First Turkish Interactive Workshop on Regulation and Approval of Similar Biotherapeutic Products/Biosimilars 2016. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):134-8. doi.10.5639/gabij.2016.0503.034
6. Generics and Biosimilars Initiative. First GCC stakeholder meeting on approval process interchangeability substitution and safety of biosimilars 2017; 20 November 2017; Riyadh, Saudi Arabia. Available from: www.gabi-journal.net/first-gcc-stakeholder-meeting-on-approval-process-interchangeabilitysubstitution-and-safety-of-biosimilars-2017
7. Alhomaidan AM, Aljuffali IA, Alnutaif FSi, AL-Howaimel NA. Pricing of biosimilars in Saudi Arabia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(1):27-9. doi:10.5639/gabij.2016.0501.007
8. Perks B. Randomized non-inferiority trial fails to find inferiority switching from infliximab originator to CT-P13 biosimilar. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):188–9. doi:10.5639/gabij.2017.0604.042
9. Rolandini Jensen A. US FDA proposals for naming of biologicals and labelling of biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):140-3. doi.10.5639/gabij.2016.0503.036
|
Author for correspondence: Professor Gianluca Trifirò, MD, PhD, Policlinico ‘G Martino’, Dipartimento di Medicina, Clinica e Sperimentale, 1 Via Consolare Valeria, IT-98125 Messina, Italy.
|
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Source URL: https://gabi-journal.net/first-gcc-stakeholder-meeting-on-approval-process-interchangeability-substitution-and-safety-of-biosimilars-2017-report.html
Copyright ©2025 GaBI Journal unless otherwise noted.
Generics and Biosimilars Initiative (GaBI)
Tel: +32 474989572 | Fax: +32 14 583 048