Aligning environmental, social, and governance to clinical development: moving towards more sustainable clinical trials
Published on 30 July 2024
Generics and Biosimilars Initiative Journal (GaBI Journal). 2024;13(3):141-50.
|
Abstract: |
Introduction
While previously, the focus was mostly on environmental issues, the current wave of the earth’s sustainability consciousness is now unprecedented [1], and commitment towards environmental, social, and governance (ESG) goals has started impacting how business operations are conducted globally. ESG goals are the non-financial metrics that companies, including pharma, have started working on to assess their governance standards, social responsibility, and environmental influence. With clients, consumers, and potential employees all gravitating towards businesses that prioritize ESG initiatives, these have become a biomarker of business purpose and success, as with a commitment towards ESG, businesses can differentiate themselves and gain competitive advantage. As the needle moves, we find that a large majority (83%) of consumers think companies should be actively shaping ESG best practices, and 86% of employees prefer to work for companies that care about these issues [2]. Science Based Targets initiatives (SBTi) guide organizations on how much and how quickly they need to reduce their greenhouse gas (GHG) emissions to prevent further climate change. This will entail more target-based actions beyond greenwashing [3]. Pharma companies are encouraged to take the climate pledge to reach net-zero carbon emissions by 2040. Many companies have committed to reducing emissions but have yet to set a target for SBTi net zero [1].
This article discusses the three principles of ESG and how drug development can be aligned to these principles. It also discusses the potential global challenges and solutions to applying these principles, considering different regulatory environments. The Low Carbon Clinical Trials Working Group has developed guidance for trialists to determine the clinical trials’ carbon footprints [4, 5]. The environment is but one of the ESG pillars, and companies need to excel in all three pillars of ESG. Inadequate performance in environmental and social issues leads to inadequate governance. Artificial intelligence (AI) and machine learning (ML) can build efficiency and enhance the governance of the trial by de-centralizing them [6, 7]. Table 1 provides the ESG values that guide companies, including the pharmaceutical industry, toward creating measurable goals [1, 4, 6-12].
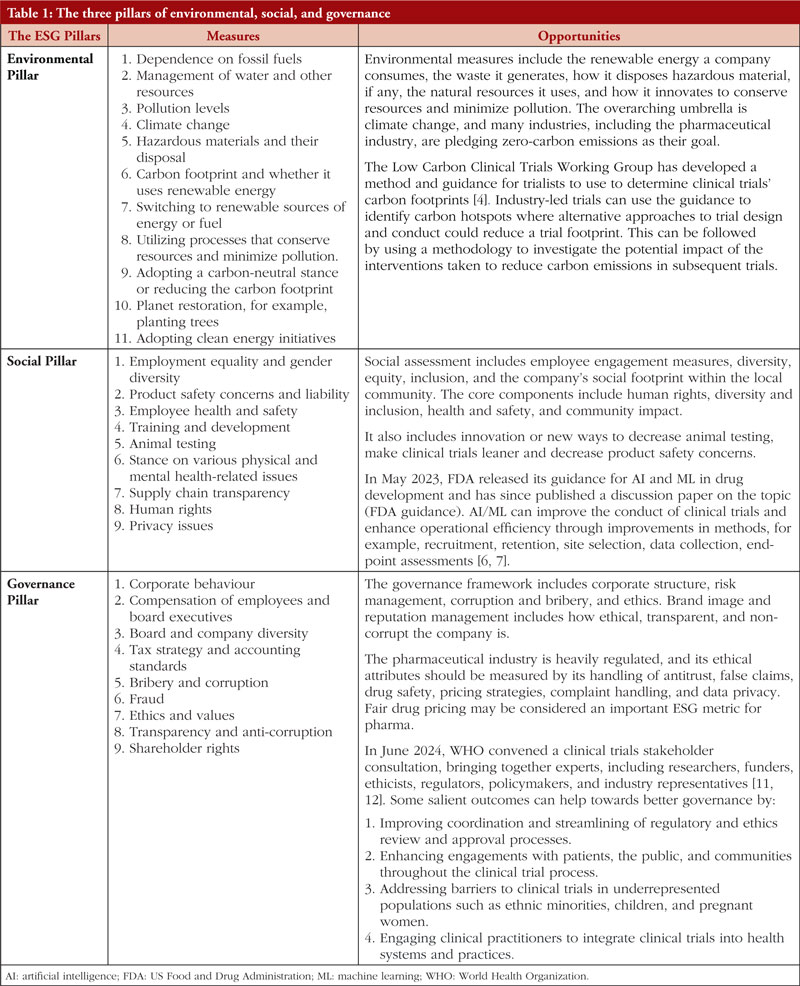
It is also pertinent to add here that policymakers and regulators are equally responsible for driving the ESG goals, as this needs to be a collective initiative. This article also discusses the recent guidelines proposed by the UK National Institute for Health Research (NIHR), US Food and Drug Administration (FDA), European Medicines Agency (EMA), and the World Health Organization (WHO) for sustainable drug development, keeping the ESG goals in focus [6, 7, 9-13].
ESG principles weaved into the governance of clinical trials
As pharmaceutical companies have pledged to zero carbon emissions, regulators and policymakers are also starting to focus on creating guidelines and policies to encourage the industry towards innovative clinical trial designs and requirements. The following section discusses the recent guidelines proposed by the EMA, FDA, NIHR, and WHO for sustainable drug development.
UK NIHR carbon reduction guidelines and recommendations
Under the Climate Change Act 2008, the United Kingdom (UK) government has committed to significantly reducing UK GHG emissions by 2050 [13]. The National Health Service (NHS) UK has, in fact, committed to becoming carbon net zero and may be the first and globally the biggest public health system in the world to adopt this approach [14]. It has a carbon footprint of about 21 million tons of CO2 annually, representing around 25% of public sector GHG emissions [13]. The NHS is committed to meeting the targets set by the Climate Change Act of 2008 by introducing the Carbon Reduction Strategy for the NHS in England [13].
The National Institute for Healthcare and Research (NIHR) Carbon Guidelines Working Group has developed Carbon Reduction Guidelines, published on 30 July 2019, for researchers [13]. These guidelines demonstrate how the principles of good carbon management and sensible study design can apply to many clinical studies, thereby reducing the carbon footprint without increasing the administrative burden on researchers, see Table 2. By implementing these guidelines, drug developers can align to ensure that average carbon emissions occur at the lowest levels possible while meeting the goal of the UK’s NHS, of becoming a carbon net zero organization [13].

ESG is integral to clinical trial strategy
The environmental and social impact of carrying out thousands of clinical trials is tremendous. Globally, 4.4%–4.6% of GHG emissions come from pharma [15]. The pharmaceutical industry is estimated to produce 50% more GHG emissions than the automotive industry operations [16]. The total global emissions of the pharma sector amounted to about 52 megatons of CO2e in 2015, more than the 46.4 megatons of CO2e generated by the automotive industry in the same year, making the 28% smaller pharma market 13% more polluting than the automotive sector [16].
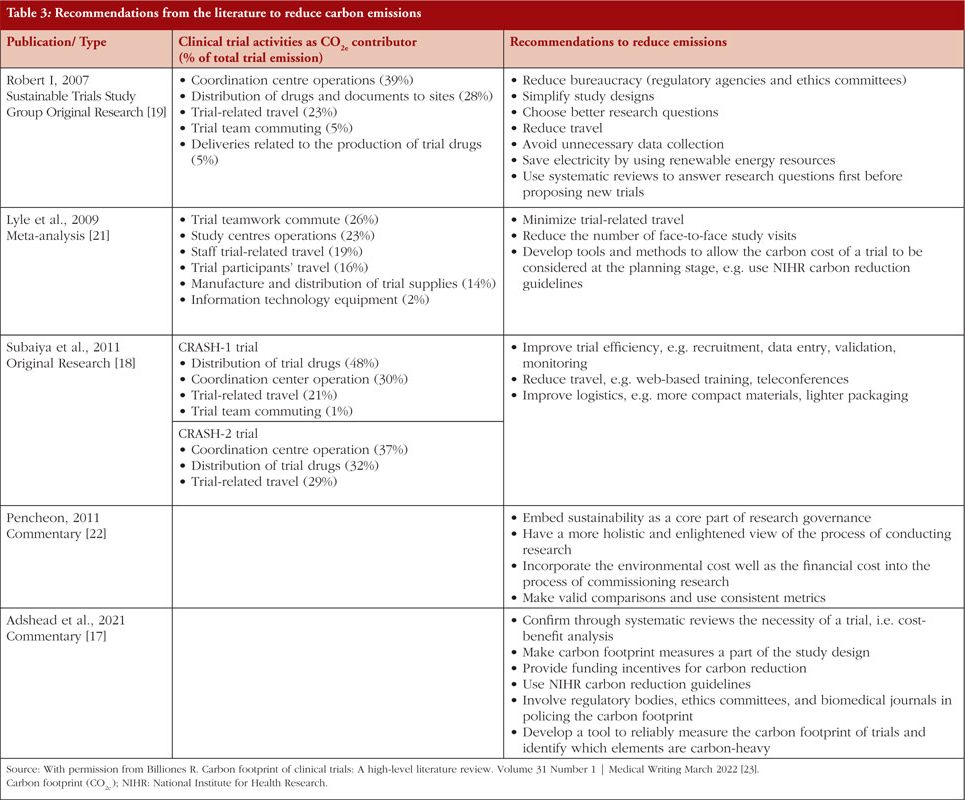
In terms of the carbon footprint of clinical trials, a recent report observes that cumulatively, up to 2021, the emissions from over 350,000 global trials have been equivalent to 27.5 million tons of GHG [17]. The CRASH 1 and CRASH 2 trials with >10,000 and >20,000 patients emitted 924.6 and 508.5 tons of GHG (CO2e) in the five-year duration that these ran [18]. The Sustainable Trials Study Group (UK NIHR) proposes methods to make clinical trials more efficient, and ESG needs to be the cornerstone of the clinical trial strategy [19].
Clinical trials are complex, and meeting the trial’s objectives while minimizing potential harmful effects on the environment and social welfare is challenging. Almost half of all clinical trials investigate novel drugs, which account for up to 20% of the carbon footprint as per NHS [15]. Both upstream (manufacturing and drug development, high transportation costs for drug distribution, manufacturing byproducts – pharmaceutical waste) and downstream processes (electricity usage, office waste disposal, drug supply and distribution of drugs and documents, trial-related travel including air travel, stays for site/hospital visits, on-site data verification, and meetings) contribute to trial-related carbon emissions.
ESG is now recognized as a critical factor in the success and sustenance of clinical research. Meeting or exceeding ESG objectives in clinical research can reduce half of the incidence of serious adverse events (SAEs) by innovative approaches that, along with patient centricity, will enable the realization of radically new solutions – more targeted interventions, reduced number of clinical trials with shorter duration and with fewer participants, and with the analysis of real-world data. This results in reduced costs, increased efficiency, reduced redundancy, reduced SAE or AE incidences, and faster delivery of novel therapies to the market.
While planning a clinical trial, it is essential to identify and assess any potential ESG risks and opportunities, including those associated with the research participants, trial site, trial infrastructure, trial management, monitoring, potential ESG impacts of the trial’s proposed interventions, the possible benefits, and risks of those interventions. A risk matrix helps identify and assess ESG risk management and incorporates both quantitative and qualitative factors. Risk assessment aims to identify and evaluate any environmental or social risks that could impact achieving the clinical trial objectives, such as the study itself, the trial population, the trial resources, or the disposal of any materials or products produced during the trial. Several tools, e.g. social and environmental impact assessment – SEIA, are used to measure ESG and integrate it into clinical trials to identify and assess a proposed clinical trial’s environmental, social, and economic impacts [20]. A summary of the literature on the carbon footprint of clinical trials and opportunities for carbon reduction is depicted in Table 3.
WHO global clinical trials forum for sustainable clinical research
In June 2024, WHO convened a clinical trials stakeholder consultation, bringing together experts, including researchers, research funders, ethicists, regulators, policymakers, and industry representatives [11]. The consultation followed the inaugural Global Clinical Trials Forum meeting held in 2023 and aimed to build on the vision to enhance global clinical research capabilities [12]. The objectives of the forum were to: (1) Develop a joint vision on strengthening clinical research capabilities aligned with the World Health Assembly resolution (WHA75.8); Strengthening clinical trials to provide high-quality evidence on health interventions and to improve research quality and coordination; (2) Discuss how we can help build, enhance, and sustain functional clinical trial capacity that is used all the time and provides utility by addressing day-to-day issues of local clinical and public health importance; (3) Provide an update on clinical research capabilities by region and globally, including key clinical research networks. Apart from the clinical researchers, regulators, ethicists, research funders, patient groups, and community engagement experts were part of the forum. Industry groups, International Federation of Pharmaceutical Manufacturers and Associations (IFPMA), Developing Countries Vaccine Manufacturers Network (DCVMN), International Generic and Biosimilar Medicines Associations (IGBA), and contract research organizations were represented.
During the two-day discussions, participants called for several priority actions for strengthening the clinical trials ecosystem, including: (1) National support for clinical trial infrastructure, including sustained domestic funding as is possible in each country; (2) Improving coordination and streamlining of regulatory and ethics review and approval processes; (3) Enhancing engagements with patients, the public, and communities throughout the clinical trial process; (4) Addressing barriers to clinical trials in underrepresented populations such as ethnic minorities, children, and pregnant women; (5) Enabling clinical trials with digital and information technologies; (6) Accelerating access to fit-for-purpose training packages for clinical trials, including innovative design; and (7) Engaging clinical practitioners to integrate clinical trials into health systems and practices [11, 12]. The anticipated outcomes from these actions would change the clinical trial landscape. Fewer, well-designed trials would generate compelling evidence for improved policy and practices that lead to improved outcomes in patients’ and the population’s health. The actions would also lead to sustained capacity-building and a streamlined learning process for stakeholders involved in clinical trials.
FDA guidance for artificial intelligence (AI) and machine learning (ML) for clinical trials and drug development
In May 2023, FDA released its guidance on AI and ML in drug development and has since published a discussion paper on the topic. Additionally, AI/ML is increasingly being integrated into areas including digital health technologies (DHTs) and real-world data (RWD) analytics [6, 7]. AI/ML has the potential to transform the design and efficiency of non-traditional trials, such as decentralized clinical trials and trials incorporating the use of RWD extracted from electronic health records (EHRs), medical claims, or other data sources, as well as analyse and interpret data collected from DHTs used in clinical studies, and even traditional trials for that matter. The following subsections highlight how AI/ML can be used during the design and conduct of clinical research.
1. Recruitment: AI/ML technology is capable of mining through extensive data sources, including clinical trial databases, announcements, social media platforms, medical publications, registries, and both structured and unstructured EHRs. This data mining helps to align participants with appropriate trials more efficiently, thus reducing recruitment timelines.
2. Selection and Stratification of Trial Participants: AI/ML can predict an individual participant’s clinical outcomes based on baseline characteristics, e.g. demographic information, clinical data, vital signs, labs, medical imaging data, and genomic data. These predictive models enrich the clinical trials, e.g. identifying high-risk participants or participants more likely to respond to the treatment. When such AI/ML algorithms are used for patient evaluation and selection before randomization, it is possible to reduce variability, increase study power, and reduce the number of study participants.
3. Dose/Dosing Regimen Optimization: AI/ML can characterize and predict pharmacokinetic (PK) profiles and study the relationship between drug exposure and response, considering confounding factors. These models optimize the dose/dosing regimen selection in special populations with limited data, e.g. rare disease studies, paediatric and pregnant populations.
4. Adherence: AI/ML can monitor and improve adherence during a clinical trial through smartphone alerts and reminders, eTracking of medication, e.g. smart pillboxes and tools for visual confirmation, and eTracking of missed clinical visits, which can trigger non-adherence alerts. Examples of AI/ML used in clinical research to improve medication adherence include applications using digital biomarkers, such as facial and vocal expressivity, to monitor adherence remotely. Adherence leads to better outcomes and thus reduces dropouts from the clinical trials.
5. Retention: AI/ML can improve participants’ access to relevant trial information through AI chatbots, voice assistance, and intelligent search. AI/ML can also reduce participants’ burden by using passive data collection techniques and extracting more information from available data generated during clinical practice or study activities. In addition, data from DHTs and similar systems can be used to develop patient profiles to predict dropouts and adverse events and potentially ensure participant retention.
6. Site Selection: Trial operations can be optimized by utilizing AI/ML to help identify which sites have the greatest success in earlier trials and to aid in identifying process gaps. For example, algorithms can be used to evaluate site performance and to help determine which sites may have a higher risk of running behind schedule based on data from other trials at that site.
7. Clinical Trial Data Collection, Management, and Analysis:
a. Data Collection: DHTs, such as wireless and smartphone-connected products, wearables, implantables, and ingestibles, are increasingly being used in clinical trials to collect objective, quantifiable, longitudinal, and continuous physiological data. This helps in decentralizing the trials efficiently.
b. Data Management: AI/ML can be used for data cleaning and curation, including duplicate participant detection and imputation of missing data, and to harmonize controlled terminology across drug development programmes.
c. Data Analysis: AI/ML has been used to analyse, for example, large volumes of diverse and complex RWD extracted from EHRs, medical claims, disease registries. Additionally, the use of AI/ML in predictive modelling and counterfactual simulation to transform clinical trial designs is being actively explored.
8. Clinical Endpoint Assessment: AI/ML can assist in assessing outcomes captured from diverse sources, e.g. DHTs, social media, during a clinical trial, including those consisting of large amounts of data for which manual review may be impractical.
9. Post-marketing Safety Surveillance, Advanced Pharmaceutical Manufacturing: AI/ML can improve the reliability of the manufacturing supply chain by forecasting product demand, analysing production schedules, forecasting and mitigating the impact of potential disruptions, and optimizing inventory. The use of AI/XML-based approaches in pharmaceutical manufacturing can cover the entire drug manufacturing life cycle, from design to commercial manufacturing.
EU recommendations for accelerating clinical trials in the European Union (ACT EU)
ACT EU aims to transform how clinical trials are initiated, designed, and run. The European Commission, the Heads of Medicines Agencies, and EMA have published recommendations that aim to facilitate the conduct of decentralised clinical trials (DCTs) while safeguarding the rights and well-being of participants and the robustness and reliability of the data collected [9, 10].
Traditionally, clinical trials have been conducted at specific sites to which patients had to travel. DCTs aim to make it easier for patients to participate in clinical trials by reducing the need to travel to central trial sites. This approach can make clinical trials available to a wider demographic of participants and improve retention. Decentralization is enabled by the advancement of digital tools, telemedicine, and more mobile and local healthcare. It includes aspects such as home health visits, remote monitoring and diagnostics, direct-to-patient shipment of study drugs, and electronic informed consent.
To facilitate innovation and, at the same time, ensure the safety, dignity, and well-being of the trial participant, a cross-disciplinary project group consisting of, amongst others, clinical trial authorization experts, ethical experts, and good clinical practice (GCP) inspectors across Member States are coming together to develop a harmonized decentralized clinical trial approach from the European Medicines Regulatory Network including ethics committee representatives [9, 10]. The DCT approach seeks to take advantage of technological and scientific progress to introduce new methodologies to the conduct of clinical trials to make clinical trials more easily accessible and participation more convenient for trial participants. Some DCT elements have already been implemented in the design and conduct of traditional clinical trials, e.g. electronic patient-reported outcomes (ePROs), electronic (e)diaries, and safety follow-up phone calls. Other DCT elements are still under development in many Member States and require further regulatory guidance and harmonization in the EU, such as home health visits and teleconsultation, direct shipment of investigational medicinal products (IMPs) to trial participants, and electronic informed consent procedures [9, 10].
Approaches to make clinical trials more ESG-effective
Driving consensus towards modernized, more sustainable clinical trials
There should be a political will to increasingly support more targeted interventions, shorter duration trials, and fewer participants, with the analysis of RWD, reduced costs, and increased efficiency where scientifically justified. Development strategies should involve broadening the inclusion criteria, using RWD, and optimizing randomized exploratory studies. We need to emphasize here that any customization in the clinical trial design must be scientifically vetted such that the safety of the patients and the efficacy of the drug are kept sacrosanct.
As discussed in earlier sections, adopting study conduct technologies that implement innovative digital solutions, such as gathering RWD through electronic health records, patient registries, and pragmatic studies, can make the research more sustainable. Collecting data from mobile and wearable medical devices and smartphones, recruiting and monitoring patients remotely through telemedicine technologies, and measuring the important quality of life indicators are some of the other measures towards enhanced sustainability in drug development operations.
A fair share of the conversation around the sustainability of clinical trials focuses on the commercial supply chain rather than the clinical supply chain [24]. In a clinical supply chain, 70% of the budget is for distribution, and almost 30% goes toward packaging. The largest cost of clinical supply chain operations is not the drugs but rather the means of getting these drugs to the patients in a safe and efficient way. The sustainability gaps where we are struggling are: (1) Out-of-date forecasting; (2) Lack of tools to measure sustainability through carbon footprinting; (3) Increased ancillary waste that contributes a significant burden on overall waste, for example, syringes, needles, and infusion kits; (4) Drug manufacturing and transportation cost [24]. Technologies brought in to help with clinical trial supply chains need to be as dynamic as the trials they are running on. Sustainability in clinical trials winds down to how responsibly resources and waste are managed. Though it is not possible to reduce waste, costs, and time lag completely, the pharma industry can take steps towards more sustainable, eco-friendly ways of running trials, and the application of AI and ML, as detailed in section 2.2.2, can help to run sustainable clinical trials.
Mitigating unsustainable trials through avoiding unnecessary studies and additional data collection: taking biosimilar development as an example
Controlled efficacy studies and interchangeability studies
The burden of clinical data requirements should be re-evaluated, and guidelines should be harmonized by the main regulatory agencies based on available systematic reviews, meta-analyses, and RWE. In general, it is necessary to identify the existing evidence body before submitting new study proposals to avoid redundancy. This is highlighted by controlled efficacy studies of biosimilar development, including interchangeability claims for low-risk, low-immunogenic molecules, e.g. secukinumab and daratumumab. The interchangeability claim granted based on the PK alone or in addition to safety and efficacy trial data, without conducting any prolonged or additional trials, can reduce the cost and time of clinical development, e.g. FDA approval of interchangeable biosimilar glargine [25].
Two systematic review switch studies involving ~14,225 and ~21,000 subjects have shown no difference in the safety, efficacy, and immunogenicity outcomes related to switching from the reference biological to the corresponding biosimilar [26-28]. This finding was supported by an analysis of switch studies of all approved biosimilars, specifically monoclonal antibodies (mAbs), published in the European public assessment reports (EPARs) [29, 30]. Hence, clinical study designs for molecules with low immunogenicity potential, such as denosumab, daratumumab, and secukinumab, may do away with the switch-arms built into the design [25-28].
On another note, drug production considerably pollutes the environment through the effluents of active pharmaceutical ingredients (APIs) that contaminate water sources. Biologicals primarily composed of amino acid components, sugars, or nucleotides can be metabolized more efficiently than synthetic APIs and do not pose environmental risks. However, biologicals require between 10 and 100 times more water during production than small molecule drugs. Thus, making clinical trials for biosimilars leaner to claim interchangeability makes more sense from the environmental perspective as well [31].
Reducing trial complexity, duration, and cost: reference product
Again, taking an example of the development of biosimilars, we propose that the use of ‘one global reference product’ as a comparator can bring down the costs in a big way, avoiding unnecessary bridging studies. A two-arm clinical PK study with three-arm bridging at the analytical level is acceptable in the EU and should be so also in all other legislations as three arms, with either of the Reference Listed Drugs, does not add relevant information and substantially increases the trial complexity, duration, and cost [25, 32]. Further, the extent of conducting a clinical trial at a global level versus limiting it to a few countries may reduce the trial management and travel costs.
Reducing trial complexity, duration, and cost: ethnic sensitivity assessments
Ethnic sensitivity assessments in the form of bridging PK studies are often needed for biosimilars of mAbs to meet regulatory requirements in regions like East Asia [33]. Bridging studies, however, are inherently limited and may not fully address the concerns and mAbs may be less likely affected by ethnic differences as they do not undergo traditional drug-related metabolism like liver and gut metabolism, unlike small molecule drugs. The current literature indicates that ethnicity generally does not influence PK, pharmacodynamic (PD), safety, and efficacy of the majority of mAbs; however, dedicated ethnic sensitivity studies (ESSs) are still routinely required for approval in countries of the East Asian region, such as China, Japan, South Korea, and Taiwan [33]. We need to revisit the requirement for running ESS trials, and the decision to mandate phase I bridging trials should be evidence-based, considering the ethnic sensitivity information available on the mAb reference drug rather than being a mandatory obligation in the clinical development of its biosimilars. Implementing such a strategy can accelerate the development of life-saving biologicals while decreasing its carbon footprint.
Manufacturing carbon-neutral devices
Parenteral drug delivery is fast moving towards disposable pen devices and pre-filled syringes (PFS), which have become popular due to increased safety, convenience, and accuracy in delivering drugs, especially biologicals. In contrast to the vials, minimal overfill volume is needed to ensure delivery of the correct dose to the patient, and the resulting savings from the reduced product volume offsets the increased cost of the PFS/auto-injector components. However, as plastic syringes are favoured over glass; in Japan, 50% of PFS are made of polymers, and as the global PFS market size is expected to reach US$16.32 billion by 2030, we must search for sustainable solutions for the materials used in PFS [34, 35]. Sustainability impact assessments show that autoinjector therapies have a very high carbon footprint due to using fossil fuel-derived plastic device parts [36]. The development of biopolymers is happening fast as a replacement for synthetic plastics due to an increased interest in environmental sustainability [37]. Biopolymers can now be made on an industrial scale, using biogenic carbon – harvested from sources such as wood pulp, anaerobic digesters (waste food compost), or waste from sugar production – to generate the chains of carbon and hydrogen required. This allows the material to be produced and decomposed without using any carbon derived from fossil fuels by using and releasing carbon within a closed-loop carbon cycle [36]. Biopolymers present essential features such as carbon neutrality, biodegradability, biocompatibility, sustainability, bioresorbability, flexibility, antibacterial activity, renewability, and stability [37].
Over 60 million inhaler items are prescribed annually in the UK, with a total carbon footprint of 1.3 billion kg of carbon dioxide (CO2),. A pressurized metered dose inhaler (pMDI) used to treat adult asthma and severe chronic obstructive pulmonary disease has been developed, which offsets all carbon emissions associated with this inhaler, enabling it to become the first carbon-neutral pMDI available to the UK market [14]. Several big pharma players are following suit and have now announced their intent to become carbon-neutral in the coming years with the initiatives they are putting into practice [14].
The Medical Device Innovation Consortium (MDIC) is seeking medical device manufacturers to participate in their Accelerate Sustainable Capability (ASC) pilot study, which was developed in collaboration with the medical device industry, the Information Systems Audit and Control Association (ISACA), and FDA. The ASC pilot study gives manufacturers insights that may help them improve product quality and safety, reach compliance quickly, structure their systems for continuous improvement, and identify metrics to monitor product safety throughout the pilot process [38].
Looping in regulatory authorities and ethics committees in the planning phase of the trials for sanctioning limited carbon emission
A cross-sectional survey in 15 countries across 4 continents done through the International Clinical Trials Center Network reported that currently, greenhouse gas emissions are neglected during the planning phase of a research project [5]. These are not addressed or assessed by default during the approval procedures by the Institutional Review Boards (IRBs), Ethical Committees (ECs), or other competent authorities. No advice or oversight from IRBs/ECs or regulatory authorities is available on controlling the carbon footprint of clinical trials. According to the International Council for Harmonisation of Technical Requirement for Pharmaceuticals for Human Use (ICH) GCP, all clinical trial protocols must be approved by IRBs or ECs. The carbon footprint assessment of a clinical trial is also an ethical subject, as climate change impacts the health status of all humans. Going forward, a long-term change will require sustained commitment towards ESG goals by clinical researchers, Pharma industries, regulatory authorities, and funding bodies. Developing biosimilars ‘smartly’ can show the effectiveness of measures to reduce carbon footprint in a shorter timeframe for others to emulate in larger trials.
Case study: quantifying the carbon footprint of the clinical trials
Using the NIHR guidelines a carbon audit of two clinical trials (the CRASH-1 and CRASH-2 trials) was done, quantifying the carbon dioxide emissions over one year [18]. Carbon emissions from the clinical trial centre, material delivery, and trial-related travel were calculated for both trials. The total emissions in carbon dioxide equivalents during the one-year audit period were 181.3 tonnes for CRASH-1 and 108.2 tonnes for CRASH-2. Overall, CRASH-1 emitted 924.6 tonnes (from 10,008 patients over 5.1 years) of carbon dioxide equivalents compared with 508.5 tonnes (20,211 patients over 4.7 years) for CRASH-2. The largest contributor to emissions in CRASH-1 was freight delivery of trial materials (86.0 tonnes, 48% of total emissions). In contrast, the largest contributor to CRASH-2 was energy use by the clinical trial centre (54.6 tonnes, 30% of total emissions). Faster patient recruitment in the CRASH-2 resulted in lower carbon emissions than CRASH-2; in addition to lighter trial materials and web-based data entry [18].
Recently, the Low Carbon Clinical Trials Working Group developed a methodology and guidance for measuring the carbon footprint (CO2e) of clinical trials [4]. A process map of the clinical trial core activities is defined in detail, such as trial set-up, Clinical Trial Unit (CTU) emissions, Trial-specific meetings and travel, treatment intervention, data collection and exchange, trial supplies and equipment, trial-specific patient assessments, samples, laboratory, and trial close-out. The activity data from the above core activities was converted to greenhouse gas emissions by multiplying it with the emission factor, which is the average emission rate of a given source relative to units of activity or processes. The method was validated with two Cancer Research UK (CRUK)-funded trials (the international randomized sarcoma trial CASPS and the UK cohort-based breast cancer trial PRIMETIME [4]. A detailed breakdown of data on the GHG emissions in different phases and tasks of CASPS and PRIMETIME trials is given in Table 4. A guidance document defining the scope, method, and assumptions is available to be applied to any publicly funded/investigator-initiated clinical trial [4].
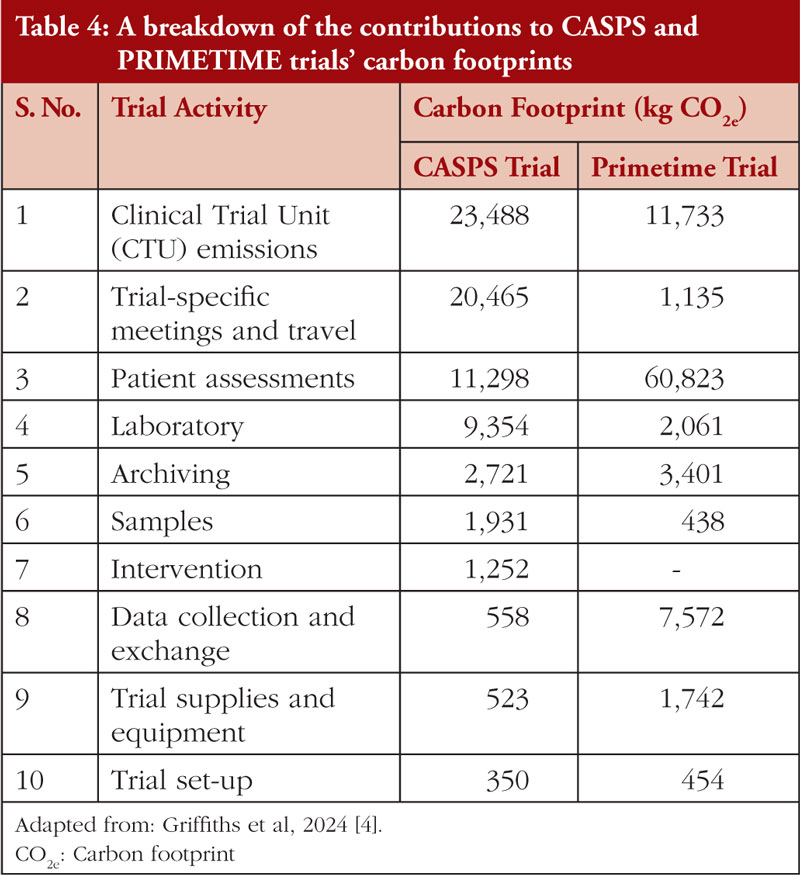
While CRASH-1, CRASH-2, CASPS, and PRIMETIME focused on emissions from non-commercial clinical trials, the extent to which these principles might be applied in pharmaceutical industry-directed trials is open for experimentation. Industry-led trials are often considerably more complex and involve much more data collection than non-commercial trials. It is likely that the carbon footprint of industry-led trials is much larger, and hence, there is a need to make emissions reductions even more important.
Conclusions
Scientifically sound and streamlined clinical development can be much in line with the global efforts to limit carbon emissions on this planet. Clinical trialists should be encouraged to pursue innovative ways to capture the estimated reduction in the carbon footprint of the planned trials while applying for grants. There remains an urgent need to develop tools capable of measuring the carbon footprint of clinical trials and assisting in identifying other trial elements that can help reach ESG goals and commitments. All relevant stakeholders, including researchers, funders, ethicists, regulators, policymakers, and industry representatives, should rally around this cause to make the pharma ESG goals attainable and sustainable.
Acknowledgements
The authors appreciate the editorial assistance provided by Geetanjali Tonpe in the earlier drafts (Biocon Biologics Ltd).
Funding sources
This research was funded by Biocon Biologics Limited.
Disclaimer
The opinions expressed in this article are personal views of the authors and should not be understood as being made on behalf of or reflecting the position of the agencies or organizations with which the authors are affiliated.
Data availability statement
The literature search results are available for sharing. All articles summarized are published. Please contact the author for more information.
Competing interests: SB and SNA are employees of Biocon Biologics Ltd (a subsidiary of Biocon Ltd). EWH is an employee of Biocon Biologics UK Ltd (a subsidiary of Biocon Biologics Ltd). SM works as a consultant with Biocon Biologics Ltd. SNA holds stocks in Biocon Ltd and Biocon Biologics Ltd. SB and EWH declare no conflict of interest.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Shivani Mittra1, MPharm, PhD
Shylashree Baraskar1, MBBS
Elena Wolff-Holz2, MD
Sandeep N Athalye1, MBBS, MD
1Clinical Development and Medical Affairs, Biocon Biologics Ltd, Bengaluru 560100, Karnataka, India
2Clinical Development and Medical Affairs, Biocon Biologics UK Ltd, London WC2B 5AH, UK
References
1. Read L. Will ESG be 2023’s hottest business topic in Bio/Pharma? Life Science Leader. Guest Column. 6th January 2023. [cited 2024 Jul 16]. Available from: https://www.lifescienceleader.com/doc/will-esg-be-s-hottest-business-topic-in-bio-pharma-0001
2. Kinghorn R, Moulavi N, O’Connell K. Beyond compliance: consumers and employees want business to do more on ESG. How business can close the expectation gap. PwC.
3. SBTi Monitoring Report 2023. Science-Based Targets. Available from: https://sciencebasedtargets.org/reports/sbti-monitoring-report-2023
4. Griffiths J, Fox L, Williamson PR; Low Carbon Clinical Trials Group. Quantifying the carbon footprint of clinical trials: guidance development and case studies. BMJ Open. 2024;14(1):e075755. doi:10.1136/bmjopen-2023-075755
5. Hoffmann JM, Bauer A, Grossmann R. The carbon footprint of clinical trials: a global survey on the status quo and current regulatory guidance. BMJ Glob Health. 2023;8(9):e012754. doi:10.1136/bmjgh-2023-012754
6. Tern. What is ESG and its three pillars? 2023.
7. U.S. Food and Drug Administration. Artificial intelligence and machine learning (AI/ML) for drug development [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.fda.gov/science-research/science-and-research-special-topics/artificial-intelligence-and-machine-learning-aiml-drug-development
8. U.S. Food and Drug Administration. Using artificial intelligence and machine learning in the development of drug and biological products. [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.fda.gov/media/167973/download?attachment
9. European Medicines Agency. Accelerating clinical trials in the EU (ACT EU). [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/clinical-trials-human-medicines/accelerating-clinical-trials-eu-act-eu
10. European Medicines Agency. ACT EU multi-annual. Workplan 2022-2026 [homepage on the Internet]. [cited 2024 Jul 16]. Available from: act-eu-multi- https://www.ema.europa.eu/en/documents/other/act-eu-multi-annual-workplan-2022-2026_en.pdfannual-workplan-2022-2026_en.pdf (europa.eu)
11. World Health Organization. WHO consults on action plan for sustainable clinical research infrastructure. 26 June 2024 [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.who.int/news/item/26-06-2024-who-consults-on-action-plan-for-sustainable-clinical-research-infrastructure
12. World Health Organization. First WHO Global Clinical Trials Forum puts forward a global vision for sustainable clinical research infrastructure. 29 November 2023 [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.who.int/news/item/29-11-2023-first-who-global-clinical-trials-forum-puts-forward-a-global-vision-for-sustainable-clinical-research-infrastructure
13. National Institute for Health and Care Research. NIHR carbon reduction guidelines. 2019 [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.nihr.ac.uk/documents/the-nihr-carbon-reduction-guidelines/21685
14. Hawkins L. Five ways pharma is reducing its carbon emissions. Pharma IQ. 2022.
15. Cuffari B. The carbon footprint of clinical trials. News Medical Life Sciences. 2021.
16. Belkhir L. Big Pharma emits more greenhouse gases than the automotive industry. The Conversation. 27 May 2019.
17. Adshead F, Al-Shahi Salman R, Aumonier S, Collins M, Hood K, McNamara C, et al. A strategy to reduce the carbon footprint of clinical trials. Lancet. 2021;398(10297):281-2. doi:10.1016/S0140-6736(21)01384-2
18. Subaiya S, Hogg E, Roberts I. Reducing the environmental impact of trials: comparing of the carbon footprint of the CRASH-1 and CRASH-2 clinical trials. Trials 2011;12:31. doi:10.1186/1745-6215-12-31
19. Sustainable Trials Study Group. Towards sustainable clinical trials. BMJ. 2007;334(7595):671-3. doi:10.1136/bmj.39140.623137.BE.
20. Socio-Economic Impact Assessment (SEIA) guidelines. March 2007. Mackenzie Valley Environmental Impact Review Board. Available from: https://reviewboard.ca/upload/ref_library/SEIA_Guidelines_Contents_and_Chapter_1.pdf
21. Lyle K, Dent L, Bailey S, Kerridge L, Roberts I, Milne R. Carbon cost of pragmatic randomised controlled trials: retrospective analysis of sample of trials. BMJ. 2009;30;339:b4187. doi:10.1136/bmj.b4187
22. Pencheon DC. Managing the environmental impact of research. Trials. 2011;12:80. Available from: https://doi.org/10.1186/1745-6215-12-80
23. Billiones R. Carbon footprint of clinical trials: a high-level literature review. Medical Writing. 2022;31(1):14-9.
24. Bendelac N. Introducing sustainability into the clinical trial supply chain. Applied Clinical Trials. 29 March 2023. Available from. https://www.appliedclinicaltrialsonline.com/view/introducing-sustainability-into-the-clinical-trial-supply-chain
25. Athalye SN, Mittra S, Ranpura AM. Biosimilars drug development: time for a paradigm shift? Generics and Biosimilars Initiative Journal (GaBI Journal). 2023;12(1):17-22. doi:10.5639/gabij.2023.1201.005
26. Barbier L, Ebbers HC, Declerck P, Simoens S, Vulto AG. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: a systematic review. Clin Pharmacol Ther. 2020;108(4):734-55. doi:10.1002/cpt.1836
27. Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463-78. doi:10.1007/s40265-018-0881-y
28. Herndon TM, Ausin C, Brahme NN, Schrieber SJ, Luo M, Andrada FC, et al. Safety outcomes when switching between biosimilars and reference biologics: a systematic review and meta-analysis. PLoS One. 2023;18(10):e0292231. doi:10.1371/journal.pone.0292231
29. Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31(2):83-91. doi:10.1007/s40259-017-0210-0
30. Kurki P, Barry S, Bourges I, Tsantili P, Wolff-Holz E. Safety, immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: a regulatory perspective. Drugs. 2021;81(16):1881-96. doi:10.1007/s40265-021-01601-2
31. Jeremias S. How biosimilars are making big pharma more eco-friendly. 24 May 2022. Am J Manag Care.
32. Webster CJ, Woollett GR. A Global reference comparator for biosimilar development. BioDrugs. 2017;31(4):279-86. doi:10.1007/s40259-017-0227-4
33. Jeon I, Kim YK, Song I et al. The necessary conduct: exploratory multiregional clinical trials in East Asia. Clin Transl Sci. 2021;14(6):2399-407. doi:10.1111/cts.13106
34. Lo C. Prefilled syringes: getting to the point. Hospital Management. 15 December 2011.
35. Pre-filled syringes: key market insights. July 2023. Fortune Business Insights.
36. Palmer-Felgate J. An eco-friendly and economical approach to autoinjectors. ONdrugDelivery. 2024;156:64-7.
37. Gheorghita R, Anchidin-Norocel L, Filip R, Dimian M, Covasa M. Applications of biopolymers for drugs and probiotics delivery. Polymers (Basel). 2021;13(16):2729. https://doi.org/10.3390/polym13162729
38. U.S. Food and Drug Administration. Accelerate Sustainable Capability (ASC) pilot study under MDIC [homepage on the Internet]. [cited 2024 Jul 16]. Available from: https://www.fda.gov/medical-devices/quality-and-compliance-medical-devices/accelerate-sustainable-capability-asc-pilot-study-under-mdic
|
Author for correspondence: correspondence: Sandeep N Athalye, MBBS, MD, Chief Development Offi cer, Biocon Biologics Ltd., Biocon House, Tower 3, Semicon Park, Plot No 29-P1 & 31-P, KIADB Industrial Area, Electronic City Phase – 2, Bangalore – 560100, Karnataka, India |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2024 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



One Reply to “Aligning environmental, social, and governance to clinical development: moving towards more sustainable clinical trials”