Assessment of the interchangeability between generics
Published on 2016/05/04
Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):55-9.
Author byline as per print journal: Luther Gwaza1,2, BPharm, MPhil; John Gordon3, PhD; Henrike Potthast4, PhD; Marc Maliepaard5, PhD; Jan Welink5, Hubert Leufkens1,5, PhD; Matthias Stahl6, MD; Alfredo García-Arieta7, PhD
|
Abstract: |
Submitted: 21 March 2016; Revised: 11 April 2016; Accepted: 11 April 2016; Published online first: 25 April 2016
Introduction
Direct comparison within well-designed and well-conducted randomized controlled trials (RCT) is the gold standard for comparing health interventions. The usual practice to obtain marketing authorization for new health interventions is to compare them to placebo, or standard of care, but not with all available health interventions. Some interventions are developed simultaneously, thus it is not feasible to perform comparison between them during this development phase to support marketing authorizations. Moreover, in some cases, there is a relatively large number of available interventions making direct comparison through RCTs between them impractical. Therefore, in these instances where multiple interventions exist, there is often lack of, or insufficient, evidence of direct comparison (head-to-head comparison) to evaluate relative effectiveness of all the available health interventions; indirect comparisons are then employed [1–3].
Indirect treatment comparison is defined as an evaluation of different health interventions using information from independent studies. This is useful when there are no data on direct comparison, or to provide supplementary evidence when the data from direct comparison are insufficient [4]. Indirect comparison can be categorized into naïve (unadjusted), informal indirect comparison, and adjusted indirect comparison [3]. Naïve indirect comparison evaluates the data from the independent studies as if the data are from the same study ignoring the between-study variance. For this reason, evidence from naïve indirect comparison is equivalent to observational studies, prone to bias and it may over- or underestimate the treatment effect; thus this approach is not recommended for analysing data from RCTs [2–4]. In informal indirect comparison, the results from the independent studies are compared directly, and relative effects or statistical significance are not formally calculated [3]. Adjusted indirect treatment comparison evaluates different treatments tested in independent studies modified based on the results of their direct comparison with a common control, partly preserving the power of RCTs [3] without the added cost of actual direct RCT comparison. However, adjusted indirect comparisons are less precise as reflected in wider confidence intervals [4], thus wherever possible direct comparisons should be performed.
Figure 1 illustrates the direct and indirect comparison of health interventions. Suppose there are four different treatments, A, B, C and D compared in three different trials. If treatment A was compared in an RCT with treatment B, treatment C in another RCT with treatment B, and treatment C with D in another RCT, adjusted indirect treatment comparison can be used to compare treatment A and C since both were tested in two independent trials with the common treatment B. Likewise, treatment B and D can be compared using adjusted indirect comparison since both were compared in direct comparison with common treatment C.
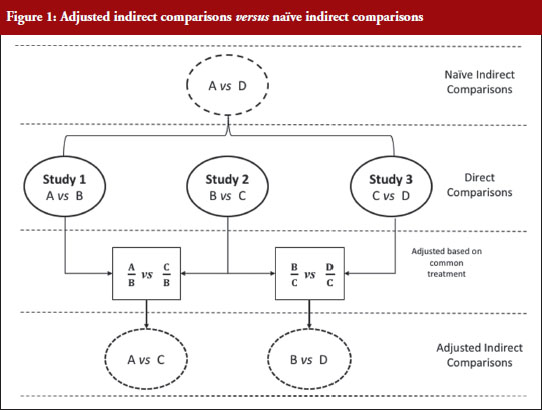
Generic medicines are approved nationally following the expiry of patents or market exclusivity period for the innovator products based on demonstration of bioequivalence with the innovator product. A bioequivalence study comparing a generic and an innovator drug is a form of direct comparison. With time, the number of approved generic medicines per active substance generally increases. Thus, requirement for direct comparison through bioequivalence studies between generics is impracticable since several generics of the same drug are licensed at various times, often without knowledge of the other generics under development. For this reason, adjusted indirect treatment comparison is a useful approach to identify those generic drug products whose interchangeability can be assured, by comparing the different generic drug products that have been demonstrated to be bioequivalent with the same reference product, to support or ensure switchability in clinical practice without concerns on efficacy or safety due to the switching.
Methods used in indirect comparisons
Several methods are available for performing indirect comparisons: (1) naïve or unadjusted indirect comparisons; (2) statistical methods using aggregate data such as simple weighted combination of separate estimates as suggested by Bucher et al. [1] (adjusted indirect comparison); (3) modelling approaches based on the individual patient data (meta-regression); and (4) mixed treatment comparisons based on Bayesian statistics (logistic regression) [2, 5].
In meta-regression analysis, the estimated difference between the groups (treatment effect) is modelled as a function of one or more study characteristics as the predictor variable. Estimated treatment effect in each study is weighted according to the inverse of its variance. A simple approach for meta-regression is weighted linear regression, and the residual heterogeneity is estimated using random effects. Meta-analysis can be performed using the fixed-effects to describe the residual heterogeneity. The key assumption for fixed effect meta-analysis is that the different trials estimated the same effect, for example, the effect of treatment A relative that of treatment B. Regression methods such as logistic regression can be used to perform indirect comparison using the generalized linear models and individual patient data. With regression modelling, one can adjust for other variables available for each study. While full individual patient data, which are required for logistic regression for indirect comparison, or the estimated treatment effect, its variance and covariates for each trial, which are necessary to perform meta-regression, are rarely publicly available, adjusted indirect comparison is performed using the summarized data available in published articles and approved product labelling. It is the simplest and most appropriate methodological approach when only two interventions are to be compared indirectly as it is the case for bioequivalence studies [6].
When using summarized data extracted from published data, first the data are extracted or calculated using appropriate summary statistics, e.g. confidence intervals, mean ratios, for each set of studies. For bioequivalence studies, the extracted data are study products, sample sizes preferably in each sequence, confidence intervals and study design (fasting or fed study, parallel or crossover, single or multiple dose studies). The confidence intervals of the bioequivalence studies are converted to log scale and used to estimate the point estimate and standard error of the treatment effects. Lastly, the data are combined to provide an overall comparison. The standard statistical result, i.e. the variance of the difference between the two independent estimates, is sum of the two variances (variance is square of the standard error), which is similar to a 2-sample t-test. Thus, using the illustration in Figure 1, if you have the two estimated effects for A υs B and B υs C as θAB and θBC, respectively, the effect of the comparison A υs C is estimated as θAC = θAB − θBC and var (θAC) = var (θAB) + var (θBC) [1]. The scale of the effect θ relates to the scale on which the data would be analysed, such as risk difference, log risk ratio, log odds ratio for binary data, the means, mean difference, mean change for continuous data and log hazard ratio for time-to-event data. In bioequivalence studies, the pharmacokinetic outcome measure is continuous data, and the effect θ is the ratio of log-transformed geometric means of the treatments, e.g. A and B (point estimate). The 90% confidence interval (CI) of the ratio of the log-transformed geometric means is the standard statistical result. Therefore, the 90% CI for the indirect comparison ![]() , where z/t in this equation is the z value of standardized normal distribution or the t value of the Student’s t distribution that corresponds to the desired level of confidence (90% in case of bioequivalence studies) and the degrees of freedom in the case of the Student’s t distribution.
, where z/t in this equation is the z value of standardized normal distribution or the t value of the Student’s t distribution that corresponds to the desired level of confidence (90% in case of bioequivalence studies) and the degrees of freedom in the case of the Student’s t distribution.
We explored the different approaches for calculating adjusted indirect comparisons [7]. In this study, we compared six methods that can be used to calculate the width of the confidence intervals based on z distribution (z0.9) or Student’s t distribution (t0.9, d.f.). Four methods that assumed small sample sizes with Student’s t distribution are: (a) Chow and Liu meta-analysis method [8], which assumes all studies had 2 × 2 crossover design and homogeneity of the distribution of reference product data in all studies; (b) homoscedastic method which assumes homogenous variances; (c) heteroscedastic method which assumes heterogeneous variances; and (d) the pragmatic approach which does not require the assumption of homogeneity of variances between studies with small sample sizes. The two methods which assumes large sample sizes with a standardized normal distribution (z0.9) are: (a) Chow and Shao meta-analysis method [9]; and (b) the z-distribution method with no assumption on homogeneity of variances [10].
We concluded that although the differences were minor, the homoscedastic method is recommended, unless there are clear differences in variances, because it is the most conservative approach for estimating the confidence intervals for adjusted indirect comparisons.
Application of adjusted indirect treatment comparison for generics
We investigated the differences in bioavailability between generics prequalified by the World Health Organization (WHO) using adjusted indirect comparisons [7, 11]. These studies investigated a diverse group of products from the antimalarials artemether/lumefantrine [7], first-line antituberculosis drugs, [11] and first-line antiretroviral drugs with a total of 394 indirect comparisons. In contrast to the ±20% acceptance range used for direct comparisons, a ±30% acceptance range is proposed for adjusted indirect comparisons [7, 11], due to the limited precision of indirect comparisons [1, 4].
First, these studies demonstrated the utility of adjusted indirect treatment comparison to compare the bioavailabilities between generic drug products that had been compared with the same reference product in direct comparisons. Second, the outcome of these comparisons indicate that antimalarial artemether/lumefantrine, first-line anti-tuberculosis, and first-line antiretroviral generics prequalified by WHO can be interchanged without any safety and efficacy concerns in clinical settings. Although some comparisons were outside the conventional acceptance limits of ±20% for direct comparisons, there were no generic–generic comparisons outside the ±30% for indirect comparisons, except one comparison for efavirenz Cmax. Failure to show equivalence within a ±30% acceptance range in one out of 394 adjusted indirect comparisons should be interpreted as insignificant number since it is less than 0.3% of the comparisons.
The results obtained with the prequalified generics are consistent with the outcomes reported elsewhere using data from other regulatory authorities [12–14]. Herranz et al. showed that exposures obtained with generic tacrolimus formulations in the Spanish market were within the ±20% acceptance range based on adjusted indirect treatment comparisons [12]. In addition, results from adjusted indirect comparisons were consistent with those from direct comparisons for multiple generic formulations of gabapentin products marketed in The Netherlands [13]. Using data from bioequivalence studies submitted to the Dutch Medicines Evaluation Board (CBG-MEB) for atorvastatin, bicalutamide, naratriptan, olanzapine, perindopril, venlafaxine, cyclosporine, tacrolimus and mycophenolate mofetil, Yu and colleagues showed that in 80% of the cases the indirect comparisons between generics fulfilled the conventional acceptance limit of ±20%, while the remainder were within ±30% [14]. A point estimate constraint in the bioequivalence studies may be relevant for drugs with a narrow therapeutic index, e.g. cyclosporine and tacrolimus, where switching between generics of these drugs is not restricted. Generally, narrow therapeutic drugs are usually assessed with a narrowed acceptance range, e.g. 90.00–111.11%, to ensure interchangeability with the reference [15–17].
We observed that assurance regarding interchangeability of two generic drug products is reduced when either the point estimate ratios in the original studies are shifted from unity by more than 5% or when the width of the 90% confidence interval is large in the direct comparisons [11]. Therefore, we investigated the influence of point estimate, variability of the pharmacokinetic parameters (Cmax and AUC), and the sample size in the original studies on the ability to demonstrate bioequivalence between generics in the adjusted indirect comparisons [18]. However, sample size and variability are not independent since the sample size is calculated based on the expected variability and the desired statistical power. Thus, statistical power is the most relevant parameter for consideration in the computations.
We calculated the outcome of adjusted indirect comparisons for 14,592 scenarios using 57 possible differences between point estimates from 0% to 14% and 16 possible study powers from 50% to 99.99%. The study results illustrated that demonstrating bioequivalence within the conventional acceptance limits of 80–125% by means of adjusted indirect comparisons is only possible if the difference between the point estimate is small (< 5%) for any sufficiently powered study (> 80%). Furthermore, even when both studies are overpowered, the difference cannot be larger than 14%. This study showed that in cases where generic–generic switching maybe of concern, regulators might consider a point estimate constraint in the original studies.
The variance for the adjusted indirect comparison is additive, ![]() , thus, the major limitation of adjusted indirect comparisons of bioequivalence studies is the reduced precision. On one hand, the inability to show bioequivalence by means of indirect comparisons is not proof of inequivalence, but it may be simply that there is not enough statistical power to make this conclusion. On the other hand, when bioequivalence is shown within the conventional acceptance limits for indirect comparisons despite the reduced statistical power, we can consider not only that the generic drug products are bioequivalent but also very similar.
, thus, the major limitation of adjusted indirect comparisons of bioequivalence studies is the reduced precision. On one hand, the inability to show bioequivalence by means of indirect comparisons is not proof of inequivalence, but it may be simply that there is not enough statistical power to make this conclusion. On the other hand, when bioequivalence is shown within the conventional acceptance limits for indirect comparisons despite the reduced statistical power, we can consider not only that the generic drug products are bioequivalent but also very similar.
The validity of indirect comparisons is dependent on the methodological quality and assumptions. Similarity of trials involved in adjusted indirect comparisons should be carefully assessed to ensure that there are no important differences between the trials under comparison in aspects that could bias the estimated formulation effect. Although standard requirements are applied on the design, conduct and analysis of results of the bioequivalence studies submitted for the prequalification of generics [15], in some cases different study designs might be employed. For example, metabolite versus parent as the analyte, or plasma versus urine as the biological fluid collected for analysis, or multiple versus single dose studies. Studies with these different study designs cannot be compared because the formulation effect cannot be expected to be the same between them. However, we consider results from conventional 2 × 2 crossover designs and replicate designs as combinable. There is no consensus on whether parallel and crossover trials should be combined in indirect comparisons [2], however, this may be possible if the participants and interventions are comparable. In all the analyses performed, all the studies were crossover trials.
In contrast to adjusted indirect comparisons of efficacy trials, confidence in the methodological quality and similarity of adjusted indirect comparisons of bioequivalence studies is assured because of the general consistency in the basic design of these studies. For instance, the characteristics of participants in bioequivalence studies are commonly defined, i.e. usually healthy adult male and/or female volunteers within 18–55 years of age, which controls for the differences in baseline characteristics between treatment groups, whereas differences in disease state in efficacy trials is of concern. The objective of bioequivalence studies is to evaluate formulation differences and external validity of the results is based on the assumption that the effect of the drug in the target populations, i.e. patients would be the same for the test and reference. Nevertheless, in some cases, subject–by–formulation interaction could occur, e.g. when one formulation has excipients that are not tolerated by specific subgroups of patients that are not present in the reference formulation. This is often mitigated by the regulatory requirement to declare such excipients on the product label. Non-randomization of the trials can confound the results if other differences between the treatment groups are linked to the outcomes. Randomization ensures that like is compared with like, i.e. that there are no differences between the groups in any factors other than the intervention itself, in this case, formulation effects. In the bioequivalence studies considered in the analysis of the prequalified generics, subjects were randomized in the allocation of sequence.
Despite the utility of the indirect comparisons, the evidence from such analyses should be interpreted with caution. The internal validity of direct comparisons should be carefully evaluated to reduce bias. In the analysis of the prequalified generics, methodological quality of the studies was not assessed as part of the adjusted indirect comparisons since only the generics that were prequalified were included in the analysis. The prequalification process entails stringent assessment including inspection of the contract research organizations at which the studies were performed, thus providing assurance of the quality of the prequalified product. Therapeutic doses are usually standardized as highest available strength, although in some cases the studies used lower doses. Nonetheless, the results are reported as mean ratios, thus the effect of dose on the outcomes is negligible. The studies were all single dose studies with the same outcome measure of pharmacokinetic parameters Cmax and AUC in all the studies, estimated using validated software. The parent compound in plasma was analysed using validated bioanalytical methods. Despite the general consistency with the bioequivalence studies used in the adjusted indirect comparisons, changes in the requirements over time encompassing several revisions of the guidelines could potentially impact on the methodological quality between studies conducted at different time points; this may be corrected by the use of the same reference product in the different bioequivalence studies. Only the studies using a common reference product as listed by the WHO Prequalification of Medicines Programme were compared in the adjusted indirect comparisons. Though US and European reference products are both accepted in the WHO Prequalification of Medicines Programme, and no distinction was made in the analysis, in some cases these are not the same due to different manufacturing sites and different excipients. However, it is assumed that these products are bioequivalent to the pivotal clinical batch used for gaining marketing authorizations in both jurisdictions.
In conclusion, adjusted indirect comparison is a useful tool to compare relative bioavailabilities between generics that have been compared with a common reference in direct comparison to ensure interchangeability between the generics. The investigated antimalarial – artemether/lumefantrine, first-line antituberculosis and antiretroviral generic drug products prequalified by WHO were considered interchangeable without safety and efficacy concerns. We have also demonstrated that the ability to show bioequivalence between generic drug products by means of indirect comparisons depends on the difference between the point estimates of the bioequivalence studies, which is the point estimate of the indirect comparison, and the power of the bioequivalence studies that are combined. In this respect, concluding equivalence in the indirect comparison within the conventional acceptance limits of 80–125% is only possible when: (a) point estimate difference between generics are low (< 5%) for any sufficiently powered study (> 80%); or (b) the differences do not exceed 14% when both studies are overpowered. Therefore, in cases where it is important to ensure generics interchangeability, the regulatory authorities may consider a point estimate constraint in the original bioequivalence studies. In the general case, due to the reduced precision of indirect comparison, a slightly wider acceptance limits (± 30%) is proposed for indirect comparisons.
Competing interests: The authors declare no conflict of interest.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Luther Gwaza1,2, BPharm, MPhil; John Gordon3, PhD; Henrike Potthast4, PhD; Marc Maliepaard5, PhD; Jan Welink5, Hubert Leufkens1,5, PhD; Matthias Stahl6, MD; Alfredo García-Arieta7, PhD
1Utrecht Institute for Pharmaceutical Sciences (UIPS), Utrecht, The Netherlands
2Medicines Control Authority of Zimbabwe, 106 Baines Avenue, Harare, Zimbabwe
3Division of Biopharmaceutics Evaluation 2, Bureau of Pharmaceutical Sciences, Health Canada, Locator 0201C, 101 Tunney’s Pasture Driveway, Ottawa, ON K1A 0K9, Canada
4Federal Institute of Drugs and Medical Devices, 3 Kurt-Georg-Kiesinger-Allee, DE-53175 Bonn, Germany
5Medicines Evaluation Board, 500 Graadt van Roggenweg, NL-3531 AH Utrecht, The Netherlands
6Group Leader Medicines Assessment Prequalification Team – Medicines, HIS/EMP/RHT, World Health Organization, 20 Avenue Appia, CH-1211 Geneva 27, Switzerland
7División de Farmacología y Evaluación Clínica, Departamento de Medicamentos de Uso Humano, Agencia Española de Medicamentos y Productos Sanitarios, 1 Calle Campezo, Edificio 8, Planta 2 Oeste, ES-28022 Madrid, Spain
References
1. Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683-91.
2. Glenny AM, Altman DG, Song F, Sakarovitch C, Deeks JJ, D’Amico R, Bradburn M, Eastwood AJ; International Stroke Trial Collaborative Group. Indirect comparisons of competing interventions. Health Technol Assess. 2005 Jul;9(26):1-134, iii-iv.
3. Song F. What is indirect comparison? What is …? series. February 2009.
4. Song F, Altman DG, Glenny AM, Deeks JJ. Validity of indirect comparison for estimating efficacy of competing interventions: empirical evidence from published meta-analyses. BMJ.2003; 326(7387):472.
5. Sutton AJ, Higgins JP. Recent developments in meta-analysis. Stat Med. 2008;27(5):625-50.
6. Schöttker B, Lühmann D, Boulkhemair D, Raspe H. Indirect comparisons of therapeutic interventions. GMS Health Technol Assess. 2009;5.
7. Gwaza L, Gordon J, Welink J, Potthast H, Hansson H, Stahl MM, García-Arieta A. Statistical approaches to indirectly compare bioequivalence between generics: a comparison of methodologies employing artemether/lumefantrine 20/120 mg tablets as prequalified by WHO. Eur J Clin Pharmacol. 2012;68(12):1611-8.
8. Chow SC, Liu J. Meta-analysis for bioequivalence review. J. Biopharm Stat. 1997;7(1):97-111.
9. Chow SC, Shao J. Bioequivalence review for drug interchangeability. J Biopharm Stat. 1999;9(3):485-97.
10. Krauss GL, Caffo B, Chang YT, Hendrix CW, Chuang K. Assessing bioequivalence of generic antiepilepsy drugs. Ann Neurol. 2011;70(2):221-8.
11. Gwaza L, Gordon J, Welink J, Potthast H, Leufkens H, Stahl M, et al. Adjusted indirect treatment comparison of the bioavailability of WHO-prequalified first-line generic antituberculosis medicines. Clin Pharmacol Ther. 2014;96(5):580-8.
12. Herranz M, Morales-Alcelay S, Corredera-Hernández MT, de la Torre-Alvarado JM, Blázquez-Pérez A, Suárez-Gea ML, et al. Bioequivalence between generic tacrolimus products marketed in Spain by adjusted indirect comparison. Eur J Clin Pharmacol. 2013;69(5):1157-62.
13. Yu Y, Teerenstra S, Vanmolkot F, Neef C, Burger D, Maliepaard M. Interchangeability of gabapentin generic formulations in the Netherlands: a comparative bioavailability study. Clin Pharmacol Ther. 2013;94(4):519-24.
14. Yu Y, Teerenstra S, Vanmolkot F, Neef C, Burger D, Maliepaard M. Investigation into the interchangeability of generic formulations using immunosuppressants and a broad selection of medicines. Eur J Clin Pharmacol. 2015;71(8):979-90.
15. World Health Organization. WHO Expert Committee on Pharmaceutical Preparations. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. in WHO Technical Report Series, No. 992. 2015, Annex 7 [homepage on the Internet]. [cited 2016 Apr 11]. Available from: http://apps.who.int/medicinedocs/documents/s21898en/s21898en.pdf
16. European Medicines Agency. Committee on Medicinal Products for Human Use (CHMP). Guideline on the investigation of bioequivalence. 20 January 2010. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr ** [homepage on the Internet]. [cited 2016 Apr 11]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
17. U.S. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Guidance for industry. Bioavailability and Bioequivalence studies for orally administered drug products — general considerations. March 2003 homepage on the Internet]. [cited 2016 Apr 11]. Available from: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3995B1_07_GFI-BioAvail-BioEquiv.pdf
18. Gwaza L, Gordon J, Potthast H, Welink J, Leufkens H, Stahl M, et al. Influence of point estimates and study power of bioequivalence studies on establishing bioequivalence between generics by adjusted indirect comparisons. Eur J Clin Pharmacol. 2015; 71(9):1083-9.
|
Author for correspondence: Alfredo García-Arieta, PhD, División de Farmacología y Evaluación Clínica, Subdirección General de Medicamentos de Uso Humano, Agencia Española de Medicamentos y Productos Sanitarios, 1 Calle Campezo, Edificio 8, Planta 2 Oeste, ES-28022 Madrid, Spain |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


