Global challenges in the manufacture, regulation and international harmonization of GMP and quality standards for biopharmaceuticals
Published on 2020/05/26
Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):52-63.
Author byline as per print journal: Adjunct Associate Professor Sia Chong Hock, BSc (Pharm), MSc; Associate Professor Sia Ming Kian, BSc (Pharm) (Hons); Chan Lai Wah, BSc (Pharm) (Hons), PhD
|
Abstract: |
Submitted: 6 May 2020; Revised: 26 May 2020; Accepted: 26 May 2020; Published online first: 8 June 2020
Introduction
Biopharmaceuticals belong to a class of medicinal products whose active pharmaceutical ingredient (API) is manufactured using living systems such as microbial, mammalian, insect, plant or animal cells. According to the Pharmaceutical Inspection Co-operation Scheme (PIC/S), a medicinal product is defined as any medicine or similar product intended for human use, which is subject to control under health legislation [1]. The API in the medicinal product is responsible for furnishing a pharmacological or other direct effect in the diagnosis, cure, mitigation, treatment or prevention of the disease, or alteration of the structure or function of the body [2].
Depending on the regulatory authorities (RAs), biopharmaceuticals may be termed as ‘biologics’, ‘biological medicines’ or ‘biotherapeutics’ [3–5]. Biotechnological methods such as ex vivo expansion [6–8], recombinant deoxyribonucleic acid (rDNA) or hybridoma technologies are typically employed to produce biopharmaceuticals. Examples of biopharmaceuticals are vaccines, insulins, monoclonal antibodies (mAbs) and other therapeutic proteins [9]. Cell-based, tissue-based or gene-based therapeutic products, also known as advanced therapy medicinal products (ATMPs) in the European Union (EU), are also considered biopharmaceuticals [10]. Biosimilars, also termed ‘subsequent-entry biologics’ and ‘similar biotherapeutic products’, are biopharmaceuticals which are highly similar in terms of safety, efficacy and quality with the innovator biopharmaceutical [11]. Such similarities are demonstrated using comparability studies with the reference product, which is the innovator biopharmaceutical that has received market authorization by the relevant RAs [12].
Frost & Sullivan has estimated that US$16.83 billion worth of biopharmaceuticals in the global market would lose their patent from 2015 to 2025. The global biosimilar market is expected to grow at a compound annual growth rate of 31.5% and reach US$66.33 billion during the same period [13]. Given the highly promising outlook of the biopharmaceutical market, manufacturers are motivated to invest in the manufacturing of biopharmaceuticals. However, there are major differences between biopharmaceuticals and the conventional chemical-based pharmaceuticals which may necessitate the use of different types of manufacturing facilities and standards.
Biopharmaceuticals consist of API molecules with a highly complex structure and very high molecular mass [14], ranging from thousands to hundred thousands of Daltons. In comparison, conventional chemical-based pharmaceuticals consist of API molecules that are significantly smaller, and possess a much simpler molecular structure, see Figure 1. As such, the characterization of biopharmaceuticals may require different analytical tools and methods such as post-translational modification characterization for recombinant therapeutic proteins [15], viral vector sequence analysis for gene-based therapeutic products [16] and reverse transcriptase polymerase chain reaction (RT-PCR) for stem cell therapies [17].
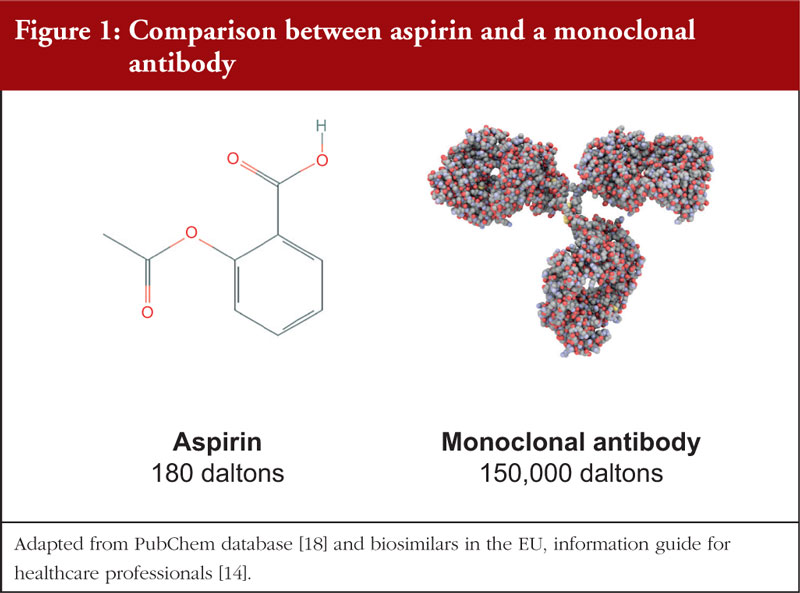
Biopharmaceuticals commonly exist as injectables [19]. This is because of the large molecular weight which hinders penetration of the molecule through the intestinal epithelium, thereby reducing systemic absorption [20]. In addition, most biopharmaceuticals are highly susceptible to degradation by the extreme pH conditions in the alimentary canal [21]. Thus, injectables remain the only viable option as they allow the molecules to bypass these obstacles. In comparison, chemical-based pharmaceuticals exist in a variety of dosage forms such as tablets, injections, nasal sprays and topical products.
Unlike conventional pharmaceuticals whose quality can be consistently assured, there exists an inherent variability in the quality of biopharmaceuticals which is largely due to their sensitivity to various conditions such as temperature, pH and mechanical stress [22]. Exposure to these factors can easily affect the quality, safety and efficacy of the end product. Therefore, monitoring these conditions is crucial to ensure that these conditions vary within appropriate specified limits. Clearly, significant challenges are encountered in the manufacture of biopharmaceuticals, and it is vital to adopt relevant GMP guidelines. According to the PIC/S, GMP ensures that ‘products are consistently produced and controlled to the quality standards appropriate for their intended use and as required by the marketing authorisation or product specification’ [23]. This perspective is also shared by the various RAs and World Health Organization (WHO) [24–26].
The increasingly globalised nature of commerce allows manufacturers to outsource activities such as procurement of raw materials overseas, where different regulatory requirements may exist. Thus, there is a need to ensure that the GMP guidelines adopted by the RAs and international organizations (IOs) are harmonized and robust. A robust set of GMP guidelines helps to safeguard public health by assuring the quality, safety and efficacy of the biopharmaceuticals [26]. To date, review on the biopharmaceutical regulatory framework has been done on western countries, such as Canada and the US, as well as some Asian countries, such as Japan and Korea [27, 28]. However, few studies have been done on the regulatory framework for biopharmaceuticals in the Association of South East Asian Nations (ASEAN), with the exception of Singapore and Malaysia [27, 29].
ASEAN provides numerous incentives to biopharmaceutical manufacturers. The low manufacturing cost in some ASEAN Member States (AMS) enables greater cost-savings in the manufacture of biosimilars [30]. In addition, ASEAN is experiencing a general epidemiological shift from communicable to non-communicable chronic diseases [31], and biopharmaceuticals play an increasing role in managing the latter. With a combined population of 600 million, the ASEAN market will provide a sizeable patient population that attracts the importation and manufacturing of biopharmaceuticals in the region [32]. Thus, there is a need to ensure that the GMP guidelines adopted are adequate in assuring the quality of biopharmaceuticals.
Therefore, the aims of this project are firstly, to understand the challenges in the manufacture of biopharmaceuticals, excluding those derived from transgenic plants and animals due to their relatively inefficient commercial scalability [33, 34]. Secondly, this project aims to analyse the GMP standards of various RAs and IOs and determine if the regulatory frameworks adopted are suitable in addressing the challenges of biopharmaceuticals. Lastly, biopharmaceuticals also present unique regulatory challenges which will be discussed in later sections. Where necessary, solutions will be proposed to promote greater harmonization of GMP standards, with the ultimate goal of improving patient safety through better regulatory capacity.
Manufacture of biopharmaceuticals – an overview
Figure 2 shows the general processes involved in biopharmaceutical manufacturing. The processes involved are generally similar and are divided into two main stages – upstream and downstream processing. The upstream processes are briefly described in 2.1 to 2.3 while the downstream processes and formulation are described in 2.4 and 2.5, respectively. For all processes, controls on process variability and contamination should be highly prioritized and their risk mitigated with appropriate strategies [35].
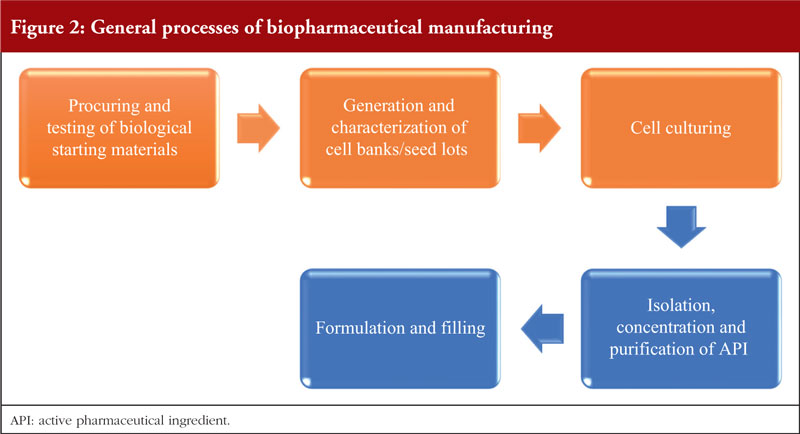
Procurement and testing of biological starting materials
Starting materials used in the production of biopharmaceuticals include culture media, buffers and expression systems such as microbial or mammalian cells, and exclude packaging materials [36, 37]. The source, origin and suitability of starting materials should be clearly defined [1]. Western blotting, capillary electrophoresis and high-performance liquid chromatography (HPLC) are common analytical tools employed to assess the identity and purity of starting materials [38]. In addition, adequate controls, such as qualification of supplier through audits, screening for adventitious agents and viral reduction strategies should be in place to assure end product safety [39]. Where the starting materials are of human or animal origin, appropriate documentation on characteristics, such as general donor health status and age [40], should be demonstrated and meet relevant national legislation [1]. This requirement is especially relevant to ATMPs such as Chimeric Antigen Receptor (CAR) T cells, where the T cells are isolated from donors via apheresis [41].
Generation and characterization of cell banks/seed lots
The development of cell banks and seed lots begin with the construction of the vector and recombinant gene. Bacterial plasmids and cells are common choices for vector construction, and bacterial gene is manipulated using enzymes such as nucleases to insert the recombinant gene. Gene delivery into the host cells is achieved via transfection with replication-defective viruses, physical or chemical means. The choice of host cells is dependent on the type of biopharmaceuticals. In general, Chinese hamster ovary (CHO) cells dominate the manufacturing of mAbs [42], microbial cells such as Escherichia coli (E. coli) are commonly applied for simpler recombinant proteins that do not require post-translational modifications [43] and human embryonic kidney 293 (HEK-293) cells are commonly used to generate viral vectors [44]. The appropriate cell lines or seed lots are selected to establish the master bank of cell lines or seed lots. Extensive characterization of the master bank is crucial as it will be used to generate the working cells or seed lots. Titre amount, growth robustness, phenotypic and genetic stability are key considerations when selecting the master bank [45]. Cryopreservation is an essential strategy for prolonged storage of cell banks and seed lots [46].
Cell culturing
This process is responsible for producing the API. It is either done in a fed-batch or continuous manner, with fed-batch being more widely employed [47]. For tissue-based ATMPs, additional considerations should be given to the scaffolds where the cells will be seeded on. These scaffolds should not be immunogenic, and because they are derived from animal or human sources, measures to prevent contamination and disease transmission are crucial [48]. In fed-batch, the culture is expanded via sequential scaling up using bioreactors of increasing volumes, up to the maximum cell density. The cell culture is terminated before the death phase and the culture medium is harvested.
In comparison, continuous cell culture begins with the scaling up of cell culture to an optimum cell density. The culture medium is continuously harvested while fresh medium is added at the same rate to maintain the cell density, which should theoretically produce API of more consistent quality [49]. Continuous cell culture typically has a smaller footprint requirement than fed-batch cell culture due to the smaller bioreactors used [50]. However, it is generally more complex and costly to operate and to validate continuous fermentation [51, 52].
Isolation, concentration and purification of API
Downstream processing is commonly done batchwise [53] and entails the recovery, intermediate purification and polishing (RIPP) stages. The general guidelines governing the design of downstream processing are outlined in Table 1 [54].

During the recovery stage, the API is separated from the harvested culture medium and subsequently concentrated. The localization of API is a crucial influence of the purification stage. For intracellular API, which is typically produced by microbial cells, cell lysis is essential for releasing the API. As such, further purification from cellular debris is necessary. In comparison, API produced by mammalian cells is typically secreted extracellularly, hence direct purification can be employed. Besides removal of impurities, viral inactivation or removal are generally necessary [55]. However, the latter is not appropriate for gene therapy API as it can damage the viral vectors [56]. Any remaining impurities are removed in the polishing stage. Table 2 lists the common methods used in each stage [57]. Where procedures that reduce bioburden cannot be applied, aseptic methods should be used [58].

Formulation and filling
At the final stage, the API is combined with excipients such as buffers, salts and preservatives to prevent product degradation or contamination [59]. In addition, biopharmaceuticals are commonly formulated as freeze-dried powders, if immediate use is not required, due to their limited stability in liquid form [60]. Furthermore, considerations must be given to the packaging materials used. In general, the packaging material should not interact with the API in a manner that jeopardises the quality, such as leaching of materials into the product or structural alteration due to adsorption of API onto the packaging material [61].
Challenges concerning manufacture of biopharmaceuticals
Extensive process and product understanding required
As the quality of biopharmaceuticals is influenced by the processing steps [62], the latter must be designed such that the critical quality attributes (CQAs) of biopharmaceuticals remain within specifications [63]. CQAs are ‘analytical measures’ associated with the quality, safety and efficacy of a biopharmaceutical, such as absence of contaminants [64]. Inappropriate processing steps can adversely impact the quality. For instance, most recombinant glycoproteins except mAbs are prone to aggregation and dimerization in prolonged residence time hence fed-batch fermentation is inappropriate for these proteins [65]. In addition, any changes to the processes or formulation must be validated to assure that these changes do not significantly jeopardise product quality. This is exemplified by the infamous pure red cell aplasia (PRCA) incident associated with Eprex® (epoetin alfa), where the insufficiently validated formulation changes are associated with a surge in PRCA incidence amongst Eprex®-treated patients [66]. Hence, an extensive knowledge on the CQAs of biopharmaceuticals, together with appropriate validation, is crucial in assuring product quality.
Inherent variability of host cells
The inherent variability of the host cells can have unpredictable effects on the quality of biopharmaceuticals. This is exemplified by the widely employed CHO cells, whose genomic plasticity allows gene manipulation to produce the desired cell lines [67]. However, this has also contributed to cell line instability such as gene silencing [68]. In addition, the requirement for cell lines to produce high titre amount places considerable metabolic stress on the host cells, resulting in spontaneous recombinant gene deletion that may be difficult to predict [69, 70]. These factors will present obstacles in ensuring consistent product quality.
Downstream processing remains a key bottleneck
Downstream processing is commonly considered to be the key bottleneck of biopharmaceutical manufacturing, with chromatography being the most commonly cited [71]. Chromatographic separation is based on the degree of association between the individual components of the culture content and the stationary columns, and the separation efficiency can be modified by altering conditions such as ionic strength, pH and polarity. The designing of a chromatographic purification process has proven challenging owing to a lack of standardization arising from the myriad of chromatography modes and equipment to consider [72]. Thus, the designing process has traditionally taken a trial-and-error approach, which can be wasteful and time consuming [72].
Review of current GMP frameworks for biopharmaceuticals
Table 3 shows a comparison of GMP principles and guidance documents adopted by selected RAs and IOs. They are chosen because most of them are key players in regulatory harmonization or biopharmaceutical manufacturing [27, 29, 73]. In general, IOs and majority of the RAs adopt similar GMP principles. They emphasize on the implementation of quality risk management (QRM) principles: (1) risk evaluation should be scientifically sound and relevant to protection of patient; and (2) the amount of resources used for risk management should be proportional to the risk level [74]. QRM also facilitates better management of the manufacturing process by identifying and prioritizing the control on critical process parameters (CPPs) [75], as their variability can impact the CQAs and consequently the product quality [76]. Most guidelines acknowledge the inherent variability of biopharmaceutical quality and recommend using in-process controls and improving the robustness of manufacturing process to control the variability [1, 37, 77]. Table 3 also shows that most RAs and IOs adopt relatively similar GMP standards for API, suggesting a significant level of harmonization is already in place. However, there are major differences in the scope of the GMP standards. For instance, PIC/S provides guidance on all types of biopharmaceuticals within Annex 2 of its GMP guide, while the European Medicines Agency (EMA) provides recommendations for ATMPs in a dedicated guidance (Eudralex, Volume 4, Part IV) [78]. However, it is noted that PIC/S is currently drafting a dedicated GMP guide for ATMPs which may be implemented in the future [79]. In addition, PIC/S provides further guidance for selected types of biopharmaceuticals in Annex 2 Part B of its GMP guide [1] while WHO does not [37].
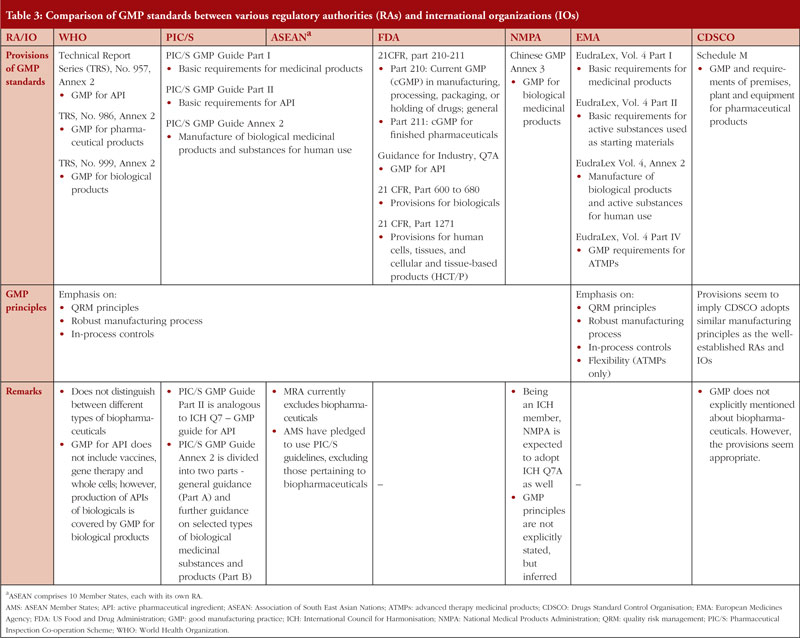
Furthermore, the National Medical Products Administration (NMPA) of China has a GMP guideline for API that is not entirely relevant to biopharmaceutical APIs. This guideline provides recommendations on API produced by classical fermentation, which typically do not employ biotechnological processes and requires less stringent control on the manufacturing processes [80]. In addition, the APIs produced by classical fermentation, such as antibiotics, amino acids and vitamins, are generally of low molecular weights [77]. Instead, GMP pertaining to biopharmaceuticals and their APIs are covered under the Chinese GMP Annex 3 only. NMPA, being a regulatory member of the International Council for Harmonisation (ICH), is expected to implement ICH Q7 guideline – GMP Guide for API [81]. Hence, NMPA’s GMP guideline on API is likely to be harmonized with international standards. However, since NMPA is not expected to implement ICH Q5 guideline – Quality of Biotechnological Products, it is difficult to ascertain whether NMPA’s GMP standards on finished biopharmaceuticals are harmonized with international standards.
The Central Drugs Standard Control Organisation (CDSCO) of India, for instance, does not explicitly mention about the inclusion of biopharmaceuticals within the scope of its GMP guideline (Schedule M) [82]. However, the provisions appear to be adequate for biopharmaceuticals and also suggest that CDSCO adopts similar GMP principles as the well-established RAs and IOs. There is an additional guideline document for biopharmaceuticals on the CDSCO website, but it was inaccessible at the time of writing this review. It is worth mentioning that both China and India are currently undergoing regulatory reforms and have expressed interest to join PIC/S [83]. There are also reports that Chinese and Indian manufacturers are improving their product quality to meet international standards [84, 85], signalling their strong commitment to GMP.
Within ASEAN, the biopharmaceutical industry is at a nascent stage. Vaccines are the main biopharmaceuticals manufactured due to the high prevalence of infectious diseases [86]. In addition, there have been reports of vaccine shortages in ASEAN which may necessitate prioritizing vaccines over other biopharmaceuticals [87]. The review of ASEAN GMP standards reveals that the majority of AMS adopt PIC/S GMP recommendations for biopharmaceuticals [88–91]. The lack of unified adoption can be attributed to the current exclusion of biopharmaceuticals from the scope of the ASEAN Mutual Recognition Arrangement (MRA) [92]. In addition, some AMS are emphasizing on generic pharmaceutical manufacturing [93, 94] and medical devices [95], which may also contribute to the lack of GMP guidelines for biopharmaceuticals. However, efforts have been made, such as the recent agreement on the ASEAN common technical requirements of biological products [96], to include biopharmaceuticals for harmonization in the future [97].
Overall, the differences in the scope of GMP standards observed is not surprising in view of the diversity of biopharmaceuticals being manufactured, and that GMP guidance is contextualised to the respective countries. It is however heartening to know that most RAs and IOs share similar GMP principles for regulating biopharmaceuticals. With concerted efforts, the outlook on harmonization is bright. Nonetheless, it must be emphasized that the adequacy of GMP and quality standards adopted by the various RAs and IOs, will ultimately depend on the extent of compliance by the biopharmaceutical manufacturers, and the robustness of enforcement of the standards by the RAs and IOs.
Challenges in the regulation of biopharmaceuticals
Resource-intensive evaluation of biosimilarity
The standard approach for approving generic conventional chemical-based pharmaceuticals, or generics, is not appropriate for biosimilars. For the approval of generics, manufacturers only need to demonstrate that the generics have identical molecular structure and is bioequivalent to the reference product [98]. However, the inherent variability of biopharmaceuticals makes it impossible for biosimilars to exactly replicate the reference product. Manufacturers may have to modify the manufacturing process based on the reference product with appropriate optimization such that the CQAs of biosimilars are highly similar to that of the reference product [99]. There will be differences, albeit slight, in the processing that can affect the end product of biological nature. Hence, a ‘totality-of-the-evidence’ approach is used to evaluate biosimilarity. This approach considers the entirety of the information submitted in the biosimilar application, such as data from analytical, preclinical, clinical studies and lot-to-lot variabilities, to evaluate the biosimilarity to the reference product [100]. The approval of biosimilars places more emphasis on extensive characterization of the API [101], with supplementary data from animal studies, clinical pharmacology or clinical trials to rule out any residual uncertainty from the characterization process [102]. Compared to chemical-based pharmaceuticals, the evaluation process clearly demands more time and expertise for the RAs. In addition, doubts have been cast on the suitability of the guidance for biosimilarity evaluation in assessing more complex biopharmaceuticals such as ATMPs [103]. Such uncertainty thus raises the need for more harmonization between different regulatory perspectives.
Differing perspectives on interchangeability
Differences exist between FDA and EMA perspectives on interchangeability. For FDA, biopharmaceuticals that are highly similar to the reference product can be classified as biosimilar product or interchangeable product. For the product to be classified as interchangeable, additional data on the safety and efficacy of switching from the reference product must be provided [104]. Once an interchangeable product is approved, the reference product may be substituted with the interchangeable product by the pharmacist without consulting the prescriber. In comparison, EMA does not require additional studies to determine if a biosimilar is interchangeable. However, EMA distinguishes the act of interchanging between reference product and biosimilar, or between biosimilars, into switching and substitution: switching is done at the prescriber level while substitution is done at the pharmacy level [14]. Differences in definition can lead to unnecessary confusion when manufacturers want their products approved for use in different countries. While the requirement for a switching study can provide better safety assurance of the interchangeable product, this also increases the production cost and possibly negate any cost savings it has over the reference product. This may also explain why there has been no interchangeable products approved by FDA currently [105, 106]. In addition, the vast clinical experience of EMA in approving biosimilars has demonstrated that biosimilars have similar efficacy and safety profiles as their reference product [107]. This is also supported by a systematic review which did not show any safety or efficacy risk from switching between reference products and biosimilars [108]. Thus, the requirement by FDA for a switching study to demonstrate interchangeability is debatable.
Growing number of data integrity lapses
‘Data integrity is the degree to which a collection of data is complete, consistent and accurate throughout the data life cycle. The collected data should be attributable, legible, contemporaneously recorded, original or a true copy, and accurate (ALCOA). Assuring data integrity requires appropriate quality and risk management systems, including adherence to sound scientific principles and good documentation practices’ [109, 110]. FDA has noted an increasing number of GMP violations pertaining to data integrity in recent years [111]. Compromised data integrity can lead to missing and inaccurate information that are vital considerations in the regulatory approval for market authorization [112], as well as jeopardising product quality assurance [113]. Lapses can be due to unintentional errors such as lack of awareness as well as inadequate standard operating procedures (SOPs) [114]. In more serious cases, deliberate data manipulations, such as data falsification instructed by upper management, have been reported [115, 116]. A review of the warning letters issued by the Center for Biologics Evaluation and Research (CBER) reveals that data manipulation can occur despite the implementation of legislative guidelines, SOPs and controls [117–119], hinting a possible lack of a quality-focused culture within these organizations.
Proposed solutions to challenges of biopharmaceuticals
Optimizing biopharmaceutical manufacturing with Industry 4.0
Industry 4.0, or the Fourth Industrial Revolution, is a broad concept that involves the amalgamation of physical and digital technologies to generate a constant flow of information, allowing real-time data access [120]. These data can then be applied to generate analytical tools such as algorithms and models to allow better process and product understanding [121]. Consequently, this allows more effective implementation of Quality-by-Design (QbD) approach in process development and optimization [122]. According to ICH, QbD is a ‘systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management’ [76]. With QbD, process capability is improved with better product and process understanding, which in turn reduces the inherent variation in quality of biopharmaceuticals [123].
Real-time data access can be achieved with process analytical technologies (PAT). FDA considers PAT as ‘a system for designing, analysing, and controlling manufacturing through timely measurements, i.e. during processing, of critical quality and performance attributes of raw and in-process materials and processes, with the goal of ensuring final product quality’ [124]. The general methodology of PAT begins with the collection of data using robust, rapid and sensitive analytical tools and sensors, such as HPLC, dynamic light scattering, pressure gauge and flow meter [125]. This is followed by the modelling of these data to generate useful process-related information and ends with the goal of using the generated data to influence the manufacturing processes [126]. For instance, PAT has been used to optimize downstream processing. This is achieved by combining the screening of previously validated chromatographic conditions with scientifically sound experiments to generate a chromatographic model that is able to predict critical process parameters for downstream optimization [127, 128]. In addition, deviations captured during routine monitoring can also be used to generate algorithms that can better categorize human errors or facilitate more effective corrective actions and preventive actions (CAPAs), thereby reducing the occurrence of failed batches as well as the cost of implementing CAPAs [129].
Industry 4.0 has allowed greater interconnectivity through platforms such as the Internet of Things (IoT), providing worldwide access to data to facilitate better process understanding. For instance, a better understanding of the CHO cell genome is achieved by pooling data from various assemblies generated by the other researchers using sequencing technologies such as short-read Illumina and single molecule real time (SMRT) sequencing [130]. With a better understanding of the genome, manufacturers may manipulate the gene more effectively and improve host-cell stability, consequently leading to more consistent product quality.
Enhanced harmonization efforts on biosimilar guidelines
With the expected influx of biosimilars due to patent expiry of the reference product, there is a need for RAs to develop guidelines that facilitate the clinical decision to choose between the reference product and biosimilars, switching between reference product and biosimilars, or switching between biosimilars as a potential therapeutic option. Although EMA does not provide recommendations on interchangeability and leave the development of substitution policies to its Member States [131], countries such as Germany, The Netherland and Scotland have endorsed the interchangeability of biosimilars [132]. In general, these countries recommend that the decision of switching should be based on shared decision-making between the patient and prescriber on the potential risks of switching, along with appropriate monitoring for early detection of adverse event [14, 133–135]. Such perspective is logically sound as it ensures that any clinical decision made is in the patient’s best interest. It is worthwhile to encourage RAs of these countries to share their regulatory experience so that other RAs can make a more informed choice when developing guidelines relating to the use of biosimilars. Such concerted efforts will promote harmonization of guidelines.
Enhancing data integrity with a culture of quality (quality culture)
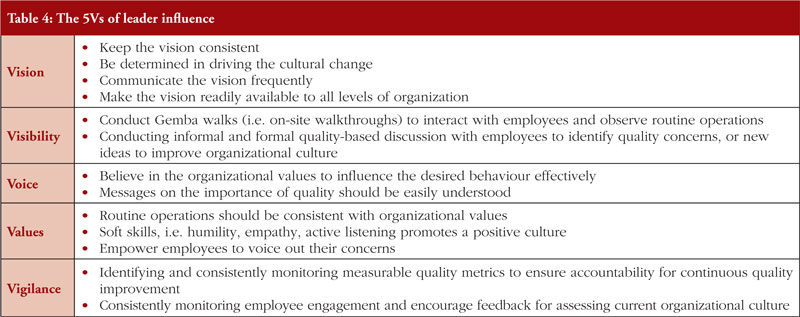
Without a culture of quality, even the simplest and preventable data integrity-related violations can occur [136]. This is because the organizational culture directly impacts routine operations which have a downstream influence on data and product quality, and senior management is responsible for creating a culture of quality [137]. A critical element of quality culture is the ‘transparent and open reporting’ of data integrity-related violations at all organizational levels [109]. Measures such as an independent reporting channel, anonymous or identifiable, or rewarding employees who report quality-related issues can help to incentivise employees to voice out their concerns [138]. A culture of quality can be created by first incorporating the ‘Leader 5Vs’ that correlate with a positive influence on quality culture [139], which are further explained in Table 4 [140]. In essence, the table emphasizes on the importance of senior management in creating a vision, leading by example, empowering their employees towards quality excellence. Senior management is encouraged to look at WHO guidance on data integrity as it provides practical recommendations on building a quality culture [138].
In facilitating a behavioural change, employers may consider the ABC (antecedent, behaviour, consequence) model: where an antecedent encourages a behaviour and leads to a consequence, which in turn influences the recurrence of behaviours [141], see Figure 3. While antecedents are essential in triggering a behaviour, it is the consequence that significantly motivates or demotivates the latter [142]. As such, in the implementation measures to effectively correct a behaviour, consequences should be emphasized over antecedents. In addition, a ratio of positive to negative consequences at 4:1 is recommended to sustain performance outcomes [141].
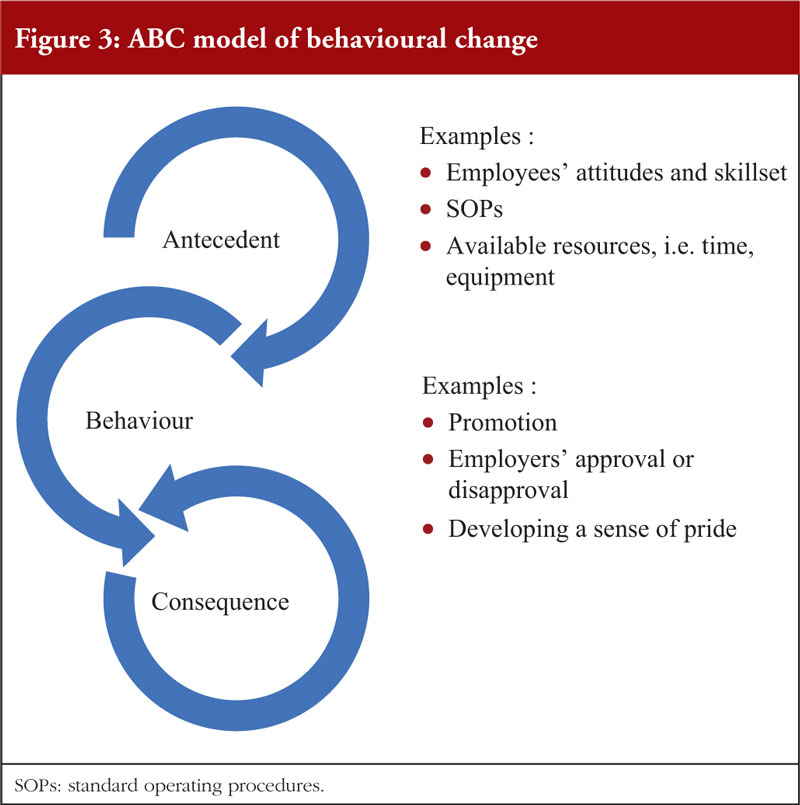
Employees are also crucial in transforming the organizational culture [143]. Training for employees should help them understand the organization’s quality objectives, SOPs and their individual role in achieving said objectives [144]. In addition, they should leverage on the ‘speak up’ culture to provide feedback on how the senior management can customise the quality culture messages to be more relevant to their work [145].
Developing a culture of quality excellence is not an instantaneous process as it requires a change of mindsets: senior management must drive the change while employees must be motivated to change. An effective collaboration at all organizational levels will ensure that the change can be expedited, and the culture remains sustainable in the long term.

Conclusion
With the patent expiry of innovator biopharmaceuticals, more biosimilars will be developed for use. In general, this paper has shown that most biopharmaceuticals share similar manufacturing processes and considerations, providing useful insights for manufacturers who are interested to include biosimilars in their pipeline. However, it is still highly advisable for manufacturers to demonstrate an extensive product and process understanding as there may be certain methods that are not suitable or relevant for their product. Due to their inherent complexity, biopharmaceuticals present challenges in assuring product quality. This can be addressed with real-time monitoring and better predictive modelling, as well as other solutions that are not discussed in this paper.
For the RAs and IOs, the outlook on GMP harmonization for biopharmaceuticals is highly promising. As countries improve and harmonize their GMP standards, there will be a greater assurance of quality and safety of biopharmaceuticals. However, more effort is needed in providing guidelines on the interchangeability of biosimilars to encourage their use. With greater collaboration among RAs and IOs, practical experience can be shared, and this can facilitate improvement of existing guidelines. The challenges presented by biopharmaceuticals, although daunting, are not insurmountable. With technological advances and better collaboration between key stakeholders, these challenges can be effectively managed.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Adjunct Associate Professor Sia Chong Hock, BSc (Pharm), MSc
Sia Ming Kian, BSc (Pharm) (Hons)
Associate Professor Chan Lai Wah, BSc (Pharm) (Hons), PhD
Department of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543
References
1. Pharmaceutical Inspection Co-operation Scheme. PIC/S GMP Guide (Related annexes). 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.picscheme.org/layout/document.php?id=1407
2. Pharmaceutical Inspection Co-operation Scheme. PIC/S GMP Guide (Part II: Basic requirements for active pharmaceutical ingredients). 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.picscheme.org/layout/document.php?id=1409
3. U.S. Food and Drug Administration. Resources for you (Biologics). 2019 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/vaccines-blood-biologics/resources-you-biologics.
4. European Medicines Agency. Biological medicine [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/glossary/biological-medicine
5. World Health Organization. Biotherapeutic products [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.who.int/biologicals/biotherapeutics/biotherapeutic-products/en/
6. European Medicines Agency. First stem-cell therapy recommended for approval in EU. 2014 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/news/first-stem-cell-therapy-recommended-approval-eu
7. Pellegrini G, Ardigò D, Milazzo G, Iotti G, Guatelli P, Pelosi D, et al. Navigating market authorization: the path Holoclar took to become the first stem cell product approved in the European Union. Stem Cells Transl Med. 2018;7(1):146-54.
8. Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;5(3):1-7.
9. Center for Biologics Evaluation and Research. What are “Biologics” questions and answers. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/about-fda/about-center-biologics-evaluation-and-research-cber/what-are-biologics-questions-and-answers
10. European Medicines Agency. Advanced therapy medicincal products: overview [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview
11. World Health Organization. WHO questions and answers: similar biotherapeutic products. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.who.int/biologicals/expert_committee/QA_for_SBPs_ECBS_2018.pdf?ua=1
12. European Medicines Agency. Biosimilar medicines: overview [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/biosimilar-medicines-overview
13. Frost & Sullivan. APAC biosimilar market, forecast 2025. [homepage on the Internet]. [cited 2020 May 26]. Available from: https://go.frost.com/APAC_PR_MTan_P9A3_Feb18
14. European Medicines Agency. Biosimilars in the EU. Information guide for healthcare professionals: 2017 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
15. Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11(7):527-40.
16. Husain SR, Han J, Au P, Shannon K, Puri RK. Gene therapy for cancer: regulatory considerations for approval. Cancer Gene Ther. 2015;22(12):554-63.
17. U.S. Food and Drug Administration. Bauer S. Development of strategies to improve cell therapy product characterization. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/development-strategies-improve-cell-therapy-product-characterization
18. National Library of Medicine. Aspirin, CID=2244 PubChem Database. 2004 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Aspirin
19. Grenha A. Systemic delivery of biopharmaceuticals: parenteral forever? Pharm Bioallied Sci. 2012;4(2):95.
20. Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11(19):905-10.
21. Vela Ramirez JE, Sharpe LA, Peppas NA. Current state and challenges in developing oral vaccines. Adv Drug Deliv Rev. 2017;114:116-31.
22. Krause ME, Sahin E. Chemical and physical instabilities in manufacturing and storage of therapeutic proteins. Curr Opin Biotechnol. 2019;60:159-67.
23. Pharmaceutical Inspection Co-operation Scheme. PIC/S Brochure. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://picscheme.org/docview/2146
24. European Medicines Agency. Good manufacturing practice [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-manufacturing-practice
25. World Health Organization. Good manufacturing practices [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.who.int/biologicals/vaccines/good_manufacturing_practice/en/
26. U.S. Food and Drug Administration. Facts about the Current Good Manufacturing Practices (CGMPs). 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/drugs/pharmaceutical-quality-resources/facts-about-current-good-manufacturing-practices-cgmps
27. Esteban E, Bustos RH, García JC, Jáuregui E. Biosimilars: an approach to some current worldwide regulation frameworks. Curr Clin Pharmacol. 2019;14(1):16-40.
28. Bas TG, Carolina Oliu C. Biosimilars in developed and developing East and Southeast Asian countries: Japan, South Korea, and Malaysia-overview, evolution, and regulations assessment. Biomed Res Int. 2016:2016:5910403.
29. Tsai W-C. Update on biosimilars in Asia. Curr Rheumatol Rep. 2017;19(8):47.
30. Tonby O, Ng J, Mancini M. Understanding ASEAN: the manufacturing opportunity. McKinsey&Company.
31. Vervisch R. The generics in Southeast Asia: market analysis and their role in shaping the future of biosimilars [thesis]. Université libre de Bruxelles; 2017.
32. Hock SC, Tribe R, Wah CL. ASEAN harmonization on GMP inspection and training of inspectors. Pharmaceut Eng. 2013;33(1):56-62.
33. Monzani PS, Adona PR, Ohashi OM, Meirelles FV, Wheeler MB. Transgenic bovine as bioreactors: challenges and perspectives. Bioengineered. 2016;7(3):123-31
34. Schillberg S, Raven N, Spiegel H, Rasche S, Buntru M. Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front Plant Sci. 2019;10:720-. Epub June 11, 2019. doi: 10.3389/fpls.2019.00720.
35. Jagschies G, Łącki KM. Chapter 4 – Process capability requirements. In: Jagschies G, Lindskog E, Łącki K, Galliher P, editors. Biopharmaceutical processing: Elsevier; 2018. p. 73-94.
36. European Commission. EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, glossary. 2004 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/pdfs-en/glos4en200408_en.pdf
37. World Health Organization. Annex 2. WHO good manufacturing practices for biological products. 2016 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.who.int/biologicals/areas/vaccines/Annex_2_WHO_Good_manufacturing_practices_for_biological_products.pdf?ua=1
38. Challener CA. Ensuring the safety, quality, and identity of biopharmaceutical raw materials. BioPharm Int. 2014;27(8):28-9,44.
39. Chen D. Safety assurance for biologics manufactured in mammalian cell cultures: a multitiered strategy. In: Zhou W, Kantardjieff A, editors. Mammalian cell cultures for biologics manufacturing. 139. Berlin, Heidelberg: Springer; 2014. p. 167-83.
40. International Conference on Harmonisation of Technical requirements for registration of pharmaceeuticals for human use. Derivation and characterisation of cell substrates used for production of biotechnological/biological products Q5D. 1997 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://database.ich.org/sites/default/files/Q5D_Guideline.pdf
41. Highfill SL, Stroncek DF. Overcoming challenges in process development of cellular therapies. Curr Hematol Malig Rep. 2019;14(4):269-77.
42. Costa AR, Rodrigues ME, Henriques M, Azeredo J, Oliveira R. Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm. 2010;74(2):127-38.
43. Lindskog EK, Fischer S, Wenger T, Schulz P. Chapter 6 – Host Cells. In: Jagschies G, Lindskog E, Łącki K, Galliher P, editors. Biopharmaceutical processing. Elsevier; 2018. p. 111-30.
44. van der Loo JCM, Wright JF. Progress and challenges in viral vector manufacturing. Hum Mol Genet. 2016;25(R1):R42-R52.
45. Ng RP. Considerations for successful upstream process development. BioPharm International. 2012;25(7):48-50.
46. Gretzinger S, Limbrunner S, Hubbuch J. Automated image processing as an analytical tool in cell cryopreservation for bioprocess development. Bioprocess and Biosyst Eng. 2019;42(5):665-75.
47. Gjoka X, Gantier R, Schofield M. Platform for Integrated continuous bioprocessing. BioPharm Int. 2017;30(7):26-32.
48. Hunsberger J, Harrysson O, Shirwaiker R, Starly B, Wysk R, Cohen P, et al. Manufacturing road map for tissue engineering and regenerative medicine technologies. Stem Cells Transl Med. 2015;4(2):130-5.
49. Walther J, Lu J, Hollenbach M, Yu M, Hwang C, McLarty J, et al. Perfusion cell culture decreases process and product heterogeneity in a head-to-head comparison with fed-batch. Biotechnol J. 2019;14(2):1-10.
50. Pollock J, Ho SV, Farid SS. Fed-batch and perfusion culture processes: economic, environmental, and operational feasibility under uncertainty. Biotechnol Bioeng. 2013;110(1):206-19.
51. Klutz S, Holtmann L, Lobedann M, Schembecker G. Cost evaluation of antibody production processes in different operation modes. Chem Eng Sci. 2016;141:63-74.
52. Li F, Vijayasankaran N, Shen A, Kiss R, Amanullah A. Cell culture processes for monoclonal antibody production. Mabs. 2010;2(5):466-79.
53. Przybycien T, Titchener-Hooker N. Continuous processing in downstream operations. Chem Eng Prog. 2015;111(12):38-44.
54. Prazeres DM. An overview of downstream processing. Plasmid Biopharmaceuticals. 2011:415-25. doi: 10.1002/9780470939918.ch14.
55. Shukla A, Aranha H. Viral clearance for biopharmaceutical downstream processes. Pharm Bioprocess. 2015;3(2):127-38.
56. Carbonell R, Mukherjee A, Dordick J, Roberts CJ. A technology roadmap for today’s gene therapy manufacturing challenges. Cell & Gene. 2019 Apri 18.
57. Aires-Barros MR, Azevedo AM. 7 – Fundamentals of biological separation processes. In: Pandey A, Teixeira JAC, editors. Current developments in biotechnology and bioengineering: Elsevier; 2017. p. 187-237.
58. Dream R, Odum J. Impact of ATMP manufacturing on process equipment and facility design. BioPharm Int. 2018;31(11):30-4.
59. Alsharabasy AM. Concise review: considerations for the formulation, delivery and administration routes of biopharmaceuticals. Arch Biotechnol Biomed. 2017;1:33-53.
60. Renteria Gamiz AG, Dewulf J, De Soete W, Heirman B, Dahlin P, Jurisch C, et al. Freeze drying in the biopharmaceutical industry: an environmental sustainability assessment. Food Bioprod Process. 2019;117:213-23.
61. Bee JS, Randolph TW, Carpenter JF, Bishop SM, Dimitrova MN. Effects of surfaces and leachables on the stability of biopharmaceuticals. J Pharm Sci. 2011;100(10):4158-70.
62. Vulto AG, Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology (Oxford). 2017;56(suppl_4):iv14-iv29.
63. U.S. Food and Drug Administration. Guidance for industry. Process validation: general principles and practices. 2011 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/media/71021/download
64. Siew A. Characterizing critical quality attributes in biopharmaceutical drug development. BioPharm Int. 2018;31(9):32.
65. Lalor F, Fitzpatrick J, Sage C, Byrne E. Sustainability in the biopharmaceutical industry: Seeking a holistic perspective. Biotechnol Adv. 2019;37(5):698-707.
66. Health Sciences Authority. Increase in antibody-mediated pure red cell aplasia (PRCA) cases with subcutaneous administration of Eprex® (epoetin alfa) in Singapore. 2013 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.hsa.gov.sg/announcements/safety-alert/increase-in-antibody-mediated-pure-red-cell-aplasia-(prca)-cases-with-subcutaneous-administration-of-eprex-(epoetin-alfa)-in-singapore
67. Wurm MF, Wurm JM. Cloning of CHO cells, productivity and genetic stability—a discussion. Processes. 2017;5(2):1-13.
68. Spencer S, Gugliotta A, Koenitzer J, Hauser H, Wirth D. Stability of single copy transgene expression in CHOK1 cells is affected by histone modifications but not by DNA methylation. J Biotechnol. 2015;195:15-29.
69. Betts Z, Croxford AS, Dickson AJ. Evaluating the interaction between UCOE and DHFR-linked amplification and stability of recombinant protein expression. Biotechnol Prog. 2015;31(4):1014-25.
70. Bort JAH, Hackl M, Höflmayer H, Jadhav V, Harreither E, Kumar N, et al. Dynamic mRNA and miRNA profiling of CHO-K1 suspension cell cultures. Biotechnol J. 2012;7(4):500-15.
71. Longer ES, Rader RA. Top trends in biopharmaceutical manufacturing, 2017: innovation speeds discovery, drives down costs, and improves productivity. Pharmaceut. Technol. 2017;41(9):58-60.
72. Wolf MW, Reichl U. Downstream processing of cell culture-derived virus particles. Expert Rev Vaccines. 2011;10(10):1451-75.
73. Han FV, Weiss K. Regulatory trends in drug development in Asia Pacific. Ther Innov Regul Sci. 2018;53(4):497-501.
74. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Quality risk management Q9. 2005 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://database.ich.org/sites/default/files/Q9_Guideline.pdf
75. Castillo FC, Cooney B, Levine HL. Biopharmaceutical manufacturing process validation and quality risk management. Pharmaceutical Engineering. 2016.
76. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Pharmaceutical development Q8 (R2). 2009 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf
77. Wheelwright SM. CFDA good manufacturing practice. Annexes. 2016 [home-page on the Internet]. [cited 2020 May 26]. Available from: https://www.slideshare.net/ScottWheelwright/cfda-good-manufacturing-practice-annexes?from_action=save
78. European Commission. Guidelines on good manufacturing practice specific to advanced therapy medicinal products. 2017 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2017_11_22_guidelines_gmp_for_atmps.pdf
79. Ph`armaceutical Inspection Co-operation Scheme. Draft Annex 2A. Manufacture of ATMP) to PICS GMP Guide For Public Consultation. 2019 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.picscheme.org/layout/document.php?id=1706
80. World Health Organization. Annex 2. WHO good manufacturing practices for active pharmaceutical ingredients. 2010 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.who.int/medicines/areas/quality_safety/quality_assurance/GMPActivePharmaceuticalIngredientsTRS957Annex2.pdf?ua=1
81. International Conference on Harmonisation of Technical requirements for registration of pharmaceeuticals for human use. Articles of Association. 2019 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://admin.ich.org/sites/default/files/2019-08/ArticlesOfAssociation_Approved_v3-0_2019_0606.pdf
82. Central Drugs Standard Control Organization. Good manufacturing practices and requirements of premises, plant and equipment for pharmaceutical products [homepage on the Internet]. [cited 2020 May 26]. Available from: https://cdsco.gov.in/opencms/opencms/system/modules/CDSCO.WEB/elements/download_file_division.jsp?num_id=MzA5
83. Tribe RW. The growing influence of PIC/S in Asia Pacific. Pharmaceutical Engineering. 2017.
84. Burke E. India: from generics to biosimilars. Pharmaceutical Engineering. 2019.
85. Fotheringham S. The rise of biopharmaceutical manufacturing in Asia. Pharmaceutical Engineering. 2018.
86. Montoya JC, Rebulanan CL, Parungao NAC, Ramirez B. A look at the ASEAN-NDI: building a regional health R&D innovation network. Infectious Diseases of Poverty. 2014;3(1). doi:10.1186/2049-9957-3-15
87. Muangchana C, Thanormwat Y, Kongsiang A. Workshops advocating ASEAN Vaccine Security and Self-Reliance (AVSSR): a regional initiative. Vaccine. 2019;37(14):2034-41.
88. Health Sciences Authority. Guidelines on good manufacturing practice standard and good distribution practice standard. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.hsa.gov.sg/therapeutic-products/dealers-licence/gmp-gdp
89. National Pharmaceutical Regulatory Agency. FAQ : good manufacturing practice/good distribution practice. 2015 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://bit.ly/2H9inlP
90. Food and Drug Administration of the Philippines. Notice on the adoption of the PIC/S guide to GMP. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov.ph/notice-on-the-adoption-of-the-pic-s-guide-to-gmp/
91. Department of Pharmaceutical Services. Ministry of Health Brunei Darussalam. Guide to application for a licence to manufacture or assemble medicinal products. 2013 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://bit.ly/35A6jTX
92. Association of Southeast Asian Nations. ASEAN sectoral mutual recognition arrangement for good manufacturing practice (GMP) inspection of manufacturers of medicinal products. 2009 [homepage on the Internet]. [cited 2020 May 26]. Available from: http://agreement.asean.org/media/download/20150119182953.pdf
93. Indonesia Healthcare Outlook. Frost & Sullivan. 2015.
94. Malaysia Healthcare Outlook. Frost & Sullivan. 2014.
95. Vietnam Healthcare Outlook. Frost & Sullivan. 2015.
96. Association of Southeast Asian Nations. ASEAN agrees on common requirements of biological products. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://asean.org/asean-agrees-common-requirements-biological-products/
97. Association of Southeast Asian Nations. Frequently asked questions on the ASEAN mutual recognition arrangement (MRA) on GMP inspection of manufacturers of medicinal products [homepage on the Internet]. [cited 2020 May 26]. Available from: https://asean.org/storage/images/archive/SnC/FAQ-GMP.doc
98. U.S. Food and Drug Administration. What is the approval process for genericdrugs? 2017 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/drugs/generic-drugs/what-approval-process-generic-drugs
99. O’Callaghan J, Barry SP, Bermingham M, Morris JM, Griffin BT. Regulation of biosimilar medicines and current perspectives on interchangeability and policy. Eur J Clin Pharmacol. 2019;75(1):1-11.
100. U.S. Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. 2015 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/media/82647/download
101. Kirchhoff CF, Wang X-ZM, Conlon HD, Anderson S, Ryan AM, Bose A. Biosimilars: key regulatory considerations and similarity assessment tools. Biotechnol Bioeng. 2017;114(12):2696-705.
102. U.S. Food and Drug Administration. Biosimilar development, review, and approval. 2017 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval
103. Tsiftsoglou AS, Ruiz S, Schneider CK. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27(3):203-11.
104. U.S. Food and Drug Administration. Biosimilar and interchangeable products. 2017 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products
105. Center for Biologics Evaluation and Research. List of licensed biological products. with (1) reference product exclusivity and (2) biosimilarity or interchangeability evaluations to date [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/media/89426/download
106. Center for Drug Evaluation and Research. List of licensed biological products with (1) reference product exclusivity and (2) biosimilarity or interchangeability evaluations to date [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/media/89589/download
107. Wiland P, Batko B, Brzosko M, Kucharz EJ, Samborski W, Świerkot J, et al. Biosimilar switching – current state of knowledge. Reumatologia. 2018;56(4):234-42.
108. Inotai A, Prins CPJ, Csanádi M, Vitezic D, Codreanu C, Kaló Z. Is there a reason for concern or is it just hype? – a systematic literature review of the clinical consequences of switching from originator biologics to biosimilars. Expert Opin Biol Ther. 2017;17(8):915-26.
109. World Health Organization. Annex 5. Guidance on good data and record management practices. 2016 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.who.int/medicines/publications/pharmprep/WHO_TRS_996_annex05.pdf
110. Medicines and Healthcare products Regulatory Agency. ‘GXP’ data integrity guidance and definitions. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/687246/MHRA_GxP_data_integrity_guide_March_edited_Final.pdf
111. U.S. Food and Drug Administration. Data integrity and compliance with drug cGMP questions and answers. Guidance for industry. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/media/119267/download
112. Scanlin A. How GMPs & data integrity align for safer products & swifter approvals. iSpeak [Internet]: Pharmaceutical Engineering. 2019.
113. Wechsler J. Data integrity key to GMP compliance. BioPharm Int. 2014;27(9).
114. Tabersky D, Woelfle M, Ruess JA, Brem S, Brombacher S. Recent regulatory trends in pharmaceutical manufacturing and their impact on the industry. Chimia (Aarau). 2018;72(3):146-50.
115. Center for Biologics Evaluation and Research. Warning letter: Instituto Bioclon, S.A. de C.V. – 04/16/2014. 2014 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/instituto-bioclon-sa-de-cv-04162014
116. AveXis. AveXis, Inc., San Diego, CA., 483 firm response dated 08/23/2019.
117. Center for Biologics Evaluation and Research. Warning letter: Irvine Stem Cell Treatment Center – 12/30/2015. 2015 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/irvine-stem-cell-treatment-center-12302015
118. Center for Biologics Evaluation and Research. Warning letter: Amniotic Therapies LLC. – 08/17/2016. 2016 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/amniotic-therapies-llc-08172016
119. Center for Biologics Evaluation and Research. Warning letter: Genetech, Inc. – 561808 – 11/29/2018. 2018 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/genetech-inc-561808-11292018
120. Sniderman B, Cottleleer M, Mahto M. Industry 4.0 and manufacturing ecosystems. 2016. Deloitte.
121. Cottleleer M, Sniderman B. Forces of change: Industry 4.0. 2017. Deloitte.
122. Kim TK, Seo K-S, Kwon S-O, Little TA, Kim M, Kim C-W. Application of the Quality-by-Design (QbD) approach for erythropoietin alpha purification. Bulletin of the Korean Chemical Society. 2019;40(7):623-33.
123. Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771-83.
124. U.S. Food and Drug Administration. Guidance for industry. PAT — a framework for innovative pharmaceutical development, manufacturing, and quality assurance. 2004 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.fda.gov/media/71012/download
125. Rathore AS, Kapoor G. Application of process analytical technology for downstream purification of biotherapeutics. J Chem Technol Biotechnol. 2015;90(2):228-36.
126. Glassey J, Gernaey KV, Clemens C, Schulz TW, Oliveira R, Striedner G, et al. Process analytical technology (PAT) for biopharmaceuticals. Biotechnol J. 2011;6(4):369-77.
127. Vicente T, Fáber R, Alves PM, Carrondo MJT, Mota JPB. Impact of ligand density on the optimization of ion-exchange membrane chromatography for viral vector purification. Biotechnol Bioeng. 2011;108(6):1347-59.
128. Vicente T, Peixoto C, Alves PM, Manuel J, Carrondo MJT. Modeling electrostatic interactions of baculovirus vectors for ion-exchange process development. J Chromatogr A. 2010;1217(24):3754-64.
129. Ko P, Stein M. Design methodologies for continuous improvement. In: Rebelo F, Soares M. editors. Advances in ergonomics in design. Springer, Cham; 2019. p431-21.
130. Rupp O, MacDonald ML, Li S, Dhiman H, Polson S, Griep S, et al. A reference genome of the Chinese hamster based on a hybrid assembly strategy. Biotechnol Bioeng. 2018;115(8):2087-100.
131. European Medicines Agency. Guideline on similar biological medicinal products. 2014 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
132. Brennan Z. Are biosimilars ‘interchangeable’ in the EU? A new perspective. 2017 Mar 31. Regulatory Focus.
133. Biosimilars Nederland. Franken AAM. The Dutch Medicines Board opinion on biosimilars. 2015 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.biosimilars-nederland.nl/wp-content/uploads/2015/09/05-Franken-IBN-030915-franken2.pdf
134. Ludwig W-D, Dicheva S. Biosimilars in Germany: guidance of the Drug Commission of the German Medical Association. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):178-80. doi:10.5639/gabij.2017.0604.037.
135. Scottish Medicines Consortium. Biosimilar medicines. 2015 [homepage on the Internet]. [cited 2020 May 26]. Available from: https://www.scottishmedicines.org.uk/media/2836/biosimilar-medicines.pdf
136. Wolf K. Why data integrity is impossible without a quality culture. Pharmaceutical Online. 2019 Feb 19.
137. Calnan NF, Davidson JG. Data integrity: beyond the lab. Pharmaceut Eng. 2019.
138. Schnedar C. 5 Steps to build a quality culture that supports data integrity. 2016 Sep 20. Pharmaceutical Online.
139. Ballman E. How leader actions and behaviors influence quality culture part 1. Pharmaceut Eng. 2016.
140. Ballman E. How leader actions and behaviors influence quality culture part 2. Pharmaceut Eng. 2016.
141. Calnan N. Leading indicators of quality: Pinpointing behaviors and measuring results. Pharmaceut Eng. 2016;36(6):64-7.
142. Walter G, Morse N, South N. The ABCs of behavioral change in biopharma manufacturing. Boston Consulting Group, 2013.
143. Giorgio AMD. Building a culture of continuous improvement & quality assurance. Pharmaceut Eng. 2016
144. Meyers C, Uydess I. Developing and sustaining a quality culture. Pharmaceutical Technology. 2011;35(12).
145. Srinivasan A, Kurey B. Creating a culture of quality. Harvard Business Review. 2014.
|
Author for correspondence: Adjunct Associate Professor Sia Chong Hock, Senior Consultant (Audit and Licensing) and Director (Quality Assurance), Health Products Regulation Group, Health Sciences Authority Singapore, 11 Biopolis Way, #11-01 Helios, Singapore 138667 |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


