Ethnic sensitivity assessments in biosimilar monoclonal antibodies clinical development programmes: necessary or not?
Published on 2023/08/08
Generics and Biosimilars Initiative Journal (GaBI Journal). 2023;12(2):61-6.
Author byline as per print journal: Sandeep N Athalye1, MBBS, MD; Dev B Baruah1, MPharm; Shivani Mittra1, MPharm, PhD; Ankit Ranpura1, MD; Kuldeep Kumar1, PhD; Elena Wolff-Holz2, MD
|
Abstract: |
Submitted: 21 June 2023; Revised: 26 July 2023; Accepted: 27 July 2023; Published online first: 9 August 2023
Introduction
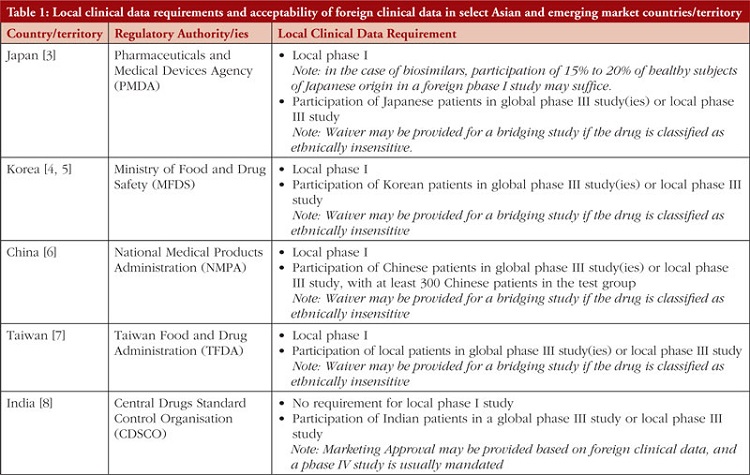
Pharmaceutical and biotechnology companies develop new drug products through multi-regional/global clinical trials to register the product in multiple geographies. In these clinical trials, various parameters such as pharmacokinetics (PK), pharmacodynamics (PD), safety and efficacy are typically evaluated to establish the drug’s effectiveness and safety profile. As variability in drug exposure and response can be affected by several complex factors, including race and ethnicity, these can sometimes become a rate-limiting step toward product registration [1]. The ICH E5 guidance deals specifically with the role of ethnic factors in the acceptability of foreign clinical data and suggests frameworks to facilitate the review and registration of global trials [2]. Often, to account for potential ethnic variability, bridging studies (e.g. a local phase I PK/PD comparability study or a phase II/III randomized controlled clinical study) or inclusion of subjects with particular ethnic backgrounds in multi-regional clinical trials (MRCTs) are needed to assess a drug’s sensitivity to ethnic factors and to support its registration in specific markets, particularly in countries/territory within East Asia, see Table 1 [3–8].
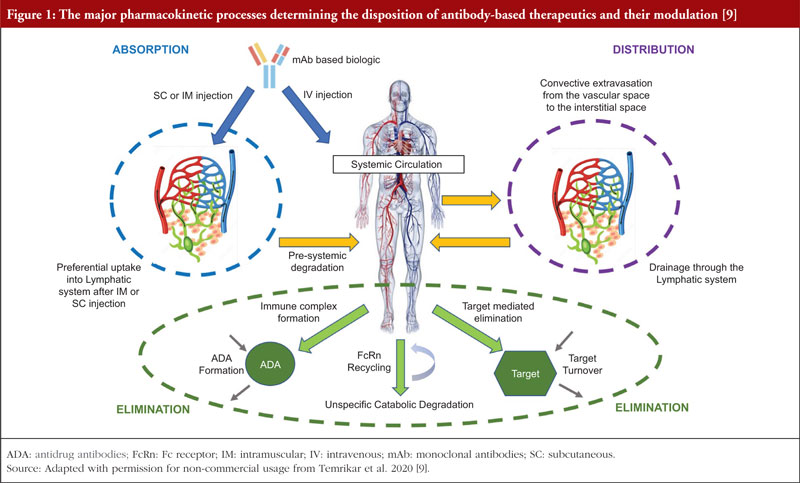
These requirements for conducting bridging studies also extend to biological products like monoclonal antibodies (mAbs), which, unlike small molecule drugs, do not undergo traditional drug-related metabolism, see Figure 1 [9]. MAbs are administered parenterally and reach systemic circulation, either directly (by intravenous (IV) route) or through the lymphatic system (by subcutaneous (SC) or intramuscular (IM) routes). MAbs being large therapeutic proteins are mostly confined to the vascular space, with substantially reduced extravascular concentrations relative to their presence in the vasculature [10]. The elimination of mAbs from the body is primarily facilitated by intracellular catabolism via lysosomal degradation after uptake into cells by either pinocytosis, an unspecific fluid-phase endocytosis, or by a receptor-mediated endocytosis process [11]. Definitive statements regarding the impact of race/ethnicity on drug exposure are often not found on biologicals labels. Body weight and body surface area are the most commonly identified covariates (82%) in population PK models for mAbs [12], and the effect of race/ethnicity or gender on PK is generally insignificant after differences in body weight are considered [13, 14].
The ICH E5 guidelines acknowledge that ethnic factors may not affect all drugs. Therefore, the guidelines recommend characterizing a drug’s potential sensitivity to ethnic factors regarding its PK, PD and therapeutic effects to determine the type of bridging study needed in a new region [2]. The guidelines specify a list of properties that would make a drug less likely to be sensitive to ethnic factors, such as minimal metabolism, high bioavailability/low susceptibility to dietary absorption effects, and little potential for food and drug interactions. As mentioned earlier, these properties are inherently related to almost all mAbs and make them less likely to be affected by ethnic differences. ICH E5 guidelines also state that in certain situations where a medicine under evaluation is categorized as ethnically insensitive and extrinsic factors in the two regions/countries are generally similar, extrapolation of clinical data might be feasible without a bridging study. Notwithstanding, pharmaceutical companies routinely carry out Ethnic Sensitivity Studies (ESS) for mAbs as a preemptive measure to comply with regulatory demands and facilitate their entrance into new markets, such as East Asia.
Based on the currently available evidence, we believe that the decision to proceed with or without ESS should be evidence-based, considering available information on the reference drug rather than being a mandatory obligation in biosimilar development programmes. This perspective is grounded in the widely accepted principle that experience with the reference product serves as the cornerstone, and the primary objective of biosimilar development is to demonstrate that the purity, potency, safety and efficacy of the biosimilar are comparable to the reference product, rather than independently establishing the safety and effectiveness of the biosimilar [15]. In this context, we assess the feasibility of excluding ethnic sensitivity assessments such as dedicated phase I bridging trials from the clinical development programmes of biosimilar mAbs.
Ethnic sensitivity in mAbs
In East Asian countries like Japan, ESSs are conducted to evaluate safety and PK in a small cohort of healthy Japanese individuals before recruiting Japanese patients into global clinical trials [1]. This process, however, affects the clinical development of proposed biosimilars for mAbs, as regulatory authorities in East Asian countries often require the inclusion of a local phase I PK/PD study as part of the biosimilar clinical development programme. An alternative approach to meeting this requirement could involve conducting a comparable phase I PK/PD study in a country with a multi-ethnic population, using a subgroup of subjects from the immigrant or expatriate population who possess the intended market’s ethnic background.
In a systematic evaluation of clinical ethnic sensitivity data for various mAbs in both approved and late-stage clinical development products, Matsushima et al. (2015) reported that exposures following a single IV or SC administration could be predicted in healthy Japanese subjects using the data from healthy non-Japanese subjects. This held true regardless of the incidence of immunogenicity, except when target-mediated disposition was involved [14].
Additionally, Matsushima et al. (2015) compared the incidence of immunogenicity between Japanese and non-Japanese healthy subjects. They noted that all mAbs, except for adalimumab, exhibited few or no instances of immunogenicity after a single administration, irrespective of the subject’s ethnicity.
There have been investigations into race as a covariate in several biologicals’ population PK models, but race has been found to be insignificant for denosumab (bone metastases from solid tumours) [16], belimumab (systemic lupus erythematosus) [17], panitumumab (advanced solid tumours) [18], anti-PD-1 mAbs such as nivolumab and pembrolizumab. [19]. Phase I PK studies of bevacizumab (advanced cancer) [20] and ustekinumab (healthy male subjects) [21] have been reported in Chinese patients, and cross-study comparisons with Caucasian patients indicate no meaningful differences across these populations. Zhou et al. (2012) reported that PK profiles of mAbs were similar in the Japanese and Caucasian populations, and no significant differences were observed in the studies. The authors suggest that clinical PK bridging studies between the two populations may not be necessary for the approval of mAbs and can be instead evaluated on a case-by-case basis, considering the mAbs’ characteristics and the target population [22]. This viewpoint is supported by similar approved dosing regimens in Japan and the US for 12 mAbs across various therapeutic areas [22].
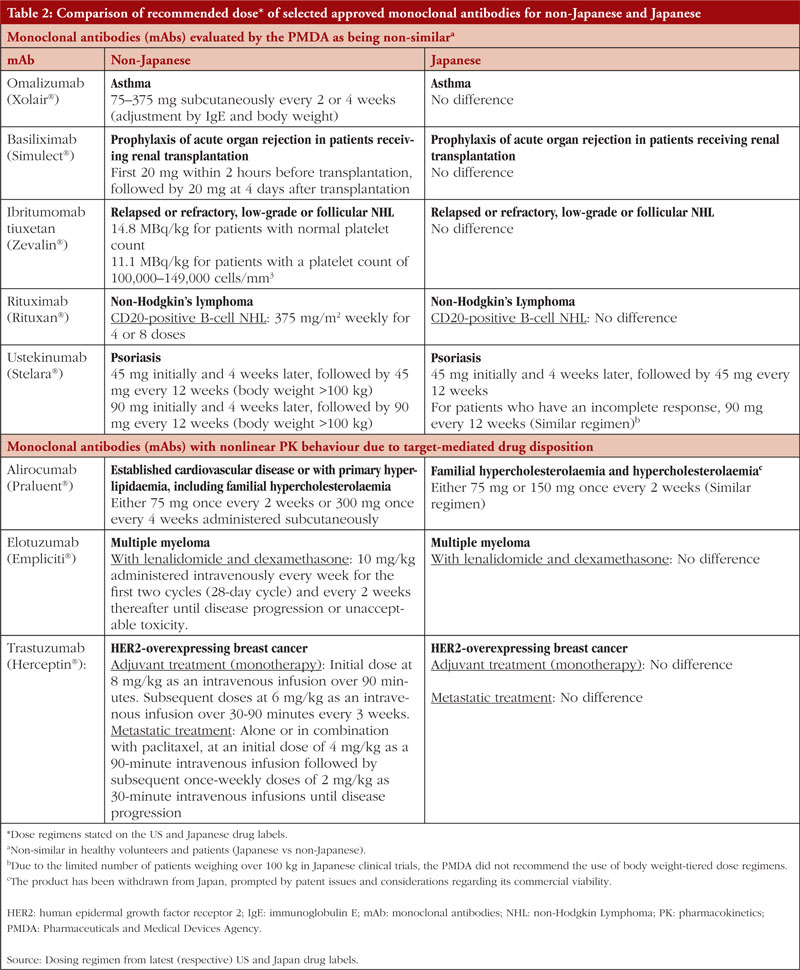
A review on the PK of mAbs approved in Japan reported that although some differences had been observed in the PK between Japanese and non-Japanese populations (mainly Caucasians), these were mainly attributed to differences in body weight and/or antigen levels [23]. Moreover, the influencing factors could be estimated without conducting regional PK/safety studies. For some mAbs that show non-linear PK due to target-mediated drug disposition, their clearance depends on the relative expression of the target antigen. Therefore, PK studies in healthy volunteers may not be appropriate in such cases, as differences between healthy volunteers and patients may arise because of variations in antigen levels. Despite this background, for 10 out of 24 mAbs approved in Japan, Japanese healthy volunteer studies were conducted before the patient studies, even though data from phase I studies in non-Japanese subjects were available. Additionally, for the mAbs (omalizumab, basiliximab, ibritumomab tiuxetan, rituximab and ustekinumab) that showed ethnic differences (Japanese vs non-Japanese) in PK profile, the doses selected in subsequent clinical studies in Japan were the same as the doses approved in the US [23], and the dose regimens stated on the US and Japan labels are essentially similar, see Table 2. Due to their small sample sizes, the inherent limitations of phase I ESS in identifying potential inter-ethnic differences in drug safety and/or PK profiles led Rajman et al. (2020) to propose a new global drug development paradigm. If relevant safety, PK, and pharmacogenetic (PG) data are available from the original phase I study population, i.e. non-Asian population, it might be possible to extrapolate these data to Asian populations, allowing for their inclusion in phase II/III trials without conducting an ESS [24].
Similarly, Jeon et al. (2021) observed that among the 43 mAb therapies approved in Japan and Korea (specific details not provided by the authors), 38 had comparable dosing regimens in both countries [25]. This implies a general absence of significant ethnic differences concerning mAbs in these East Asian populations, suggesting that clinical trial results from one country may be extrapolated to others in the region. Additionally, the authors noted that variations in approved dosing regimens for some drugs across the four Asian countries/territory (Korea, Japan, China and Taiwan) could be attributed to legislative differences rather than ethnic distinctions. For example, differences in dosage and regimen for mAb therapy could be due to local clinical trials conducted in each country in which the study population and dosing were different. Jeon et al. (2021) advocated for creating an Asia Clinical Pharmacology study network comprised of Asian investigators specializing in clinical pharmacology and therapeutics to facilitate the adoption of ICH E17 guidelines. They believed this would result in a growing body of evidence concerning ethnic sensitivity in MRCTs and ultimately expedite drug development efficiency in Asian countries [25].
Some studies have indicated that genetic polymorphisms, such as Fc receptor variants, may contribute to variations in clinical response to biologicals, particularly for cancer immunotherapy and other immune diseases among different ethnic groups [26–28]. Fc receptors play a pivotal role in the PK and PD of mAbs, and alterations in the expression levels or activity of the neonatal Fc receptor (FcRn) due to polymorphisms in the encoding gene FCGRT could result in interindividual differences [1]. However, these differences do not become evident on a population level, which is likely why there is limited research further exploring the association between these genetic variations and racial or ethnic factors in the context of clinical response to biologicals.
Target-mediated drug disposition (TMDD) of mAbs and ethnic factors
TMDD is a phenomenon in PK where the PK behaviour of a drug, particularly mAbs, is significantly influenced by its molecular target [29]. MAbs exhibit linear PK profiles, but a small percentage shows nonlinear PK behaviour due to TMDD. According to a study by Ryman and Meibohm (2017) [30], around 15% of mAbs (specifically alirocumab, elotuzumab, evolocumab, ofatumumab, panitumumab, tocilizumab and trastuzumab) demonstrate nonlinear PK profiles at therapeutic doses. It is worth noting that among these seven mAbs, only alirocumab (targeting PCSK9), elotuzumab (targeting SLAMF7) and trastuzumab (targeting HER2) have reported racial/ethnic variations in PK or efficacy profiles based on clinical studies. However, these variations did not lead to notable differences in dosing regimens between Japanese and non-Japanese populations for the same indications, see Table 2.
Hence, it can be concluded that most mAbs are administered at doses that result in target saturation, making TMDD non-measurable or overridden by non-target mediated clearance.
Robustness of biosimilar clinical studies
Biosimilars and their reference products contain highly similar versions of the same active substance, and the permitted differences are similar to those that are acceptable after manufacturing changes. Therefore, extensive clinical studies, such as ESSs, may not be required for biosimilars, as they are for new biologicals. This is supported by several systematic reviews and meta-analyses that have evaluated the clinical outcomes of biosimilars and their reference products in oncology. For example, a systematic review and meta-analysis evaluating 31 studies on three oncology reference products and their biosimilars, involving 12,310 patients distributed across the globe, reported rigorous clinical evaluation of the biosimilars, with the clinical outcomes statistically indistinguishable from those of the original products across drugs and cancer types irrespective of race/ethnicity [31]. Another systematic review and meta-analysis evaluating the efficacy and safety of three anticancer biosimilars compared to their reference biologicals [32] identified 23 randomized controlled trials (RCTs), of which eight RCTs investigated rituximab biosimilars (N = 1,534), six evaluated bevacizumab biosimilars (N = 1,897), and nine evaluated trastuzumab biosimilars (N = 4,953). Using the Grading of Recommendations, Assessment, Development and Evaluations (GRADE) approach to rate the quality of the evidence, the study concluded that the biosimilars of mAbs had highly comparable efficacy and safety profiles to their reference biologicals irrespective of race/ethnicity [32].
Limitation of this article
We deliberated on mAbs, that stand as the largest class within biological drugs with at least 100 approved mAb therapies globally as of 2022. We did not delve into other biological drug classes such as fusion proteins, recombinant proteins, vaccines, and gene and cell therapies. This was based on factors such as limited available information in certain cases, e.g. fusion proteins, and the consideration of PK, efficacy, or safety behaviours with regards to race and ethnicity, e.g. interindividual and inter-racial/ethnic differences in efficacy for insulins. These areas hold potential for future discussions.
Concluding remarks
Our literature review and analysis suggest that the ethnic sensitivity of mAbs can be determined by using the original data from previously studied population(s) without conducting additional studies. Therefore, we propose that the ESS should be waived-off for biosimilar mAbs, as they are highly similar to the reference products and have already demonstrated quality, safety and efficacy in various regions. We recommend that regulatory authorities in East Asia consider harmonizing the legislation and/guidance on ethnic sensitivity to facilitate biosimilar drug development and ensure patient access without compromising its safety and efficacy requirements.
Funding sources
Biocon Biologics Ltd paid for the Article Processing Charges.
Disclaimer
The opinions expressed in this article are personal views of the authors and should not be understood as being made on behalf of or reflecting the position of the agencies or organizations with which the authors are affiliated.
Competing interests: SNA, DBB, AMR, SM and KK are employees of Biocon Biologics Ltd (a subsidiary of Biocon Ltd). EWH is an employee of Biocon Biologics UK Ltd (a subsidiary of Biocon Biologics Ltd). SNA, DBB, and SM hold stocks in Biocon Ltd and/Biocon Biologics Ltd.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Sandeep N Athalye1, MBBS, MD
Dev B Baruah1, MPharm
Shivani Mittra1, PhD
Ankit Ranpura1, MD
Kuldeep Kumar1, PhD
Elena Wolff-Holz2, MD
1Clinical Development and Medical Affairs, Biocon Biologics Ltd, Biocon House, Tower 3, Semicon Park, Plot No. 29-P1 & 31-P, KIADB Industrial Area, Electronic City Phase – 2, Hosur Road, Bengaluru 560100, Karnataka, India
2Clinical Development and Medical Affairs, Biocon Biologics UK Ltd, London WC2B 5AH, UK
References
1. Davda J, Krishna R. The importance of collecting race/ethnicity information in clinical trials for biologics: a clinical pharmacology perspective. Certara 2021. https://www.certara.com/app/uploads/2021/11/Certara_WP_Clinical-Trials-for-Biologics.pdf
2. European Medicines Agency. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Topic E 5 (R1). Ethnic factors in the acceptability of foreign clinical data. September 1998. Updated 1998 [homepage on the Internet]. [cited 2023 Jul 26]. Available from: https://database.ich.org/sites/default/files/E5_R1__Guideline.pdf
3. Ministry of Health, Labour and Welfare (MHLW), Japan. Basic principles on global clinical trials. Notification 0928010. September 28, 2007 [homepage on the Internet]. [cited 2023 Jul 26]. Available from: https://www.pmda.go.jp/files/000153265.pdf
4. Ministry of Food and Drug Safety (MFDS), The Republic of Korea. Biosimilar product evaluation guideline, English Version, (2022.7) [homepage on the Internet]. [cited 2023 Jul 26]. Available from: https://www.mfds.go.kr/eng/wpge/m_37/de011024l001.do
5. Ministry of Food and Drug Safety (MFDS), The Republic of Korea. Biosimilar regulation on approval and review of biological products [homepage on the Internet]. [cited 2023 Jul 26]. Available from: https://www.mfds.go.kr/eng/brd/m_27/down.do?brd_id=eng0005&seq=70201&data_tp=A&file_seq=1
6. National Medical Products Administration (NMPA), China. Regulations on overseas data and waiving local clinical trials (NMPA-No35-2017, NMPA-No52-2018). [homepage on the Internet]. [cited 2023 Jul 26]. Available from: http://english.nmpa.gov.cn/
7. Taiwan Food and Drug Administration, (TFDA); Center for Drug Evaluation, Taiwan. Inspection and registration of biosimilar drugs (Accessed 15 Jun 2023). Retrieved from: https://www.cde.org.tw/law/law_more?id=42
8. Central Drugs Standard Control Organisation (CDSCO), NDCT rules 2019, India. [homepage on the Internet]. [cited 2023 Jul 26]. Available from: https://cdsco.gov.in/opencms/export/sites/CDSCO_WEB/Pdf-documents/NewDrugs_CTRules_2019.pdf
9. Temrikar ZH, Suryawanshi S, Meibohm B. Pharmacokinetics and clinical pharmacology of monoclonal antibodies in pediatric patients. Paediatr Drugs. 2020;22(2):199-216.
10. Meibohm B. Pharmacokinetics and pharmacodynamics of therapeutic peptides and proteins. In: Crommelin DJA, Sindelar RD, Meibohm B, editors. Pharmaceutical biotechnology: fundamentals and applications. 5th ed. Springer: New York; 2019. p. 105-37.
11. Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1-110.
12. Thomas VA, Balthasar JP. Understanding inter-individual variability in monoclonal antibody disposition. Antibodies (Basel). 2019;8(4):56.
13. Zhao L, Ren TH, Wang DD. Clinical pharmacology considerations in biologics development. Acta Pharmacol Sin. 2012;33(11):1339-47.
14. Matsushima S, Huang Y, Suzuki H, Nishino J, Lloyd P. Ethnic sensitivity assessment – pharmacokinetic comparability between Japanese and non-Japanese healthy subjects on selected mAbs. Expert Opin Drug Metab Toxicol. 2015;11(2):179-91.
15. U.S. Food and Drug Administration. Department of Health and Human Services. Scientific considerations in demonstrating biosimilarity to a reference product. 2015 [homepage on the Internet]. [cited 2023 Jul 26]. Available from: https://www.fda.gov/media/82647/download
16. Gibiansky L, Sutjandra L, Doshi S, Zheng J, Sohn W, Peterson MC, et al. Population pharmacokinetic analysis of denosumab in patients with bone metastases from solid tumours. Clin Pharmacokinet. 2012;51(4):247-60.
17. Struemper H, Chen C, Cai W. Population pharmacokinetics of belimumab following intravenous administration in patients with systemic lupus erythematosus. J Clin Pharmacol. 2013;53(7):711-20.
18. Ma P, Yang BB, Wang YM, Peterson M, Narayanan A, Sutjandra L, et al. Population pharmacokinetic analysis of panitumumab in patients with advanced solid tumors. J Clin Pharmacol. 2009;49(10):1142-56.
19. Shang J, Huang L, Huang J, Ren X, Liu Y, Feng Y. Population pharmacokinetic models of anti-PD-1 mAbs in patients with multiple tumor types: a systematic review. Front Immunol. 2022;13:871372.
20. Wu JY, Wu XN, Ding L, Zhao YB, Ai B, Li Y, et al. Phase I safety and pharmacokinetic study of bevacizumab in Chinese patients with advanced cancer. Clin Med J (Engl). 2010; 123(7):901-6.
21. Zhu Y, Wang Q, Frederick B, Bouman-Thio E, Marini JC, Keen M, et al. Comparison of the pharmacokinetics of subcutaneous ustekinumab between Chinese and non-Chinese healthy male subjects across two Phase 1 studies. Clin Drug Investig. 2013;33(4):291-301.
22. Zhou H, Tsukamoto Y, Davis HM. Should clinical pharmacokinetic bridging studies between Caucasian and Asian populations be required for approval of monoclonal antibodies? J Clin Pharmacol. 2012;52(8):1273-6.
23. Chiba K, Yoshitsugu H, Kyosaka Y, Lida S, Yoneyama K, Tanigawa T, et al. A comprehensive review of the pharmacokinetics of approved therapeutic monoclonal antibodies in Japan: are Japanese phase I studies still needed? J Clin Pharmacol. 2014;54(5):483-94.
24. Rajman I, Hirano M, Honma W, Zhao S. New paradigm for expediting drug development in Asia. Drug Discov Today. 2020;25(3):491-6.
25. Jeon I, Kim YK, Song I, Yoon DY, Huh KY, Jin X, et al. The necessary conduct: exploratory multiregional clinical trials in East Asia. Clin Transl Sci. 2021;14(6):2399-407.
26. Kim J, Lee JY, Kim HG, Kwak MW, Kang TH. Fc receptor variants and disease: a crucial factor to consider in the antibody therapeutics in clinic. Int JMol Sci. 2021;22(17):9489.
27. Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol. 2009:157(2):244-54.
28. Kaifu T, Nakamura A. Polymorphisms of immunoglobulin receptors and the effects on clinical outcome in cancer immunotherapy and other immune diseases: a general review. Int Immunol. 2017;29(7):319-25.
29. Mager DE, Krzyzanski W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res. 2005;22(10):
1589-96.
30. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576-88.
31. Bloomfield D, D’Andrea E, Nagar S, Kesselheim A. Characteristics of clinical trials evaluating biosimilars in the treatment of cancer: a systematic review and meta-analysis. JAMA Oncol. 2022;8(4):537-45.
32. Yang J, Yu S, Yang Z, Yan Y, Chen Y, Zeng H, et al. Efficacy and safety of anti-cancer biosimilars compared to reference biologics in oncology: a systematic review and meta-analysis of randomized controlled trials. BioDrugs. 2019;33(4):357-71.
|
Author for correspondence: Sandeep N Athalye, MBBS, MD, Chief Medical Offi cer, Biocon Biologics Ltd, Biocon House, Tower 3, Semicon Park, Plot No. 29-P1 & 31-P, KIADB Industrial Area, Electronic City Phase – 2, Hosur Road, Bengaluru 560100, Karnataka, India |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2023 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


