Pharmacovigilance, traceability and building trust in biosimilar medicines
Published on 2017/06/12
Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):135-40.
Author byline as per print journal: Professor Teun van Gelder, MD, PhD; Benedicte Lunddahl, DVM; Professor Barbara OM Claus, PharmD, PhD
|
Abstract: |
Submitted: 22 May 2017; Revised: 21 June 2017; Accepted: 21 June 2017; Published online first: 4 July 2017
Introduction
The Biosimilar Medicines Group (formerly EBG) held a satellite symposium entitled ‘Biosimilar medicines in clinical practice – important role for hospital pharmacists!’ on 22 March 2017 at the 22nd Congress of the European Association of Hospital Pharmacists (EAHP) in Cannes, France.
The Biosimilar Medicines Group represents the leading pharmaceutical companies that are developing, manufacturing and marketing generic and biosimilar medicines across Europe. It is a sector group of Medicines for Europe (previously the European Generic medicines Association).
Biosimilar medicines have been used successfully in the European Union (EU) for over 10 years and there are now 32 approved biosimilar medicine products on the market in the EU today [1]. This number has been steadily rising, with 12 products being added to the market last year [2]. Biosimilar medicines improve access to modern therapies for millions of European patients receiving both chronic and acute care. They offer more affordable treatment pathways, increase medicine accessibility and make healthcare funds available for other uses [3].
Traceability of biologicals
Dr Benedicte Lunddahl, (Head of Pharmacovigilance at the Danish Medicines Agency, Lægemiddelstyrelsen, DKMA), gave a presentation that highlighted how the agency was tackling the reporting of adverse events and traceability for both originator biological and biosimilar medicines.
In Denmark, there is a national recommendation for the use of biosimilar medicines that was initiated in 2015. This involves a nationwide tender process whereby biosimilar medicines that win the tender are those supplied to hospital pharmacies. Both treatment naïve patients, and those in established treatment regimens are treated with biosimilar medicines. Infliximab was the first biosimilar medicine to win a tender which has led to high uptake of this medicine in Denmark when compared to other EU countries. In 2016, see Figure 1, biosimilar infliximab won the tender, which has led to similar high usage of this medicine.

Denmark works under the legal framework set out by European pharmacovigilance legislation and EU Good Pharmacovigilance Practices (GVP) modules [4], in addition to Danish law and possible executive orders. This framework incorporates the need to trace all biological molecules, as initially outlined in EU pharmacovigilance legislation of 2010 [5]. This states that all EU Member States are required to ensure that biological medicine products can be identified and traced, and gives countries the power to impose specific obligations on healthcare professionals to ensure this. Despite this legislation being in place, Dr Lunddahl noted that there are issues associated with implementing product tracking in the hospital setting [6]. She added that similar traceability obligations can be imposed on Marketing Authorisation Holders to ensure the collection of Adverse Drug Reaction (ADR) data and any follow-up data. In the Individual Case Safety Reports of biological medicines, it is obligatory that the batch number is recorded, see Table 1.

To address patients’ concerns and inform physicians about biosimilar medicines, the DKMA set up an Action plan on Biological Medicines, which aims to:
- Encourage monitoring of biological medicines at the product level, thus improving traceability
- Raise awareness about biosimilar medicines
- Promote IT solutions to ease reporting of ADRs
- Focus on the surveillance of biological and biosimilar medicines
To improve monitoring of biological medicines in Denmark two executive orders came into effect in January 2016. These require healthcare professionals to record the brand name and batch number when prescribing a biological or biosimilar medicine. They also make it mandatory for healthcare professionals to include, where possible, the brand name and batch number when reporting suspected ADRs [7].
To facilitate the monitoring of biological medicines, the DKMA has made an electronic list of biological medicines and their alternative biosimilar medicine products. This is to be updated when new biosimilar medicines become available on the Danish market. There are also electronic reporting forms available to physicians and consumers so that ADRs can be easily recorded. The form available to physicians has popups to remind the physician to enter the brand name and batch number of the products prescribed. The patient’s form has a specific field for these details to be added.
The DKMA has concluded that their ADR data collected supports the equal safety profiles of biosimilar medicines and reference biological medicines. This data also shows no correlation between batches and reported ADRs. In addition, scientific literature, including the Norwegian NOR-SWITCH study, which is investigating the efficacy, safety and immunogenicity when switching patients from the reference biological to biosimilar infliximab, supports the similar safety profiles of reference biological and biosimilar medicines [8].
Dr Lunddahl noted that raising awareness of biosimilar medicines amongst physicians and patients is key to increasing their uptake. In Denmark, initial meetings with patient organizations were held to outline what information needed to be supplied. Targeted communication routes were then created, which included information leaflets handed out to patients by physicians designed to initiate patient–physician dialogue; a clear and straightforward Q&A on the DKMA website, and information videos available online in which patients ask questions and have these answered by regulators.
Building trust in biosimilar medicines
A presentation by Professor Teun van Gelder (internist, nephrologist and clinical pharmacologist at Erasmus MC in Rotterdam, The Netherlands), highlighted how to build trust in biosimilar medicines.
In The Netherlands, generic medicines are procured via a tendering system whereby prescribed drug products have the potential to be changed every six to 12 months as they are replaced by the lowest price alternative. This means that patients may receive a different formulation time and time again. Professor van Gelder highlighted that it is likely they will be switched from the expensive originator, to a generic, and then to another cheaper generic medicine, and so on. If not managed properly by the physician and/or pharmacist, this can lead to patient confusion and ultimately also to reduced adherence or to duplicate treatment. In this scenario, physicians have lost control over the actual medication that a patient receives. It is possible for physicians to apply for a specific formulation to be dispensed, but only if they provide medical justification. This perceived lack of control over drug formulations dispensed has instigated concerns over the introduction of biosimilar medicines in The Netherlands.
When it comes to biosimilar medicines, there are three generations of therapeutic proteins, see Table 2.

There are a number of reasons that can impede the penetration of biosimilar medicine usage in a given EU country. In some cases, physicians may lack education and understanding of the concept of biosimilarity [9] and, even when knowledgeable on biosimilar medicines, they may not trust in their safety and efficacy [10].
Professor van Gelder described a scenario in which there were side effects associated with an approved biological medicine. This example was chosen to emphasize that there can be issues related to such approved medicines when products are manufactured, stored or handled incorrectly. Here, he explained that erythropoietin (EPO) is a hormone secreted by the kidneys that increases the rate of production of red blood cells in response to falling oxygen levels in tissues. In patients suffering from anaemia due to chronic kidney disease, the decreased production of EPO with declining renal mass is considered the primary etiologic factor. Anaemia is associated with symptoms that include fatigue, weakness and dyspnea, as well as worsening quality of life and performance status. To treat this condition, the first biological recombinant human EPO produced was epoetin alfa. He explained that, when trying to attain a normal patient haemoglobin level by increasing the dose of this biological EPO, patients could develop hypertension and higher risk of cardiovascular events. There were also increased reports of epoetin-associated pure red cell aplasia [11, 12] when Eprex was the formulation treatment (this was one of a number of approved formulations available on the market at the time). After procedures were adopted to ensure appropriate storage, handling and administration of Eprex to patients with chronic kidney disease, the exposure-adjusted incidence decreased by 83% worldwide. Professor van Gelder noted that the main concern expressed in the case of biosimilar medicines is that of immunogenicity, as when compared to the original biological medicine these products undergo different manufacturing processes and processing and purification techniques and have different master cell lines, inert ingredients and packaging.
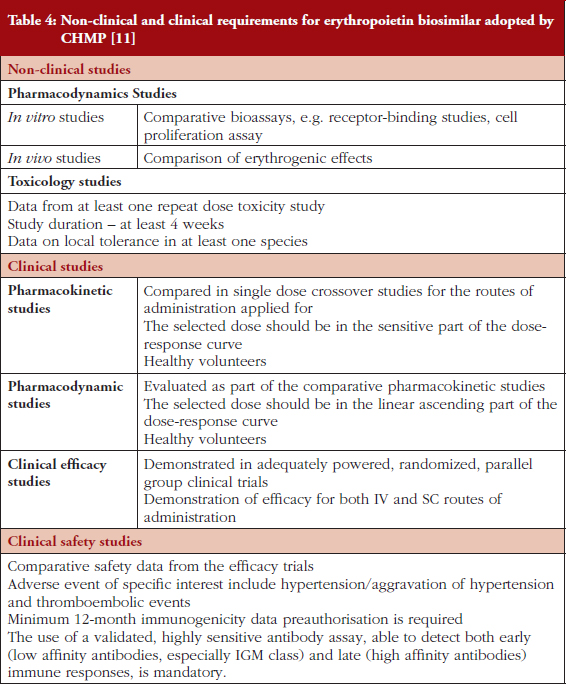
Despite the issues related to first-generation innovator, Eprex, highlighted, there are currently a number of epoetin biosimilars on the market in Europe, see Table 3. These have undergone a registration procedure that includes demonstration of safety and efficacy through clinical studies [11], see Table 4. To date, there is no evidence for an increase in pure red cell aplasia or any other adverse events associated with biosimilar epoetins. However, despite the apparent safety of these biosimilar medicines, the extent of their uptake by patients in EU countries is widely varied.

Professor van Gelder’s example emphasized the need for doctors and patients to be made aware that, within the life cycle of an originator biological medicine, there will be numerous changes in the manufacturing processes, including its purification, inert ingredients and packaging. These changes can also lead to the induction of an unexpected immune response, such as that which occurred after the formulation of Eprex was changed [12, 13].
Professor van Gelder noted that doctors would rather stick to what they know, especially as there is no therapeutic advantage, but only a cost benefit in using a biosimilar medicine. It will therefore take a very long time to build trust in these products. This is where regulators have an important role to play in passing on their knowledge. They need to inform doctors that, over the past 10 years, there has not been a single serious incident involving a biosimilar medicine, that the regulatory system is working as expected, and that the mistrust of biosimilar medicines is unjustified. As a result, prescribers should be convinced that biosimilar medicines offer a (financial) benefit to society without compromising the quality of treatment. Prescribers need to be well informed to increase their trust in biosimilar medicines.
Professor van Gelder concluded by noting that at the Erasmus Medical Centre in The Netherlands, pharmacists, doctors and representatives from drug producers meet and discuss the pros and cons of introducing biosimilar medicines when they become available. This improves transparency and increases trust. Overall, despite the proven safety and efficacy of biosimilar medicines, medical doctors may still feel uncertain or reluctant to use biosimilar medicines and we need to:
- Teach them
- Increase transparency by identifying push and pull factors
- Involve them
- Avoid repetitive substitutions among biosimilar medicines
- Be aware that incentives work
Pharmacists have a key position and can act as independent educators for prescribers and patients.
The role of hospital pharmacists
Professor Barbara Claus, a pharmacist from the University Hospital Ghent, Belgium, gave a presentation highlighting what needs to be considered by hospital pharmacists when biosimilar medicines enter the hospital.
Professor Claus highlighted the importance of pharmacists’ understanding and knowledge of biosimilar medicines. They should be able to answer questions from other healthcare professionals on biosimilar medicines. This means that they need to be aware of the regulatory approval process for biosimilar medicines and how this process differs from that for originator biological medicines:
Originator medicine: phase I–III trials to demonstrate a positive risk–benefit balance
Biosimilar medicine: physicochemical and functional characterization and pharmacokinetic/pharmacodynamics (PK/PD) studies to demonstrate comparability
The approval of biosimilar medicines is based on an inverse triangle, see Figure 2, with most evidence of biosimilarity being required for structural and physicochemical characterization. Biosimilar trials are then based on sensitive endpoints, e.g. in oncology ‘response rate’, rather than hard endpoints, such as ‘overall survival rate’.

The pharmacist’s knowledge should also extend to the meaning of the black triangle ▼, which is used to indicate that the medicine is part of a risk management programme for biological medicines and that traceability is mandatory. The European Medicines Agency (EMA) published a new, GVP module in 2016 (EMA/168402/2014) [4].
The role of the hospital pharmacist is important for the interchangeability, pharmacovigilance and traceability of biosimilar medicines, as well as in communication during the hospital uptake process.
Pharmacists should know that interchangeability does not mean automatic substitution. At present, there is confusion over interchangeability among physicians and pharmacists. It is thought that this may be because the EU and US definitions are not the same, and across the EU interchanging with a biosimilar medicine is a Member State decision. In the US, interchangeability includes automatic substitution, which can lead to switching at the pharmacy level without prescribers’ consent. However, Professor Claus emphasized that switching should be a controlled and informed action guided by a traceable process. It should be carried out in agreement with the prescriber with the aim of achieving the same clinical goal in a patient. Professor Claus noted that switches do currently occur and this is being monitored. Switching patterns are being recorded to track any possible issues, although none appear obvious at present.
To facilitate pharmacovigilance, as stated by Dr Lunddahl, Professor Claus reinforced the fact that pharmacists should collect both brand names and batch numbers in patient records. EAHP has suggested that pharmacists track by scanning barcodes of single units. However, increasing traceability by implementing bar-code scanning is not universal at present and manual recording can be time consuming.
Communication between all stakeholders (physicians, pharmacists, pharmacy technicians and patients) and their overall education needs to be improved. Incentivization for all healthcare providers is proving beneficial and, once a decision is made to use a specific biosimilar medicine, educational material should be available to all, so that patients are informed and are encouraged to report any adverse effects. In The Netherlands, such an initiative quickly resulted in a controlled switch of over 90% of patients to a biosimilar medicine, without an increase in meaningful ADRs.
In addition, Professor Claus noted that the landscape is changing and that the 25 top pharmaceutical companies are now developing biological and biosimilar medicines.
In conclusion, she added that pharmacists, companies, hospital boards, formulary committees and physicians need to merge ideas and exchange information. The final goal is to provide consistent information to patients and to create sustainable treatment regimes now, and in the future. In the end, the continuity of product will produce better health and well-being for patients.
Discussion: audience Q&A
Following the main presentations, the three speakers answered questions from the audience and Chair, Ms Sue Naeyaert of Merck Biosimilars.
Professor van Gelder stated that formulary committees need to involve medical doctors to avoid resistance to biosimilars and comply with traceability initiatives. It is beneficial to have doctors meet with representatives from pharmaceutical companies at an early stage of negotiations. Professor Claus added that it is important to discuss tendering and for all to understand the criteria for this, so that transitions are smooth and additional discussions are not required.
Professor Claus noted that pharmaceutical companies are coming up with incentives and services, other than reducing prices, to boost sales and the uptake of biosimilar medicines. These include 24-hour access lines for patients, travel kits and services, such as drug transport to hospital and discussions with patients. Professor van Gelder said that it takes time for a medical doctor to explain the aspects of administration to patients. When drug providers offer a service to explain the aspects of the drug administration processes to patients, doctors’ time is saved and they can go on to assess and treat more patients.
Dr Lunddahl discussed the executive order enforced when infliximab entered the Danish market. This was brought about by a medical council expert group that recommended its approval. In Denmark, it is known that when a drug product is recommended, regulatory councils will not compromise safety and efficacy of treatment when approving products. This led to significant use of biosimilar medicines in Denmark, as physicians have high levels of confidence and trust in the councils. Professor Claus added that, in Belgium, uptake was low by comparison. There is now a close follow-up of the results of the tendering processes and physicians need to consider biosimilar medicines in the same way as originator products.
Conclusion
The Biosimilar Medicines Group satellite symposium, ‘Biosimilar medicines in clinical practice – important role for hospital pharmacists?’ saw Ms Sue Naeyaert, Dr Benedicte Lunddahl, Professors Teun van Gelder and Barbara Claus, discuss the traceability of biosimilar medicines, how to build trust in biosimilar medicines, and the role that hospital pharmacists can play in improving their acceptance and usage.
Dr Lunddahl’s presentation highlighted how the Danish Medicines Agency was tackling the reporting of adverse events and traceability for both originator biological and biosimilar medicines. Their action plan aimed to encourage monitoring of biological medicines at the product level to increase traceability, raise awareness of biosimilarity, promote IT solutions to ease reporting of ADRs, and focus on the surveillance of biological and biosimilar medicines. This has led to the high usage of biosimilar medicines relative to other countries across the EU.
Professor van Gelders’s presentation highlighted the key concerns that medical doctors may have when prescribing biosimilar medicines. The main issues he highlighted are that physicians have a lack of trust in safety and efficacy of biosimilar medicines and that they do not want to relinquish their control over the prescribing of biological products. He noted that prescribers’ concerns are not validated by clinical evidence and that they need to be better informed. Discussions between physicians, pharmaceutical companies and pharmacists can lead to increased transparency and higher levels of trust.
Professor Claus’s presentation highlighted considerations that should be made by hospital pharmacists when biosimilars enter the hospital. She noted the importance of pharmacists’ understanding and knowledge of biosimilar medicines. They should be able to answer questions from other healthcare professionals on biosimilar medicines and need to be aware of the regulatory process for the approval of biosimilar medicines. In addition, improvements to healthcare systems need to be made to increase traceability.
Overall, emphasis by the speakers was on the need for increased education to improve trust in biosimilar medicines, and the need for traceability of biosimilar medicines. They demonstrated this by describing the scenarios occurring in different EU countries and determining which tactics have led to increased penetration of biosimilar medicines in Member States.
Acknowledgement
The authors wish to thank Alice Rolandini Jensen, MSci, GaBI Journal Editor, in preparing this meeting report.
Disclosure of financial and competing interests: The satellite symposium was organized by the Biosimilar Medicines Group, a sector group of Medicines for Europe. Fees were paid to the organization for the conference slot, however, none of the speakers received direct payments from Medicines for Europe.
Professor Teun van Gelder has been a member of the speakers’ bureau or has participated in advisory boards for Astellas, Chiesi, Novartis, Sandoz and Teva. He has also received research funding from Chiesi, and travel support from Roche.
Professor Barbara OM Claus and Dr Benedicte Lunddahl declare that there is no conflict of interest to report.
Provenance and peer review: Not commissioned; internally peer reviewed.
Authors
Professor Teun van Gelder, MD, PhD, Internist – nephrologist/clinical pharmacologist, Departments of Internal Medicine and Clinical Pharmacology, Erasmus University Medical Center, PO Box 2040, NL-3015 CE Rotterdam, The Netherlands
Benedicte Lunddahl, DVM, Danish Medicines Agency, Division of Pharmacovigilance and Medical Devices, 1 Axel Heides Gade, DK-2300 Copenhagen S, Denmark
Professor Barbara OM Claus, PharmD, PhD, Pharmacy Department, Ghent University Hospital, 185 De Pintelaan, BE-9000 Ghent, Belgium
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jun 21]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
2. Ekman N, Vulto AG, Cornes P. Reducing healthcare costs and building trust in biosimilar medicines. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):84-8. doi:10.5639/gabij.2016.0502.020
3. GaBI Online – Generics and Biosimilars Initiative. EMA finalises pharmacovigilance guidance [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jun 21]. Available from: www.gabionline.net/Guidelines/EMA-finalises-pharmacovigilance-guidance
4. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP) Product – or Population-Specific Considerations II: Biological medicinal products. EMA/168402/2014. 4 August 2016 [homepage on the Internet]. [cited 2017 Jun 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/08/WC500211728.pdf
5. Giezen TJ, Straus SMJM. Pharmacovigilance of biosimilars: challenges and possible solutions. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):118-9. doi:10.5639/gabij.2012.0103-4.033
6. Claus BOM, Somers A, Bauters T. Generics and Biosimilars Initiative Journal (GaBI Journal). Pharmacovigilance of biosimilars and other biologicals within the hospital: current practices and future challenges. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):24-6. doi:10.5639/gabij.2017.0601.005
7. Lunddahl B. Pharmacovigilance on biologicals and biosimilars: a Danish perspective. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):123-4. doi:10.5639/gabij.2016.0503.030
8. GaBI Online – Generics and Biosimilars Initiative. NOR-SWITCH study finds biosimilar infliximab not inferior to originator [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jun 21]. Available from: www.gabionline.net/Biosimilars/Research/NOR-SWITCH-study-finds-biosimilar-infliximab-not-inferior-to-originator
9. Kurki P. Biosimilars for prescribers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):33-5. doi:10.5639/gabij.2015.0401.008
10. Annese V, Avendaño-Solá C, Breedveld F, Ekman N, Giezen TJ, Gomollón F, et al. Roundtable on biosimilars with European regulators and medical societies, Brussels, Belgium, 12 January 2016. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):74-83. doi:10.5639/gabij.2016.0502.019
11. European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins (Revision). EMEA/CHMP/BMWP/301636/2008 Corr.* 18 March 2010 [homepage on the Internet]. [cited 2017 Jun 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089474.pdf
12. Giezen TJ, Schneider CK. Safety assessment of biosimilars in Europe: a regulatory perspective. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):180-3. doi:10.5639/gabij.2014.0304.041
13. GaBI Online – Generics and Biosimilars Initiative. Epoetin alfa and pure red cell aplasia [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Jun 21]. Available from: www.gabionline.net/Biosimilars/Research/Epoetin-alfa-and-pure-red-cell-aplasia
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


