Assessing biosimilarity and interchangeability of biosimilar products under the Biologics Price Competition and Innovation Act
Published on 2012/10/19
Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1):20-5.
Author byline as per print journal: Professor Shein-Chung Chow, PhD, Christine Ju, MSc
|
Abstract: |
Submitted: 15 October 2012; Revised: 20 November 2012; Accepted: 21 November 2012; Published online first: 9 January 2013
Introduction
For small molecule drug products, when an innovative (brand-name) drug product is going off patent, pharmaceutical and/or generics companies may seek regulatory approval of the generic copies of the brand-name drug. In 1984, the US Food and Drug Administration (FDA) was authorized to approve generic drug products under the Drug Price Competition and Patent Term Restoration Act, which is also known as the Hatch-Waxman Act. For approval of generic drug products, FDA requires that evidence in relative bioavailability in terms of the rate and extent of drug absorption be provided through the conduct of bioequivalence studies [1–3]. Similar requirements exist in the EU. In the case of biosimilar products, the European Commission reached the conclusion that, unlike small molecule generics, copies of biologicals can only be similar – but not identical – to innovator biologicals. Thus, the generic versions of biological products are similar biological drug products (SBDP). Similar biological drug products are usually referred to as biosimilars by the European Medicines Agency (EMA) of the EU, follow-on biologics by FDA, and subsequent entered biologics by the Public Health Agency of Canada.
For small molecule generic drug products, reductions in price of around 80% have been observed after the first six months to a year of generics entry to the market in countries such as Germany, UK and the US [4, 5]. Unlike small molecule generic drug products, the price difference between follow-on biologics and originator biological products is likely to be smaller than that observed between originator and generics. The concept for development of SBDP, which are made of living cells, is very different from that of small molecule generic drug products. Given that follow-on biologics incur higher research, development, and manufacturing costs due to these differences, they are generally priced about 30% less than the originator product. To assist sponsors in development of SBDP, EMA issued a series of general guidelines and product class-specific guidelines for biosimilar erythropoietin, granulocyte colony-stimulating factor, insulin, human growth hormone, interferon alpha, and low molecular weight heparin [6–11]. EMA has been successful in devising a system for authorizing the marketing of biosimilar product, and has approved 14 biosimilars, 13 of which are currently on the market in major countries of the EU [12]. In the US, the Biologics Price Competition and Innovation (BPCI) Act (as part of the Affordable Care Act) was written into law on 23 March 2010, which has given FDA the authority to approve SBDP.
In the past couple of years, regulatory approval pathway for assessment of follow-on biologics has received much attention in the US. Standard methods for assessment of bioequivalence for regulatory approval of generic drug products are not appropriate due to some fundamental differences between small molecule generics and large molecule biosimilar products [13]. For example, biological products are made of living cells and have heterogeneous structure (usually mixtures of related molecules, differences in the tertiary structure, different glycosylation pattern) which is difficult to characterize. In addition, biological products are often variable and sensitive to environmental conditions such as light and temperature. A small change or variation at any critical stage of a manufacturing process of a biological product could result in a change in molecular structure and therefore clinical outcomes.
The purpose of this paper is to review and address (if possible) within the current US situation scientific factors that are commonly encountered when assessing biosimilarity and interchangeability of follow-on biologics. In the next two sections, scientific factors for assessing biosimilarity and interchangeability of biosimilars are discussed. The section on ‘recent development in the US’ provides recent development regarding approval pathway of biosimilars in the US. Brief remarks are given in the last section on ‘conclusions’.
Biosimilarity
Definition and basic principles
As indicated earlier, on 23 March 2010, the BPCI Act (as part of the Affordable Care Act) was written into law which has given FDA the authority to approve biosimilar products. In the BPCI Act, a follow-on biologic is defined as a product that is highly similar to the reference product notwithstanding minor differences in clinically inactive components and without clinically meaningful differences in terms of safety, purity, and potency (in the EU, the sponsors should demonstrate comparable quality, safety and efficacy) [14]. Based on this definition, we would interpret that a biological medicine is biosimilar to a reference biological medicine if it is highly similar to the reference in safety, purity and potency, where purity may be related to some important quality attributes at critical stages of a manufacturing process and potency has something to do with the stability and efficacy of the biosimilar product. Though recently issued draft guidances do begin to clarify the issue, little or no discussion regarding how similar is considered highly similar is given in the BPCI Act [14].
The BPCI Act seems to suggest that a biosimilar product should be highly similar to the reference drug product in all spectrums of good drug characteristics such as identity, strength (potency), quality, purity, safety, and stability as described in the US Pharmacopeia and National Formulary [15]. In practice, however, it is almost impossible to demonstrate that a biosimilar product is highly similar to the reference product in all aspects of good drug characteristics in a single study. Thus, to ensure a biosimilar product is highly similar to the reference product in terms of these good drug characteristics, different biosimilarity studies may be required. For example, if safety and efficacy is a concern, then a clinical trial must be conducted to demonstrate that there are no clinically meaningful differences in terms of safety and efficacy between a biosimilar product and the innovator biological product. Additionally, to demonstrate a product is highly similar to a reference product despite differences in the manufacturing process, important quality attributes at critical stages of a manufacturing process, assay development/validation, process control/validation, and product specification of both the reference and follow-on products are necessarily established, see Editor’s comments. This requirement holds true for changes between a reference product’s and follow-on product’s manufacturing process, as well as for changes within the original reference’s manufacturing process. This is extremely important because biological products are known to be sensitive to a small change or variation in environmental factors such as light and temperature during the manufacturing process, so larger changes in the process itself could result in undesirable differences. In some cases, if a surrogate endpoint such as pharmacokinetic (PK), pharmacodynamic (PD), or genomic marker is predictive of the primary efficacy/safety clinical endpoint, then a PK/PD or genomic study may be used to assess biosimilarity between a follow-on biologic and the reference product.
It should be noted that current FDA regulatory requirements are guided based on a case-by-case basis by the following basic principles of: (1) the extent of the physicochemical and biological characterization of the product; (2) nature or possible changes in the quality and structure of the biological product due to changes in the manufacturing process (and their unexpected outcomes); (3) clinical/regulatory experiences with the particular class of the product in question; and (4) several factors that need to be considered for comparability [16]. Most recently, in its draft guidance on scientific considerations in demonstrating biosimilarity to a reference product, FDA suggests totality-of-the-evidence be provided for assessing biosimilarity of biosimilar products. This indicates that FDA (as does EMA) is interested in demonstrating global similarity in all aspects related to safety, purity, and potency of the biosimilar products.
Criteria for biosimilarity
The BPCI Act defines a biosimilar product as a biological product that is highly similar to the reference drug product. However, no criteria for assessing biosimilarity were mentioned. Statistically, one could refer to similarity between two drug products as similarity in average, variability, or distribution of the response of a specific study endpoint of interest between drug products. In practice, the assessment of similarity in average of the response of a specific study endpoint is often considered. A typical example is the assessment of average bioequivalence for regulatory approval of generic drug products. In this paper, unless otherwise stated, we will focus on biosimilarity in average response of the study endpoint of interest for a given biosimilar study.
In practice, the terms biosimilarity (similarity), bioequivalence (equivalence), and comparability (consistency) are used interchangeably in pharmaceutical research and development. For comparison between drug products, some criteria for assessment of bioequivalence (e.g. drug absorption profiles comparison), similarity (e.g. dissolution profiles comparison), and comparability or consistency (e.g. comparison between product protein structures and function) are available in either regulatory guidelines/guidances or literature. These criteria, however, can be classified into either: (1) absolute change versus relative change, (2) aggregated versus disaggregated, or (3) moment-based versus probability-based. In this section, different categories of criteria are briefly reviewed.
Absolute change versus relative change
In clinical research and development, for a given study endpoint, post-treatment absolute change from baseline or post-treatment relative change (% change) from baseline is usually considered for comparison between treatment groups. A typical example would be the study of weight reduction in obese patient population. In practice, it is not clear whether a clinically meaningful difference in terms of absolute change from baseline can be translated to a clinically meaningful difference in terms of relative change from baseline. Sample size calculation based on power analysis in terms of absolute change from baseline or relative change from baseline could lead to a very different result.
For generic drug approval, current US regulation adopts one size-fits-all criterion based on relative change for bioequivalence assessment. In other words, we conclude (average) bioequivalence between a test product and a reference product if the 90% confidence interval for the ratio of means of the primary endpoint (e.g. pharmacokinetic response such as area under the blood or plasma-concentration time curve) between the two drug products is totally within 80% and 125%. Note that regulatory agencies suggest that a log-transformation be performed before data analysis for assessment of bioequivalence.
Aggregated versus disaggregated
As indicated by Chow and Liu (2008), bioequivalence can be assessed by evaluating differences in averages, intra-subject variabilities, and variance due to subject-by-formulation interaction between drug products separately [17]. Individual criteria for assessment of differences in averages, intra-subject variabilities, and variance due to subject-by-formulation interaction between drug products are referred to as the disaggregated criteria. If the criterion is a single summary measure composed of these individual criteria, it is called the aggregated criterion.
For assessment of bioequivalence in average bioavailability, most regulatory agencies recommend the use of disaggregate criterion based on average bioavailability. That is, bioequivalence in regards to bioavailability is concluded if the average bioavailability of the test formulation is within a certain confidence interval (80%, 125%) of that of the reference formulation, with a certain assurance. Note that FDA (2001) and World Health Organization (WHO) (2005) use the same equivalence criterion of 80% to 125% for the log-transformed PK responses such as area under the blood or plasma concentration time curve (AUC) [18, 19]. However, for maximum or peak concentration (Cmax), in cases of highly variable drugs, EMA and WHO allow a wider interval of 69.84% to 143.19% for the ratio of average bioavailability to address any safety and efficacy concerns for patients switched between formulations [20]. If a wider interval is used, it must be pre-specified in the protocol. More details can be found in Chow and Liu (2009) [13].
For aggregated criteria, FDA proposes the use of individual bioequivalence (IBE) criterion (IBC) for addressing drug switchability and population bioequivalence (PBE) criterion (PBC) for addressing drug prescribability [18]. For assessment of IBE, the IBC, denoted by ?i can be expressed as
 (1)
(1)
where 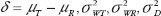 are the true difference in means, intra-subject (within-subject) variabilities of the test product and the reference product, and variance due to subject-by-formulation interaction between drug products, respectively.
are the true difference in means, intra-subject (within-subject) variabilities of the test product and the reference product, and variance due to subject-by-formulation interaction between drug products, respectively. 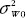 is the scale parameter specified by the user. Similarly, the PBC for assessment of population bioequivalence, denoted by ?P suggested in FDA guidance is given by
is the scale parameter specified by the user. Similarly, the PBC for assessment of population bioequivalence, denoted by ?P suggested in FDA guidance is given by
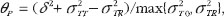 (2)
(2)
where 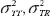 are the total variances for the test product and the reference product, respectively and
are the total variances for the test product and the reference product, respectively and 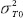 is the scale parameter specified by the user [18].
is the scale parameter specified by the user [18].
A typical approach is to construct a one-sided 95% confidence interval for ?I(?P) for assessment of individual (population) bioequivalence. If the one-sided 95% upper confidence limit is less than the bioequivalence limit of ?I(?P) we then conclude that the test product is bioequivalent to that of the reference product in terms of individual (population) bioequivalence. More details regarding individual and population bioequivalence can be found in the paper by Chow and Liu (2009) [13].
Moment based criteria versus probability based criteria
Schall and Luus (1993) proposed the moment-based and probability-based measures for the expected discrepancy in PK response between drug products [21]. The moment-based measure suggested is based on the following expected mean-squared differences [21]:
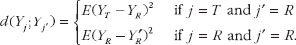 (3)
(3)
For some pre-specified positive number r, one of probability-based measures for the expected discrepancy is given as
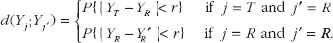 (4)
(4)
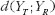 measures the expected discrepancy for some pharmacokinetic metric between test and reference formulations, and
measures the expected discrepancy for some pharmacokinetic metric between test and reference formulations, and 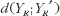 provides the expected discrepancy between the repeated administrations of the reference formulation [21]. The role of
provides the expected discrepancy between the repeated administrations of the reference formulation [21]. The role of 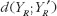 in formulation of bioequivalence criteria is to serve as a control. The rationale is that the reference formulation should be bioequivalent to itself. Therefore, for the moment-based measures, if the test formulation is indeed bioequivalent to the reference formulation, then
in formulation of bioequivalence criteria is to serve as a control. The rationale is that the reference formulation should be bioequivalent to itself. Therefore, for the moment-based measures, if the test formulation is indeed bioequivalent to the reference formulation, then 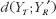 should be very close to
should be very close to 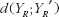 It follows that if the criteria are functions of the difference (or ratio) between
It follows that if the criteria are functions of the difference (or ratio) between  and
and 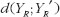 bioequivalence is concluded if they are smaller than some pre-specified limit. On the other hand, for probability-based measures, if the test formulation is indeed bioequivalent to the reference formulation, as compared with
bioequivalence is concluded if they are smaller than some pre-specified limit. On the other hand, for probability-based measures, if the test formulation is indeed bioequivalent to the reference formulation, as compared with 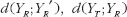 should be relatively large. As a result, bioequivalence is concluded if the criteria based on the probability-based measure are greater than some pre-specified limit.
should be relatively large. As a result, bioequivalence is concluded if the criteria based on the probability-based measure are greater than some pre-specified limit.
Chow et al. (2010) compared the moment-based criterion with the probability-based criterion for assessment of bioequivalence or biosimilarity under a parallel group design [22]. The results indicate that the probability-based criterion is not only a much more stringent criterion, but also has sensitivity to any small change in variability. This justifies the use of the probability-based criterion for assessment of biosimilarity between follow-on biologics if a certain level of precision and reliability of biosimilarity is desired.
Interchangeability
As indicated in the Subsection (b)(3) amended to the Public Health Act Subsection 351(k)(3), the term interchangeable or interchangeability in reference to a biological product that is shown to meet the standards described in subsection (k)(4), means that the biological product may be substituted for the reference product without the intervention of the healthcare provider who prescribed the reference product [14]. Along this line, in what follows, definition and basic concepts of interchangeability (in terms of switching and alternating) are given.
Definition and basic concepts
As indicated in the Subsection (a)(2) amends the Public Health Act Subsection 351(k)(3), a biological product is considered to be interchangeable with the reference product if: (i) the biological product is biosimilar to the reference product; and (ii) it can be expected to produce the same clinical result in any given patient. In addition, for a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biosimilar and the reference product is not greater than the risk of using the reference product without such alternation or switch [14]. A truly interchangeable product should be able to be substituted or alternated by a pharmacist, without intervention or even necessarily notification of the prescribing doctor, whereas a strictly biosimilar product may yield a comparable outcome as the reference, but may require transitioning or input by a healthcare provider in order to be switched or alternated with the reference (or not be able to at all) due to other factors such as excipients.
Thus, there is a clear distinction between biosimilarity and interchangeability. In other words, biosimilarity does not imply interchangeability which is much more stringent. However, interchangeability is expected to produce the same clinical result in any given patient, which can be interpreted as that the same clinical result can be expected in every single patient. In reality, conceivably, lawsuits may be filed if adverse effects are recorded in a patient after switching from one product to another.
It should be noted that when FDA declares the biosimilarity of two drug products, it may not be assumed that they are interchangeable. Therefore, labels ought to state whether for a follow-on biologic which is biosimilar to a reference product, interchangeability has or has not been established. However, payers and physicians may, in some cases, switch products even if interchangeability has not been established.
Switching and alternating
Unlike small molecule drug interchangeability (in terms of prescribability and switchability), FDA has a slightly different perception of drug interchangeability for biosimilars [3]. From FDA’s perspectives, interchangeability includes the concept of switching and alternating between an innovative biological product (R) and its follow-on biologics (T). The concept of switching is referred to as not only the switch from ‘R to T’ or ‘T to R’ (narrow sense of switchability), but also ‘T to T’ and ‘R to R’ (broader sense of switchability). As a result, in order to assess switching, biosimilarity for ‘R to T’, ‘T to R’, ‘T to T’, and ‘R to R’ need to be assessed based on some biosimilarity criteria under a valid study design.
On the other hand, the concept of alternating is referred to as either the switch from T to R and then switch back to T (i.e. ‘T to R to T’) or the switch from R to T and then switch back to R (i.e. ‘R to T to R’). Thus, the difference between ‘the switch from T to R’ then ‘the switch from R to T’ and ‘the switch from R to T’ then ‘the switch from T to R’ needs to be assessed for addressing the concept of alternating.
Study design
For assessment of bioequivalence for chemical drug products, a standard two-sequence, two-period (2 x 2) crossover design is often considered, except for drug products with relatively long half-lives. The present approved biosimilars all demonstrated bioequivalence in a crossover design. For monoclonal antibodies and other biological products with relatively long half-lives, it is suggested that a parallel group design should be considered. However, parallel group design does not provide independent estimates of variance components such as inter-subject and intra-subject variabilities and variability due to subject-by-product interaction. Thus, it is a major challenge for assessing biosimilarity and interchangeability (in terms of the concepts of switching and alternating) of biosimilar products under parallel group designs.
For assessment of switching, a switching design should allow the assessment of biosimilarity for the switch from ‘R to T’, ‘T to R’, ‘T to T’, and ‘R to R’ in order to determine whether there is a risk when a switch occurs. For this purpose, the Balaam’s 4 x 2 crossover design (i.e. TT, RR, TR, RT) may be useful. Similarly, for addressing the concept of alternating, a two-sequence, three-period dual design (i.e. TRT, RTR) may be useful since the design allow the assessment of the switch from T to R and then back to T (i.e. ‘T to R to T’) and from R to T and then back to R (i.e. ‘R to T to R’). For addressing both concepts of switching and alternating for drug interchangeability of biosimilars, a modified Balaam’s crossover design (i.e. TT, RR, TRT, RTR) is then recommended.
Remarks
With small molecule drug products, bioequivalence generally reflects therapeutic equivalence. Drug prescribability, switching, and alternating are generally considered reasonable. With biological products, however, variations are often higher (other than pharmacokinetic factors may be sensitive to small changes in conditions). Thus, often only parallel-group design rather than crossover kinetic studies can be performed. It should be noted that very often, with follow-on biologics, biosimilarity does not reflect therapeutic interchangeability. Therefore, switching and alternating should be pursued with extreme caution.
Recent development in the US
Following the passage of the BPCI Act, in order to obtain input on specific issues and challenges associated with the implementation of the BPCI Act, FDA conducted a two-day public hearing on Approval Pathway for Biosimilar and Interchangeability Biological Products held on 2–3 November 2010 at FDA in Silver Spring, Maryland, USA. Several scientific factors were raised and discussed at the public hearing. These scientific factors include, but are not limited to, the definition of ‘highly similar’ including the degree of similarity; criteria for assessing biosimilarity (e.g. average versus variability and moment-based versus probability-based criteria); study design (e.g. crossover versus parallel design); statistical methods (e.g. the potential use of Bayesian approach); and tests for comparability in quality attributes of manufacturing process and/or immunogenicity [23]. These issues primarily focus on the assessment of biosimilarity. The issue of interchangeability in terms of the concepts of alternating and switching were also mentioned and discussed. These scientific factors have generated a lot of research interest. On 9 February 2012, FDA released three draft guidances about demonstrations of biosimilarity. These draft guidances are: (1) Scientific Considerations in Demonstrating Biosimilarity to a Reference Product; (2) Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product; (3) Biosimilars: Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation (BPCI) Act of 2009. As stated, these guidances are not only: (1) intended to assist sponsors in demonstrating that a proposed therapeutic protein product is biosimilar to a reference product for purpose of the submission of a marketing application under section 351(k) for the Public Health Service (PHS) Act; but also (2) to describe FDA’s current thinking on factors to consider when demonstrating that a proposed protein product is highly similar to a reference product licensed under section 351(a) of the PHS Act for purpose of submitting a marketing application under section 351(k) of the PHS Act. In addition, the guidances provide answers to common questions from sponsors interested in developing proposed biosimilar products; biologics license application holders, and other interested parties regarding FDA’s interpretation of the Biologics Price Competition and Innovation (BPCI) Act of 2009 [23].
In the draft guidances, FDA suggests the use of stepwise approach for assessing biosimilarity. The stepwise approach is to evaluate biosimilarity step by step. The idea is well understood. However, little or no information regarding criteria for assessment of biosimilarity to be used at each step was mentioned. Some scientific issues regarding the suggested stepwise approach are raised. These issues include: (1) How many steps are required? (2) Does the order of the steps matter? (3) Should each step carry the same weight (in other words, are some steps more important than others)? and (4) How to control the overall type 1 error rate? These issues need to be addressed for feasibility and validity of the stepwise approach. In the draft guidance, FDA also introduces the concept of totality-of-the-evidence. FDA seems to suggest a scoring system for measuring the totality-of-the-evidence in order to account for: (1) the distinction between local biosimilarity and global biosimilarity; (2) degree of biosimilarity which may vary from domain to domain; and (3) each domain may carry different weights. In the current FDA draft guidance, little or no information regarding interchangeability was mentioned.
Conclusions
In summary, according to the definitions given in the BPCI Act, there is a clear distinction between biosimilarity and interchangeability. Although the recent FDA draft guidances did provide some insights regarding the assessment of biosimilarity of follow-on biologics, many scientific issues such as the definition of highly similar, criteria for biosimilarity, the feasibility of the stepwise approach, statistical tests for comparability in quality attributes, and the assessment of totality-of-the-evidence still remain unresolved for meeting regulatory expectations within the US. Regarding drug interchangeability, in practice, it is difficult, if not impossible, to demonstrate the expected ‘same clinical result in any given patient’. However, it is possible to demonstrate ‘same clinical result in any given patient with certain assurance’. Under a given study design, the biosimilarity index and/or totality biosimilarity index proposed by Chow (2011) [24] and Chow et al. (2011) [25] may be considered to develop an alternating index and/or switching index for addressing interchangeability in terms of alternating and/or switching. However, further research is necessary. It should be noted that in order to obtain input and comments on the draft guidances and to discuss the issue of interchangeability, FDA hosted a public hearing on 11 May 2012 at FDA, Silver Spring, Maryland, USA. It is our hope that more specific guidances that cover design and analysis for assessment of biosimilars and discussion of the unresolved scientific issues would be developed for implementation of BPCI Act. The purpose of this paper and the upcoming special issue on Biosimilarity and Interchangeability is an attempt to address some of the scientific factors and practical issues regarding the assessment of biosimilarity and interchangeability discussed at the 2010 FDA Public Hearing (held between 2–3 November 2010), the 2011 FDA public meeting (held on 16 December 2011), and the 2012 FDA Public Hearing (held on 11 May 2012). In addition, it is our intention to raise awareness to the scientific community that many of these scientific factors and practical issues still remain unresolved within the US, and more research on these topics is needed.
Editor’s comments
FDA and EMA do not require comparability in the manufacturing process. Manufacturing process could be different. EMA allows total different manufacturing processes even using different cell lines. Comparability of the active substance in the presented formulation should be shown.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Professor Shein-Chung Chow, PhD
Christine Ju, MSc
Department of Biostatistics and Bioinformatics, Duke University School of Medicine, 2424 Erwin Road, Hock Suite 1102, Durham, North Carolina 27710, USA
References
1. United States Food and Drug Administration [homepage on the Internet]. Guidance for Industry. Bioavailability and bioequivalence studies for orally administrated drug products – General Considerations. 2003 March [cited 2012 Nov 21]. Available from: http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070124.pdf
2. European Medicines Agency [homepage on the Internet]. Draft guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1. 2008 July [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003011.pdf
3. Chow SC, Hsieh E, Chi E, Yang J. A comparison of moment-based and probability-based criteria for assessment of follow-on biologics. J Biopharm Stat. 2010;20(1):31-45.
4. Simoens S, Verbeken G, Huys I. Market access of biosimilars: not only a cost issue. Oncologie. 2011;13(5):218-21.
5. GaBI Online – Generics and Biosimilars Initiatives. Generics grab 80% share of US market and fill 78% of prescriptions [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2012 Nov 21]. Available from: www.gabionline.net/Reports/Generics-grab-80-share-of-US-market-and-fill-78-of-prescriptions
6. European Medicines Agency [homepage on the Internet]. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant erythropoietins. EMEA/CHMP/BMWP/94526/2005 Corr. 2006 March [cited 2012 Nov 21]. Available from: http://www.tga.gov.au/pdf/euguide/bmwp9452605en.pdf
7. European Medicines Agency [homepage on the Internet]. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant granulocyte-colony stimulating factor. EMEA/CHMP/BMWP/31329/2005. 2006 February [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003955.pdf
8. European Medicines Agency [homepage on the Internet]. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant human soluble insulin. EMEA/CHMP/BMWP/32775/2005. 2006 February [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003957.pdf
9. European Medicines Agency [homepage on the Internet]. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing somatropin. EMEA/CHMP/BMWP/94528/2005. 2006 February [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003956.pdf
10. European Medicines Agency [homepage on the Internet]. Draft guideline on similar medicinal products containing recombinant interferon alpha. EMEA/CHMP/BMWP/102046/2006. 2007 Oct [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003931.pdf
11. European Medicines Agency [homepage on the Internet]. Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins. EMEA/CHMP/BMWP/118264/2007. 2009 March [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003927.pdf
12. GaBI Online – Generics and Biosimilars Initiatives. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2012 Nov 20]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
13. Chow SC, Liu JP. Statistical assessment of biosimilar products. J Biopharm Stat. 2009;20(1):10-30.
14. Biologics Price Competition and Innovation Act of 2009, Pub. L. 111-148, Sect. 7001-7003, 124 Stat. 119. Mar. 23, 2010.
15. United States Pharmacopeia and National Formularly XXI. United States Pharmacopeia 24 and National Formulary 19. Rockville, Maryland: United States Pharmacopeial Convention, Inc.; 2000.
16. United States Food and Drug Administration [homepage on the Internet]. Guidance for industry. Scientific considerations in demonstrating biosimilarity to a reference product. 2012 Feb [cited 2012 Nov 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
17. Chow, Shein-Chung, Liu JP. Design and analysis of bioavailability and bioequivalence studies. Boca Raton: Chapman & Hall/CRC Press; 2008.
18 United States Food and Drug Administration [homepage on the Internet]. Guidance for Industry. Statistical approaches to establishing bioequivalence. 2001 Jan [cited 2012 Nov 21]. Available from: http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070244.pdf
19. World Health Organization [homepage on the Internet]. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. QAS/04.093/Rev.4. 2005 Nov [cited 2012 Nov 21]. Available from: http://www.who.int/medicines/services/expertcommittees/pharmprep/QAS04_093Rev4_final.pdf
20. European Medicines Agency [homepage on the Internet]. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr. 2010 January [cited 2012 Nov 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
21. Schall R, Luus HG. On population and individual bioequivalence. Stat Med. 1993;12(12):1109-24.
22. Chow SC, Hsieh E, Chi E, Yang J. A comparison of moment-based and probability-based criteria for assessment of follow-on biologics. J Biopharm Stat. 2010;20(1):31-45.
23. United States Food and Drug Administration [homepage on the Internet]. FDA issues draft guidance on biosimilar product development. 2012 Feb [cited 2012 Nov 21]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm291232.htm
24. Chow SC. Quantitative evaluation of bioequivalence/biosimilarity. J Bioequiv Bioavailab S1. 2011; doi:10.4172/jbb.S1-002
25. Chow SC, Endrenyi L, Lachenbruch PA, Yang LY, Chi E. Scientific factors for assessing biosimilarity and drug interchangeability of follow-on biologics. Biosimilars. 2011;1:13-26.
|
Author for correspondence: Christine Ju, MSc, Department of Biostatistics and Bioinformatics, Duke University School of Medicine, 2424 Erwin Road, Hock Suite 1102, Durham, North Carolina 27710, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
Pharmacovigilance of biosimilars: challenges and possible solutions


