Pharmacokinetic bioequivalence studies of a new Etoricoxib tablet formulation developed using proprietary MiST technology — risk assessment and mitigation using GastroPlus software
Published on 28 November 2023
Generics and Biosimilars Initiative Journal (GaBI Journal). 2024;13(1):14-21.
Author byline as per print journal: Dhananjay Panigrahi1, MPharm; Aditya Murthy2, MPharm, PhD; Shubham Jamdade2, MPharm; Manoj Gundeti2, MPharm; Nagarjun Rangaraj1, MPharm, PhD; Anup Avijit Choudhury1, MPharm; Tausif Ahmed2, MPharm, PhD; Venkat Ramana Naidu1, MPharm, PhD
|
Introduction: In this work, we present model` guided development of a new Etoricoxib tablet formulation using a proprietary technology. Application of absorption modelling using GastroPlus to guide the product development process is presented. An integrated approach of using in vitro, modelling and in vivo pharmacokinetics (PK) data to demonstrate bioequivalence between newly developed formulation and the commercial formulation is presented. |
Submitted: 26 May 2023; Revised: 9 November 2023; Accepted: 15 November 2023; Published online first: 28 November 2023
Introduction
Developing new drug products is a resource intensive process. The model-informed drug development (MiDD) is an approach where biological, mathematical, and statistical models are applied to the drug development process to save time and cost of development [1]. On a broader level, this approach has three fundamental pillars: (1) Understanding of pharmacokinetics (PK) and pharmacodynamics (PD) of the drug; (2) Developing mathematical models that integrate data from PK, PD (preclinical and clinical studies) and in vitro studies; and (3) Applying the knowledge from the models to make decisions at various stages of development [1]. This is applicable to development of both innovator and generic drug products. It can accelerate development and help in effective regulatory decision-making [2].
The objectives of this work are presented below
- Develop a new, cost-effective tablet formulation for Etoricoxib with a proprietary MiST technology; further, the new formulation must have improved dissolution rate and must be bioequivalent to a commercially available reference product with early onset of action in fed state.
- Utilize existing (literature), in-house PK data and in vitro dissolution data to develop a physiologically based biopharmaceutics model (absorption model) using GastroPlus®.
- Validate the model with available data and employ the model for population PK and virtual bioequivalence (VBE) predictions that can aid in making informed decisions during product development.
Etoricoxib (ETR) is an orally active [5-chloro-2-(6-methylpyridin-3-yl)-3-(4-methylsulfonylphenyl) pyridine] derivative, see Figure 1, that selectively inhibits the cyclooxygenase-2 (COX-2) [3]. It belongs to the selective COX-2 inhibitors of the class non-steroidal anti-inflammatory drugs (NSAIDS) [4]. Compared to the other COX-2 inhibitors like rofecoxib, valdecoxib and celecoxib, ETR displays a higher COX-1 to COX-2 binding ratio (IC50 of 1.1 ± 0.1 μM for COX-2 and 116 ± 8 μM for COX-1) [5]. Thus, ETR is highly selective towards the COX-2 and has superior gastrointestinal safety and tolerability compared to other drugs in the same therapeutic class. The ETR has become drug of choice in the treatment of acute pain, acute gouty arthritis, chronic low back pain, primary dysmenorrhea, and chronic treatment for the signs and symptoms of osteoarthritis and rheumatoid arthritis [6].
The ETR is a weakly basic drug (pKa 4.6) and classified as BCS class II. It demonstrates pH dependent solubility (soluble in acidic media) [7]. It is soluble in at pH <1.2, but insoluble at pH >3. Gastrointestinal transit of ETR from gastric to intestinal region results in precipitation of the drug [8]. Hence, its oral absorption displays high fast-fed variability. Administration after a high-fat meal results in a 36% reduction of the maximum concentration (Cmax) and increase in maximum time (Tmax) by 2 h (Tmax in fasting state 1h, under fed it is 3h) [9]. Increase in Tmax under fed state results in delayed absorption of the drug, thus delaying onset of action [10]. Significant differences in the exposure are observed between fasting and fed states.
The primary objective of this work was to develop a new, cost-effective formulation of ETR and to evaluate its in vivo performance using in vitro (dissolution) and in silico (absorption modelling) tools. In the past, many research groups have explored approaches to develop more effective ETR by various approaches like amorphous solid dispersion [11], complexation with -cyclodextrin [6] and co-solvency approach [12]. Additionally, nanoparticulate formulations like niosomes and solid lipid nanoparticles have also been evaluated [13] and [14]. However, none of them have demonstrated the scalability of the proposed technologies. In the current work, we have developed a proprietary formulation technology (MiSTTM) that is a combination of homogenization and top spray granulation approaches. This technology is easily scalable and can be readily translated to commercial product. This work details the application of proprietary MiSTTM technology to develop ETR tablets and subsequent in vitro and in silico evaluation.
Materials and methods
Materials
Etoricoxib was procured from in-house source, Polymeric stabilizer was obtained from Dow Chemicals, India. Wetting agent was obtained from BASF, India. Lactose monohydrate was procured from DFE Pharma, India; microcrystalline cellulose (MCC) was procured from FMC Bio, India. Aerosol 200 was obtained from Evonik, India.
Methods
Formulation development
Quality by design approach (QbD)
The QbD is a systematic approach that enables researchers to develop a formulation with in-built quality attributes. Unlike the traditional approaches where the quality is tested after the product is developed, in the QbD approach, quality is built into the product. Development of a product using QbD approach involves utilizing prior knowledge, design of experiments (DoE) risk and knowledge management. Product development via QbD pathway involves establishing quality target product profile (QTPP), identifying critical quality attributes (CQAs), critical material attributes (CMAs), critical process parameters (CPPs) all of which can be bundled as critical bioavailability attributes (CBAs) [15].
Homogenization and manufacture of spray dispersion
The drug, stabilizer, wetting agent and disintegrant were dispersed in water. This dispersion was homogenized using high shear homogenizer (Ultraturrax T-25, IKA, Germany). The homogenization time was optimized based on the particle size of the dispersion obtained [16].
Particle size analysis
Particle size analysis by dynamic light scattering
The homogenized dispersion (before top spray granulation) was subjected to particle size analysis using Zetasizer Nano ZS (Malvern Instrument Ltd., Worcestershire, UK). The dispersion was diluted ~10X with Milli-Q water and analysed for particle size [15].
Top spray granulation
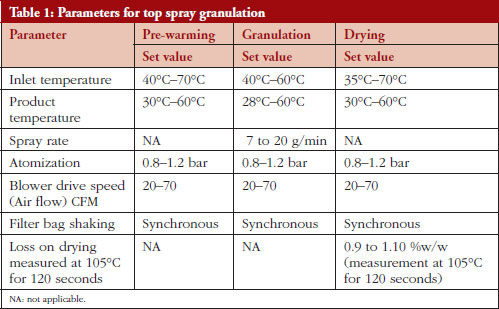
The core material (lactose monohydrate and microcrystalline cellulose) were loaded into top spray bowl and the drug dispersion was top sprayed onto the core material. Granulation was continued using fluid bed processor (GPCG 1.1, Glatt, Germany) using parameters presented in Table 1. The granules were blended, lubricated and compressed into spherical-shaped tablets.
In vitro drug release evaluation
The objective of this work was to develop a novel test formulation using novel MiST Technology that can be administered under fed conditions. The new product had to be bioequivalent with the reference ETR tablets under fed conditions. Hence, dissolution studies were performed in the media that mimic human fed physiology. Dissolution studies were performed with N = 6 units in the following media considering fed relevant conditions:
a) 10 mM acetate buffer, pH 4.5 ± 0.1
b) 10 mM acetate buffer, pH 5.5 ± 0.1
Absorption modelling and simulation using GastroPlus®
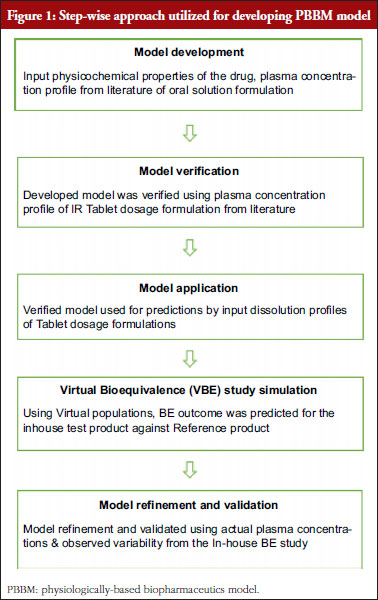
GastroPlus® version 9.8 (Simulations Plus Inc, USA) was employed to build a physiologically-based biopharmaceutical model (PBBM) for ETR tablets 120 mg. The population PK model was utilized to predict drug plasma concentrations using dissolution inputs from physiologically relevant media under fed conditions. The stepwise approach followed to build the model for ETR tablets is presented in Figure 1.
Physicochemical and biopharmaceutical properties of Etoricoxib
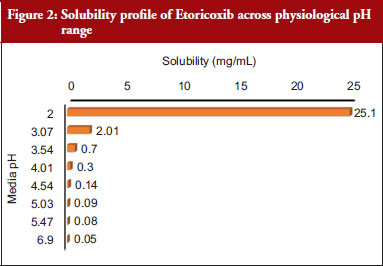
The physicochemical and biopharmaceutical properties of ETR were defined using a combination of literature-based inputs and optimized inputs from the simulations. In case of unavailability of literature parameters, ADMET predicted parameters from GastroPlus® were considered. ETR is a BCS Class II compound, and it exhibits pH dependent solubility – low solubility in alkaline pH and highly soluble in the acidic pH [7]. Solubility profile of ETR across physiological pH range is presented in Figure 2.
Pharmacokinetics of Etoricoxib
Orally administered ETR is well absorbed. The absolute bioavailability is approximately 100%. Peak plasma concentration under fasting is 1 h; under high fat meal it is 2 h [17]. ETR is extensively metabolized with <1% of a dose recovered in urine as the parent drug. The principal metabolite is the 6’-carboxylic acid derivative of ETR. It is approximately 92% bound to human plasma protein over the range of concentrations of 0.05 to 5 μg/mL. The volume of distribution at steady state (Vdss) was approximately 120 L in humans. Following administration of a single 25-mg radiolabelled intravenous dose of ETR to healthy subjects, 70% of radioactivity was recovered in urine and 20% in feces, mostly as metabolites. Less than 2% was recovered as unchanged drug. Dosing with food (a high-fat meal) had no effect on the extent of absorption of ETR after administration of a 120-mg dose. The rate of absorption was affected, resulting in a 36% decrease in Cmax and an increase in Tmax by 2 hours. These data are not considered clinically significant. The PK of ETR is linear across the clinical dose range (30 mg–120 mg) [18].
Model development and verification
The ACAT model was developed using following inputs: (1) PK profile of oral solution formulation product from the literature [19]; (2) solubility data across physiological pH range was generated from literature and used as an input in the model [17]; (3) disposition PK calculated from oral solution profile. Two compartment model was fitted as best fit model; and (4) Model was verified with literature reported data [16].
The verified model was then employed to assess VBE between the new test formulation against the reference product. For this, dissolution data for the respective formulation were used as inputs. The verified model was employed to predict VBE between novel MiST formulation against the reference product.
Results
Formulation development
In the MiSTTM technology, drug is homogenized with addition of wetting agent. This process has dual benefits: (1) increase in the surface area of the API due to homogenization; and (2) improved wetting of the API due to use of surfactant during the process. Particle-size reduction was achieved by homogenization of the dispersion using high speed homogenizer. The initial particle size of the API was 16 ± 4 μm, which was further micronized to 3.3 ± 3 μm using the homogenization process. The polymeric stabilizer used to stabilize the drug suspension also helps in preventing Ostwald ripening [18].
Characterization of Etoricoxib tablets
In vitro evaluation
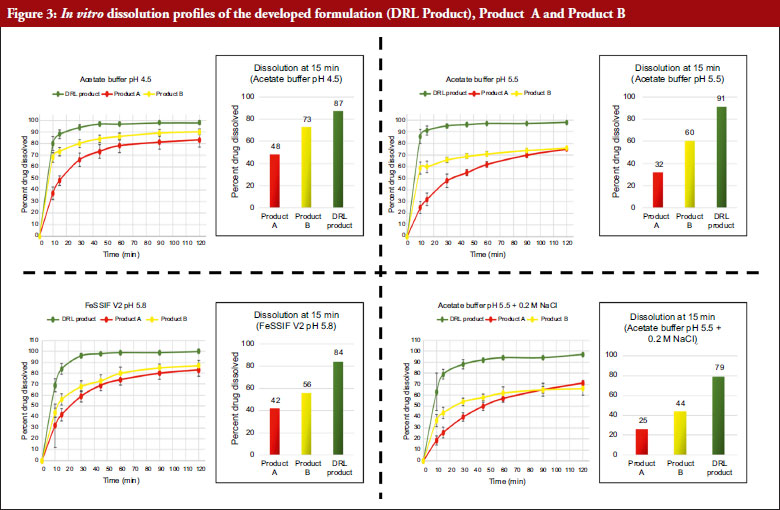
In our current study, BE study evaluation was performed under fed condition. Therefore, for dissolution comparison, fed state relevant dissolution media were employed. Comparative dissolution profiling was performed between novel ETR formulation and few commercial formulations in the following buffers: pH 4.5 acetate buffer, pH 5.5 acetate buffer, FeSSIF v2 (pH 5.8) and pH 5.5 acetate buffer with 0.2 M sodium chloride. Results of the dissolution studies are illustrated in Figure 3. As seen from the figure, the novel ETR formulation with MiST technology demonstrated significantly faster dissolution profile in all the tested media compared to other commercial products. ETR in the commercial product (reference) demonstrated pH dependent solubility; however, the novel ETR product manufactured with proprietary technology mitigated the pH dependent behaviour. This formulation enablement is particularly useful for molecules that demonstrate significant pH and food effect. Mitigating the same with enabled formulation is beneficial in improving both patient convenience and compliance.
Dissolution data used for the simulations
Dissolution data indicated significant improvement in dissolution for the new test product when compared to marketed formulations of Product A and Product-B (reference products) in all the fed relevant media, i.e. pH 4.5 Acetate Buffer, pH 5.5 Acetate Buffer with or without 0.2M NaCl and FeSSIF v2 media. However, for the modelling purpose, considering the Tmax, absorption region of the drug and fed physiology, dissolution in pH 4.5 Acetate Buffer media, pH 5.5 Acetate Buffer media were used with Z-factor fitting in GastroPlus®.
Model development and verification
Human PK parameters were estimated by fitting 100 mg oral solution data to a two-compartment model using PKPlusTM module [19]. The best-fit compartment model was selected. The parameters used as input in the model are presented in Table 2.
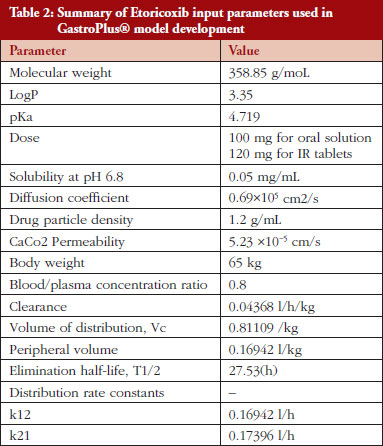
Virtual BE study simulations were performed by using input in vitro dissolution data. The simulation outcomes are presented in Table 3 and Figure 4. Based on this initial risk assessment, the test formulation was selected for PK-BE studies. Equation 1 was used for calculating PE%.
PE% = ((Predicted value-Observed value)/
(Observed value)) × 100 – Equation 1
Acceptance criteria: ± 15% for individual PK parameters and ± 10% for mean PK parameters
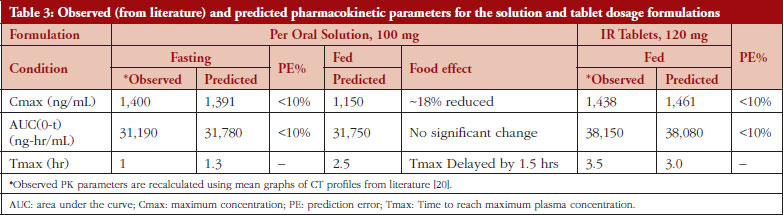
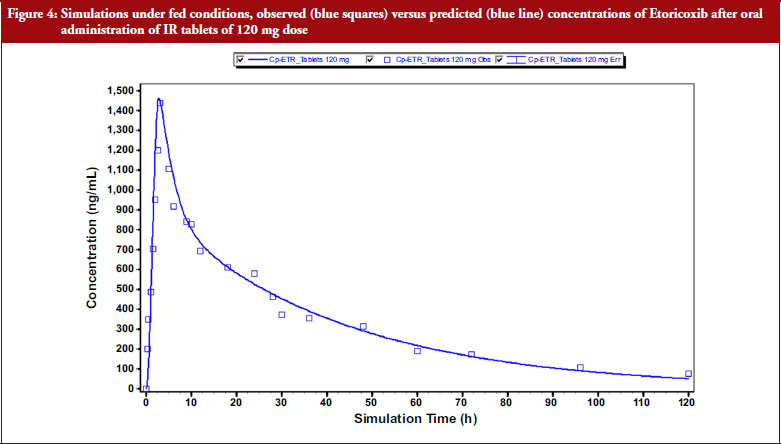
Virtual BE simulations from GastroPlus®
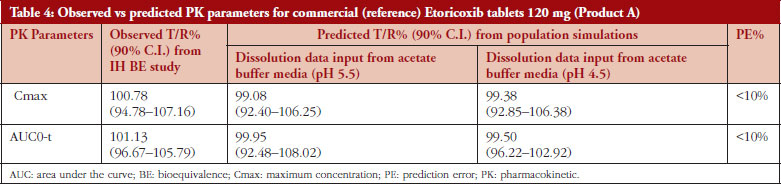
Simulation studies were performed using developed and verified model under fed physiology. The modelling outcomes are presented in the Supplementary Material. The results are tabulated in Table 4.
Model refinement using in-house study data
Based on the in-house data obtained from the PK-BE study, the earlier model (developed from literature data) was further optimized to accurately predict the absolute values of PK parameters. The observed vs predicted outcome from the refined model is presented in Table 5.
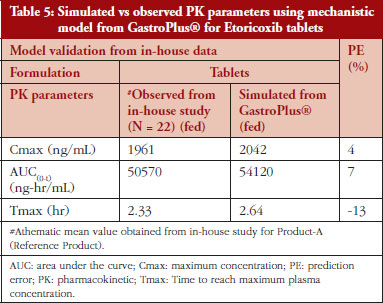
Figure 5 demonstrates observed and simulated PK profile for the commercial formulation (Product A) 120 mg tablets.
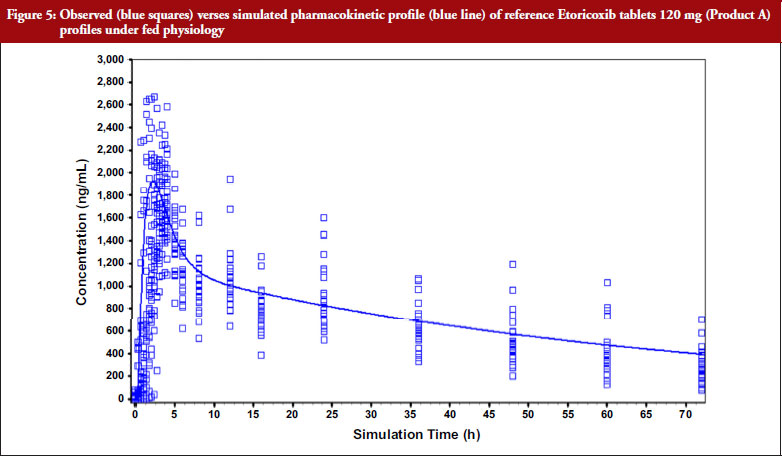
The results demonstrate that the observed PK parameters for the reference product (Product A) from in-house study was close to the simulated PK parameters predicted parameters. PE of less than 10% for both Cmax and AUC was obtained.
Utilizing this refined and more accurate model, population simulations using demographic data from the BE study were performed for both reference and new test product. VBE simulation was performed to validate the model with respect to actual BE results and 90% confidence interval for both Cmax and AUC was computed. The simulated VBE outcome was computed against obtained BE outcome. This comparison is presented in Table 6.
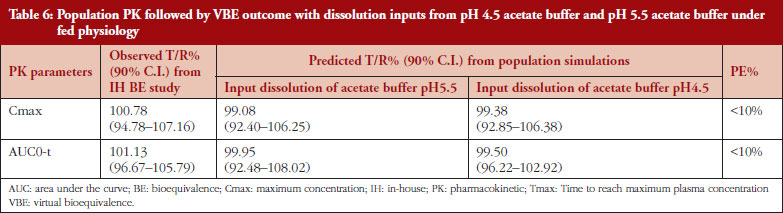
From Table 6, it is evident that the optimized model was well predictive of the obtained BE results. Further, both pH 4.5 Acetate Buffer and pH 5.5 Acetate Buffer media were indicative of the in vivo performance of ETR absorption. The IVIVR between in vitro dissolution studies in these media and the in vivo absorption is presented in Figure 6.
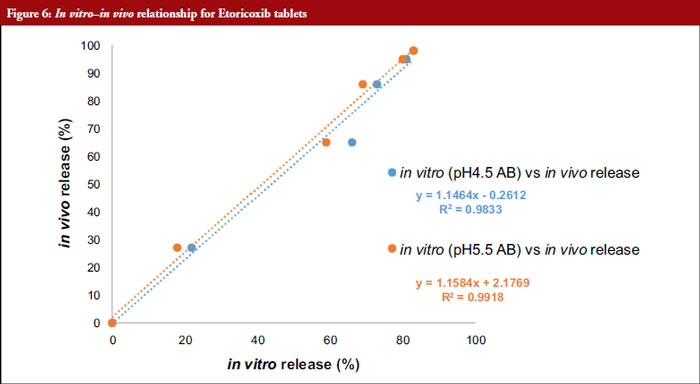
An IVIVR between in vitro dissolution and in vivo fraction absorbed indicates that, both the dissolution media (Acetate Buffer pH4.5 & pH5.5) were well-predictive of in vivo absorption for ETR.
Based on in vitro in vivo relationship for the commercial (reference) Product-A, these media can be considered bio reflective for ETR tablets under fed conditions. The ETR demonstrates low solubility across pH 4.5 to 7.0; however, the ADMET predicted permeability is high (5.23 x 10-5 cm/sec). It is absorbed rapidly and extensively from the upper part of the small intestine, reaching maximum serum concentrations within few hours after oral administration. Oral bioavailability of ETR from tablet dosage form is ~90–100%. Hence, dissolution in pH conditions relevant to upper GIT are more critical for predicting the in vivo performance of this product.
Discussion
As ETR is reported to be a BCS class II with pH dependent solubility, the prime objective of this study was to reduce the pH dependency on dissolution. It was hypothesized that reduction of pH dependent solubility behaviour could reduce the fasting vs fed variability seen with this drug. A proprietary MiST technology was employed in the manufacture of novel ETR tablets. The novel formulation had faster dissolution in fed relevant dissolution media compared with the marketed formulation. This enhanced drug release from the developed formulation could be explained by enhanced improved solubility of the API across pH conditions. Particle size reduction and addition of a wetting agent along with process parameters could contribute to enhanced drug dissolution in the novel product. Particle size reduction would create new surfaces and also increases the effective surface area for a poorly wetting drug like ETR [20]. As per Ostwald-Freundlich equation, decrease in the radius causes an increase in the surface area to volume ratio leading to enhanced solubility [19]. Incorporation of wetting agent (surfactant) is important to enhance the solubility of poorly soluble drugs. These surfactants are amphiphilic molecules with both hydrophilic and hydrophobic regions. Enhancement of ETR solubility by incorporation of SLS has been reported earlier [6].
The homogenized drug dispersion is uniformly top sprayed onto the core material. Hence, this approach yielded granules with good dissolution [6]. This uniform coating of drug around the core material would also lead to increased surface area and better solubility.
Dissolution testing is employed to assess the impact of formulation and manufacturing variables on the performance of the drug product. In case of novel ETR formulation, the rate of dissolution was faster compared with the conventional commercial formulations. Advances in PBBM and simulation help in integrating in vitro dissolution data with physiological factors to predict in vivo performance of the drug product [21]. In some of the earlier work from our group, we have demonstrated various applications of PBBM to predict in vivo performance of the drug product. In a publication by Swati et al., virtual BE was utilized to understand the effect of dissolution specification changes on the drug product performance [22]. In another recent article, we have demonstrated utilization of PBBM to understand the impact of faster dissolution profile on the in vivo performance of the drug product [23]. Numerous articles on the application of PBBM to understand the impact of dissolution on the product performance have been published. Farhan et al., have demonstrated the use of PBPK model to accelerate product development and improve the success of BE for generic products [24]. In another recent publication on Vildagliptin MR product, utilization of PBBM to propose clinically relevant dissolution specifications has been demonstrated [25]. However, despite the advances in PBPK modelling for predicting in vivo performance of new drug products, there are some known limitations. For example, Wu et al. report the concept of finite absorption time in PBPK modelling. It is known that, in vivo, the drug absorption terminates after a specific point of time. Nevertheless, in the current commercially available software, the models assume continued absorption across the GIT, often leading to over-predicted plasma drug concentrations [26].
In the present study, we have demonstrated utilization of PBBM in predicting the in vivo performance of the novel ETR drug product. We have utilized PBBM as a bridge to connect in vitro dissolution with in vivo performance. From this study, it has been demonstrated that the novel formulation manufactured by proprietary MiST technology showed faster dissolution profile, independent of the media pH conditions. It also demonstrated BE with the reference product. The simulations also demonstrated that the novel formulation could achieve bioequivalence compared to conventional commercial formulations without compromising on the extent of absorption. An IVIVR was also established to demonstrate the relevance of dissolution in pH 4.5 & pH 5.5 AB to predict the in vivo performance of the drug product.
Conclusion
In this work, we have presented a novel enablement approach that could be used to mitigate pH effect, especially for BCS II drugs. Further, through appropriate dissolution data inputs in the model, we have demonstrated that the novel drug product can be bioequivalent to commercial formulation (reference product). This work provides an integrated approach of using an enabled formulation, in vitro and in silico tools to overcome the limitations of ETR formulation. This approach can be extended to other BCS II drugs and has potential to accelerate the drug development process, thus reaching patients faster. In this work, an integrated MiDD approach was utilized to assess the in vivo performance risk for the new test product and to accelerate product development. Although we have presented a simple case here, the same approach could be effectively utilized for the aiding the development of more complex drug products in the future.
Acknowledgements
The authors would like to thank Dr Reddy’s Laboratories Ltd for providing an opportunity to publish this work.
DRL Publication Number: PUB00628-23.
Funding sources
This work was funded by Dr Reddy’s Laboratories Ltd. No other funding was received for this work.
Author contributions
DP, AM, AAC, TA and VRN are involved in concept and design, writing manuscript and manuscript review. SJ, NR, MG are involved in concept and design and writing manuscript.
Disclaimer
The data presented in this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Compliance with ethical standards
The study protocol was approved by MAARG independent Ethics committee with meeting No: MIEC-052-21 & Informed consent was confirmed by the ethics committee.
Competing interests: All the authors (DP, AM, SJ, MG, NR, AAC, TA and VRN) are employees of Dr Reddy’s Laboratories Ltd and declare that they have no conflict of interest. The authors alone are responsible for the content and writing of this article.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Dhananjay Panigrahi1, MPharm
Aditya Murthy2, MPharm, PhD
Shubham Jamdade2, MPharm
Manoj Gundeti2, MPharm
Nagarjun Rangaraj1, MPharm, PhD
Anup Avijit Choudhury1, MPharm
Tausif Ahmed2, MPharm, PhD
Venkat Ramana Naidu1, MPharm, PhD
1BRaIN-Formulation R&D-DF, Dr Reddy’s Laboratories Ltd, Hyderabad, India
2Global Clinical Management, Dr Reddy’s Laboratories Ltd, Hyderabad, India
References
1. Madabushi R, Seo P, Zhao L, Tegenge M, Zhu H. Role of model-informed drug development approaches in the lifecycle of drug development and regulatory decision-making. Pharm Res. 2022;39(8):1669-80.
2. Bi Y, Liu J, Li L, Yu J, Bhattaram A, Bewernitz M, et al. Role of model-informed drug development in pediatric drug development, regulatory evaluation, and labeling. J Clin Pharmacol. 2019;59:S104-S111.
3. Riendeau D, Percival MD, Brideau C, Charleson S, Dube D, Ethier D, et al. Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J Pharmacol Exp Ther. 2001;296(2):558-66.
4. Matsumoto AK, Cavanaugh Jr, PF. Etoricoxib. Drugs Today (Barc). 2004;40(5):395-414.
5. Cochrane DJ, Jarvis B, Keating GM. Etoricoxib. Drugs. 2002;62(18):2637-51.
6. Babu S. A factorial study to evaluate the individual and combined effects of [beta]-cyclodextrin and tween 80 on the solubility and dissolution rate of aceclofenac. Asian J Chem. 2010;22(6):4281.
7. Mitra A, Kesisoglou F, Dogterom P. Application of absorption modeling to predict bioequivalence outcome of two batches of etoricoxib tablets. AAPS PharmSciTech. 2015;16(1):76-84.
8. Gonzalez-Alvarez I, Bermejo M, Tsume Y, Ruiz-Picazo A, Gonzalez-Alvarez M, Hens B, et al. An in vivo predictive dissolution methodology (IPD methodology) with a BCS class IIb drug can predict the in vivo bioequivalence results: Etoricoxib products. Pharmaceutics. 2021;13(4):507.
9. Agrawal NG, Porras AG, Matthews CZ, Rose MJ, Woolf EJ, Musser BJ, et al. Single-and multiple-dose pharmacokinetics of etoricoxib, a selective inhibitor of cyclooxygenase-2, in man. J Clin Pharmacol. 2003;43(3):268-76.
10. Sapkal SB, Adhao VS, Thenge RR, Ashok R, Darakhe SAS, Shrikhande VN. Formulation and characterization of etoricoxib solid dispersion using natural polymers. Turk J Pharm. 2020;17(1):7-19.
11. Dani P, Puri V, Bansal AK. Solubility advantage from amorphous etoricoxib solid dispersions. Drug Dev Ind Pharm. 2014;40(1):92-101.
12. Nayak AK, Panigrahi PP. Solubility enhancement of Etoricoxib by cosolvency approach. International Scholarly Research Notices. 2012.
13. Kesharwani R, Sachan A, Singh S, Patel D. Formulation and evaluation of solid lipid nanoparticle (SLN) based topical gel of etoricoxib. J Appl Pharm Sci. 2016;6(10):124-31.
14. Ravalika V, Sailaja AK. Formulation and evaluation of etoricoxib niosomes by thin film hydration technique and ether injection method. Nano Biomed Eng. 2017;9(3):242-8.
15. Rangaraj N, Reddy Pailla S, Chowta P, Sampathi S. Fabrication of ibrutinib nanosuspension by quality by design approach: intended for enhanced oral bioavailability and diminished fast fed variability. AAPS PharmSciTech. 2019:20(8):326.
16. Alizadeh MN, Shayanfar A, Jouyban A. Solubilization of drugs using sodium lauryl sulfate: experimental data and modeling. J Mol Liq. 2018;268:410-14.
17. European Medicines Agency. Arcoxia [homepage on the Internet]. [cited 2023 Nov 9]. Available from: https://www.ema.europa.eu/en/documents/referral/arcoxia-article-6-12-referral-annex-i-ii-iii-iv_en.pdf
18. Müller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int J Pharmaceutics. 1998;160(2):229-37.
19. Rodrigues AD, Halpin RA, Geer LA, Cui D, Woolf EJ, Matthews CZ. Absorption, metabolism, and excretion of etoricoxib, a potent and selective cyclooxygenase-2 inhibitor, in healthy male volunteers. Drug Metab Dispos. 2003;31(2):224-32.
20. Saleh, Mubarak Abdullah, Salam A. Mohammed, Ezzat C. Abdullah, and Laith A. Hashim. Enhancement the dissolution rate and solubility of poorly soluble drugs. In Advanced Materials Research. Vol. 701, pp. 234-238. Trans Tech Publications Ltd, 2013.
21. Wu D, Li M. Current state and challenges of physiologically based biopharmaceutics modeling (PBBM) in oral drug product development. Pharm Res. 2023;40(2):321-36.
22. Jaiswal S, Ahmed T, Kollipara S, Bhargava M, Chachad S. Development, validation and application of physiologically based biopharmaceutics model to justify the change in dissolution specifications for DRL ABC extended release tablets. Drug Dev Ind Pharm. 2021;47(5):778-9.
23. Aishwarya R, Murthy A, Ahmed T, Chachad S. A novel approach to justify dissolution differences in an extended release drug product using physiologically based biopharmaceutics
|
Author for correspondence: Tausif Ahmed, MPharm, PhD, Head Biopharmaceutics and Bioanalytical, Global Clinical Management, Dr Reddy&rquo;s Laboratories Ltd, IPDO, Bachupally Campus, Qutbullapur Mandal, Hyderabad – 500090, Telangana, India |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2024 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


