Pharmacokinetics and relative bioavailability of sitagliptin hydrochloride and sitagliptin phosphate tablets formulations: a randomized, open-label, crossover study in healthy male volunteers
Published on 2023/01/05
Generics and Biosimilars Initiative Journal (GaBI Journal). 2023;12(1):12-6
Author byline as per print journal: Chuei Wuei Leong1, PhD; Elton Sagim1, BBiomedSc, Kar Ming Yee1, BPharm; Muhammad Shalhadi Saharuddin1, BSc; Sharifah Radziah Syed Abd Rahim1, MSc; Khairil Sabri1, BSc; Mohd Zulhairi Jamaluddin1, BSc; Shahnun Ahmad1, MBBS; Atiqah Amran1, BSc; Rabab F Tayyem2, PhD
|
Introduction/Study Objectives: The present study aimed to evaluate the comparative bioavailability of a new generic sitagliptin formulation. |
Submitted: 15 September 2022; Revised: 29 November 2022;Accepted: 30 November 2022; Published online first: 20 December 2022
Introduction/Study Objectives
Diabetes mellitus is a highly prevalent disease affecting over half a billion people, accounting for over 10% of the world’s adult population, with an anticipated 46% increase by 2045 and a drastic economic and health burden [1]. Most diabetic patients present with type 2 diabetes mellitus (T2DM) [2], characterized by impaired insulin sensitivity resulting in hyperglycaemia [3]. Furthermore, T2DM is associated with both microvascular and macrovascular complications that are the major causes of its morbidity and mortality [4, 5]. As T2DM progresses, pancreatic β cells progressively deteriorate, requiring effective glycaemic control that can be achieved by lifestyle modifications and treatment with any of a number of hypoglycaemic agents [3].
Sitagliptin is an oral hypoglycaemic agent that selectively inhibits the dipeptidyl peptidase-4 (DPP-4) enzyme [6] responsible for the inactivation of the glucagon-like peptide-1 (GLP-1) and the glucose-dependent insulinotropic polypeptide (DPP-4). DPP-4 inhibition results in an incremental prolongation of incretin activity leading to a glucose-dependent boost of insulin secretion and a decrease in glucagon secretion [7]. Sitagliptin’s glycaemic control effect is characterized by lowering of both fasting and postprandial glucose concentrations [8]. DDP-4 inhibitors (DPP-4i) are well tolerated when given either alone or in combination with other hypoglycaemic agents and its use is associated with a clinically insignificant risk of hypoglycaemia or weight gain [9]
The absorption of sitagliptin is rapid, with a median time to the maximum concentration (Tmax) of 1–4 hours and an apparent half-life (T1/2) of 8–14 hours [10, 11]. Sitagliptin is metabolized via oxidation catalyzed mainly by cytochrome P450 (CYP) 3A4 isoenzymes in the liver, with a small contribution from CYP2C8 [12]. Sitagliptin area under the curve (AUC) is increased in a relatively dose-dependent fashion in healthy volunteers [11] and its absolute bioavailability was found to be 87%. The majority of a sitagliptin dose is excreted unchanged in urine (87%) and feces (13%) [12, 13]. The renal elimination of sitagliptin is by active tubular secretion. Its renal clearance has been reported to be 388 mL/min [11]. Therefore, renal function is expected to be a significant factor affecting the pharmacokinetics (PK) of sitagliptin [14]. However, no dose adjustment is required in patients with only mild renal impairment; that is patients with a creatinine clearance of equal to or more than 50 mL/min but less than 80 mL/min. Minor decreases in renal function were found to have a clinically insignificant impact on sitagliptin PK. [14].
Different sitagliptin salts, e.g. hydrochloride, malate, and tartrate, have judged to have similar bioavailabilities compare to the reference sitagliptin phosphate product (Januvia®) with similar safety and efficacy profiles when they were given market authorization [15]. In a recent study comparing the tablet formulations of sitagliptin hydrochloride with sitagliptin phosphate, the uniformity of dosage form as measured by weight variability was found to be superior in sitagliptin hydrochloride tablets. Furthermore, sitagliptin hydrochloride tablets were shown to have superior chemical stability compared to sitagliptin phosphate and were therefore considered a better option [16]. Similar dissolution profiles were also demonstrated for both sitagliptin phosphate and hydrochloride salts, with an acceptable similarity factor, indicating that both salts have similar in vivo behaviour [17]. This was explained by sitagliptin’s high bioavailability and solubility [18]. This study aimed to evaluate the relative bioavailability of a test formulation (containing 100 mg of sitagliptin in the form of sitagliptin hydrochloride) with a reference tablet formulation (containing 100 mg of sitagliptin in the form of sitagliptin phosphate).
Methods
Study design
The relative bioavailability of a single dose of a test formulation with a reference formulation, both containing 100 mg sitagliptin, was evaluated in an open-label, randomized, balanced, two-period, two-way, two-sequence crossover study design in healthy, fasting adult volunteers. The washout period was one week between the two study periods.
The study was carried out in the ACDIMA BioCentre in Amman, Jordan in accordance with the International Conference on Harmonization–Good Clinical Practice, the Declaration of Helsinki, and the ASEAN (Association of Southeast Asian Nations) guideline for the conduct of bioequivalence (BE) studies. Approval was obtained from the ACDIMA BioCenter’s Institutional Review Board (IRB) (Research ID: 940-2019) on 15 September 2020 and the Jordan Food and Drug Administration (JFDA) (Research ID: 6/BIO/20) on 5 February 2020. Informed consents were obtained from all participants prior to enrolment. The study was also registered in the Thai Clinical Trials Registry (TCTR20220628001) on 28 June 2022.
Study population
Participants were recruited based on the fulfilment of the inclusion criteria of: 1) healthy as confirmed by the medical evaluation [basic medical check-up, medical history, electrocardiogram (ECG), and laboratory findings (complete blood count, biochemical tests, serological tests, and urine analysis] performed on admission; 2) body mass index (BMI) of 18.5 to 30 kg/m2 (weight more than 59 kg); 3) adults aged between 18 and 50 years; 4) not receiving any medications for the last two weeks prior to the commencement of the study; 5) non-pregnant women; 6) with fasting blood glucose concentration of more than 70 mg/dL prior to dosing at the start of each period. Exclusion criteria included: 1) heavy smokers (more than 10 cigarettes per day); 2) presence of any contraindications or allergies to sitagliptin; 3) consumption of grapefruits products in the prior week or caffeinated beverages within two days of the study date; and 4) abuse of alcohol or illicit drugs which was confirmed by laboratory testing (alcohol saliva test and urine test for benzodiazepines, amphetamine, marijuana, cocaine and opiates).
Based on sample-size calculation to achieve a power of 80%, considering an intra-subject and inter-subject coefficient of variation (ISCV) for sitagliptin AUC following a 100 mg single dose found in the literature to be around 5.8% and 15.1%, respectively [19], a minimum of 12 subjects was required to be recruited; therefore, thirty subjects from the Jordan population were enrolled for this study.
Study products and administration
The BE of Fortreas® tablets (containing 100 mg of sitagliptin in the form of sitagliptin hydrochloride) manufactured by Duopharma, Malaysia (test formulation) was compared with Januvia® tablets (containing 100 mg of sitagliptin in the form of sitagliptin phosphate) manufactured by Merck Sharp & Dohme Ltd, England (reference formulation) in the present study. Volunteers were assigned randomly to ingest the reference or test tablet with 240 ± 2 mL of water after overnight fasting of 10–12 hours. Subjects were instructed to stay in an upright position for four hours after dosing. Standardized meals were served to all volunteers at a fixed schedule throughout the study where on Day 1, standardized dinner (chicken scallop sandwich) was served 12 hours before dosing and on Day 2, standardized lunch (chicken rice and bread) was served 4 hours after dosing, a snack 8 hours after dosing, and a standardized dinner (chicken scallop sandwich) 12 hours after dosing. Fluid restrictions were placed only on Day 2 of the study. Subjects were not allowed to drink water 1 hour before dosing and 4 hours after, except for: 120 ± 2 mL of water was served 1 hour before dosing, 240 ± 2 mL of water with the dose and 120 ± 2 mL of water 2 and 3 hours after dosing.
Sampling
A total of 22 blood samples (7 mL each) were taken from each volunteer at pre-dose and 0.3, 0.7, 1, 1.3, 1.7, 2, 2.3, 2.7, 3, 3.3, 3.7, 4, 4.3, 4.7, 5, 6, 8, 10, 12, 23, and 48 hours post-dose. Lithium heparin tubes were used for blood samples collection; then, centrifugation at 4,000 RPM for 5 min at 10°C was carried out. Plasma was separated and stored in the freezer (-70°C), then transferred to the bioanalytical laboratory site of the on-site ACDIMA BioCentre–Bioanalytical Unit and stored in a freezer (-70°C) until the bioanalytical analyses were performed.
Subject monitoring
Throughout the study, clinical assessments and laboratory investigations were performed in an attempt to evaluate both protect the safety of all participants and report and assess any adverse events observed during the study.
Determination of sitagliptin plasma concentrations
An analytical method for the estimation of sitagliptin in human plasma was developed and validated using reversed-phase liquid chromatography and tandem mass spectrometry (LC-MS/MS) with positive ion electrospray ionization (Agilent, USA). Plasma was extracted through protein precipitation where 200 μL of each plasma sample was spiked with 25 μL Sitagliptin D4 as an internal standard; then 1 mL of the precipitation solvent, acetonitrile, was added, and the mixture was vortexed and centrifuged for 5 min at 6,000 RPM at 5°C. Afterward, 10 μL was injected via the autosampler, and separation was performed using an Agilent Eclipse XDB CN (150 × 4.6 mm, i.d.: 5 μm) at 35°C and a flow rate of 1 mL/min of the mobile phase, which was composed of acetonitrile and 50 mM ammonium formate (50:50, v/v) with 0.5 mL formic acid. Quantitation of the analyte was done on a triple-stage quadrable mass spectrometer. Sitagliptin and internal standard were monitored at the molecular ion 408.2–412.1 m/z and MS/MS (daughter) 235.0–239.0 m/z, respectively. Validation of the method was carried out in the range of concentrations between 1 ng/mL to 750 ng/mL with good linearity of r2 equals 0.999. The method achieved intra-and inter-day precision of less than 5% and accuracy of 98.8%–103.5%. The analyte and the internal standard recoveries were 82%–88%. The limit of quantification was found to be 1 ng/mL. Between-and within-day CV% were below 3.3% and 4.5%, respectively.
Pharmacokinetic and statistical analysis
The randomization schedule was carried out using SAS software. The data analysts were blinded for the randomization, and the bioanalytical laboratory conducting the PK analysis was blinded for the identity of the test and reference product ingested to avoid any bias arising from the open-label study design. Pharmacokinetic parameters were estimated by performing non-compartmental PK analysis using the Phoenix WinNonlin version 8.1 (Pharsight Corporation, USA) in terms of maximum concentration (Cmax), AUC, 0 to infinity (AUC0–inf), AUC, 0 to 48 hours (AUC0–48), elimination constant (Ke), Tmax, and T1/2 of the drug. Statistical analysis was carried out using SAS software version 9.4 (SAS Institute Inc, Cary, North Carolina) by performing multivariate analysis of variance (ANOVA) on the Ln-transformed and untransformed PK parameters Cmax, AUC0–48, and AUC0–inf using the linear mixed effects model with sequence and period X treatment were included as the fixed effects and subjects as the random effect. The test formulation BE with the reference product was established based on an alpha value of 0.05 (within a 90% confidence interval (CI)).
Results and discussion
The relative bioavialability of a test formulation with a reference formulation in 30 healthy male volunteers under fasting conditions was investigated in the current study in an attempt to meet regulatory requirements for declaring the test formulation to be bioequivalent to the reference product. The participation of four volunteers was terminated due to the violation of the study protocol during the second period (positive results of illicit drugs). One volunteer withdrew during the second period due to personal reasons. While women meeting the inclusion and exclusion criteria were eligible to be enrolled, only male volunteers were successfully recruited. Data obtained from 25 male volunteers who completed the study were included in the analysis with a mean ± standard deviation BMI of 25.0 ± 2.7 kg/m2 and age of 27.6 ± 9.5 years, as summarized in Table 1. Mild headache was reported in one subject, while the white blood cells count (WBC) was above the normal range for two (13.5 and 15.3 × 109 cells/L) for two participants 48 hours after the dose. However, these changes were not considered to be clinically significant.
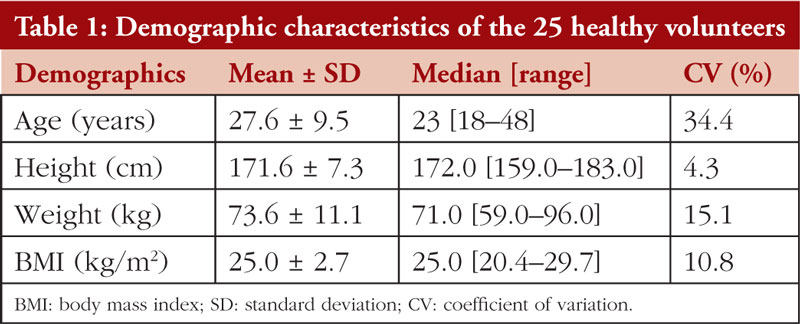
A one week washout period, sufficient to cover at least ten elimination half-lives of sitagliptin, was included in the crossover study design between the two phases to avoid any possible carryover phenomenon. The PK parameters of the reference and test formulations, i.e. AUC0-inf, AUC0-48, Cmax, Tmax, and T1/2, are presented in Table 2. Cmax, AUC0-inf, and AUC0-48 values were comparable between the test and reference formulations. Furthermore, intrasubject variabilities in Cmax were found to be 20.3% and 23.6%, AUC0-48, 14.3% and 15.6%, and AUC0-inf, 14.4% and 15.4% for the test and reference formulation, respectively. Tmax achieved comparable results with those reported in previous BE studies of a single dose of sitagliptin 100 mg [20] or 50 mg [21] tablet in American Hispanic/Latino and non-Hispanic/Latino volunteers. All PK parameters were identical to those recently reported in a Malaysian population following a 100 mg single dose of sitagliptin tablet [22]. Cmax, Tmax, and T1/2 were comparable in an Asian population receiving a single dose of sitagliptin 100 mg or 500 mg tablets alone or in combination with metformin [23–26]. Sitagliptin safety profile, tolerability, PK, and pharmacodynamics (PD) were evaluated in single doses ranging from 1.5 mg to 600 mg. The PK parameters found in these studies were in line with current results [11].
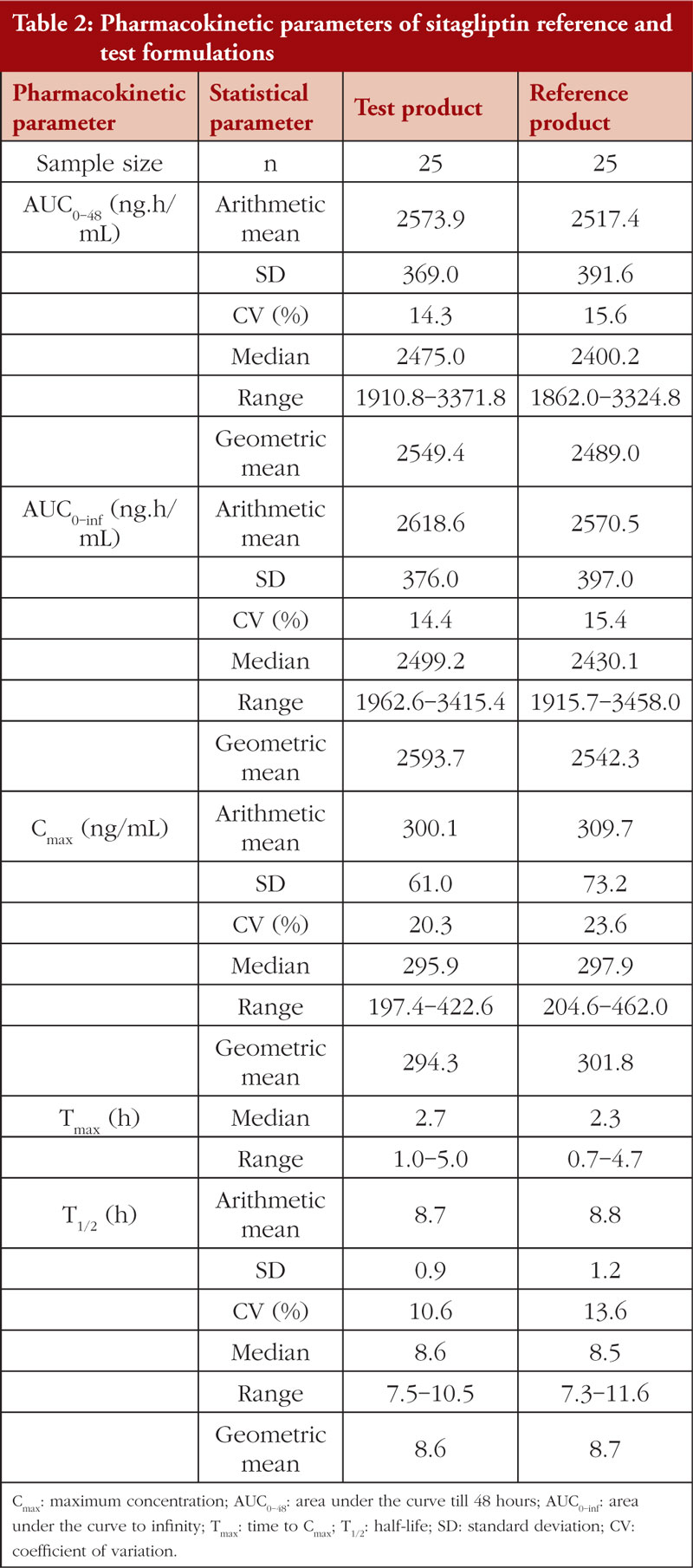
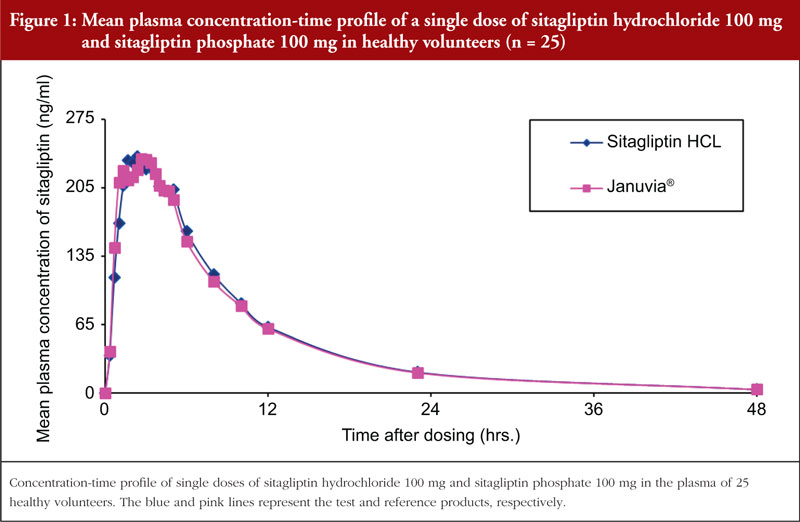
A comparison of the mean plasma concentration/time profiles of sitagliptin test and reference formulations in these 25 healthy male volunteers is represented in Figure 1 (all data points below the limit of quantification were entered in as zero and were included as zero in the calculation of means). The current results showed that the median Tmax was comparable for both formulations. Sitagliptin elimination occurred gradually over the sampling interval of 48-hours. No significant difference was found in Cmax, AUC0–inf, or AUC0–48 between the test and reference formulations based on the multivariate ANOVA statistical analysis. The 90% CIs of the Ln-transformed Cmax, AUC0–inf, and AUC0–48, were 89.2%–06.0%, 100.4%–103.5%, and 100.8%–104.0%, respectively. The two-sided 90% CIs of Ln-transformed Cmax, AUC0-inf, and AUC0–48 of sitagliptin were within the BE acceptance range of 80%–125%, as defined by the ASEAN Guideline for the Conduct of Bioequivalence Studies [27]. Therefore, the test formulation successfully met the acceptance criteria for a BE study.
The current study involved only Caucasian participants. This might influence the generalizability of the study findings in other populations. Furthermore, only male volunteers were recruited, limiting the ability to extrapolate the PK parameters to the female population. Inter-individual variability was accounted for in the randomized crossover study design, therefy limiting its potential impact on the BE of the study products. With these potential limitations, it was concluded that the PK profiles of study products in terms of absorption, distribution, metabolism, and elimination were comparable. However, no investigation of drug PD was carried out in the current study. This would be a potential topic for future studies involving racially and sexually diverse actual diabetic patient populations.
Conclusions
The present study found that single doses of the test and reference formulations, each containing 100 mg sitagliptin in the form of sitagliptin hydrochloride monohydrate and sitagliptin phosphate monohydrate, respectively produced drug concentration/time curves under fasting conditions that met regulatory requirements for declaring the products to be bioequivalent.
Funding sources
This work was financially supported by the Duopharma Biotech Berhad.
Competing interests: CWL, ES, KMY, MSS, SRSAR, KS, MZJ, SA, and AA are employees of Duopharma, RFT is an employee of ACDIMA BioCenter that was paid to perform the study for Duopharma.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Chuei Wuei Leong1, PhD
Elton Sagim1,BBiomedSc
Kar Ming Yee1, BPharm
Muhammad Shalhadi Saharuddin1, BSc
Sharifah Radziah Syed Abd Rahim1, MSc
Khairil Sabri1, BSc
Mohd Zulhairi Jamaluddin1, BSc
Shahnun Ahmad1, MBBS
Atiqah Amran1, BSc
Rabab F Tayyem2, PhD
1Duopharma Innovation Sdn Bhd, Selangor,
No. 2, Jalan Saudagar U1/16, Zon Perindustrian Hicom Glenmarie, Seksyen U1, Shah Alam 40150, Darul Ehsan, Malaysia
2ACDIMA BioCenter, Amman 11190, Jordan
References
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
2. Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X, et al. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci Rep. 2020;10(1):14790.
3. Vijayakumar TM, Jayram J, Meghana Cheekireddy V, Himaja D, Dharma Teja Y, Narayanasamy D. Safety, efficacy, and bioavailability of fixed-dose combinations in type 2 diabetes mellitus: a systematic updated review. Curr Ther Res Clin Exp. 2017;84:4-9.
4. Stumvoll M, Goldstein BJ, Van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333-46.
5. Albitar O, Harun SN, Abidin NE, Tangiisuran B, Zainal H, Looi I, et al. Predictors of recurrent ischemic stroke in obese patients with type 2 diabetes mellitus: a population-based study. J Stroke Cerebrovasc Dis. 2020;29(10):105173.
6. Dhillon S. Sitagliptin: a review of its use in the management of type 2 diabetes mellitus. Drugs. 2010;70(4):489-512.
7. Scott LJ. Sitagliptin: a review in type 2 diabetes. Drugs. 2017;77(2):209-24.
8. Karasik A, Aschner P, Katzeff H, Davies MJ, Stein PP.Sitagliptin, a DPP-4 inhibitor for the treatment of patients with type 2 diabetes: a review of recent clinical trials. Curr Med Res Opin. 2008;24(2):489-96.
9. Pfeiffer AF, Klein HH. The treatment of type 2 diabetes. Dtsch Arztebl Int. 2014;111(5):69-82.
10. Bergman AJ, Stevens C, Zhou YY, Yi B, Laethem M, De Smet M, et al. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin Ther. 2006;28(1):55-72.
11. Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther. 2005;78(6):675-88.
12. Vincent SH, Reed JR, Bergman AJ, Elmore CS, Zhu B, Xu S, et al. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab Dispos. 2007;35(4):533-8.
13. Bergman A, Ebel D, Liu F, Stone J, Wang A, Zeng W, et al. Absolute bioavailability of sitagliptin, an oral dipeptidyl peptidase-4 inhibitor, in healthy volunteers. Biopharm Drug Dispos. 2007;28(6):315-22.
14. Bergman AJ, Cote J, Yi B, Marbury T, Swan SK, Smith W, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care. 2007;30(7):1862-4.
15. Charoo NA, Abdallah DB, Bakheit AA, Haque KU, Hassan HA, Abrahamsson B, et al. Biowaiver monograph for immediate-release solid oral dosage forms: sitagliptin phosphate monohydrate. J Pharm Sci. 2022;111(1):2-13.
16. Zakowiecki D, Edinger P, Papaioannou M, Hess T, Kubiak B, Terlecka A. Exploiting synergistic effects of brittle and plastic excipients in directly compressible formulations of sitagliptin phosphate and sitagliptin hydrochloride. Pharm Dev Technol. 2022;27(6):702-13.
17. Boddu R, Vadla HC, Prathap VR, Kothamasu U, Rallabandi BC, Gannu R, et al. Development of an in vitro-in vivo correlation for sitagliptin and metformin prolonged-release tablet formulations. Turk J Pharm Sci. 2021;18(2):233-41.
18. Tsume Y, Mudie DM, Langguth P, Amidon GE, Amidon GL. The Biopharmaceutics Classification System: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur J Pharm Sci. 2014;57:152-63.
19. Drugs.com. Januvia [homepage on the Internet]. [cited 2022 Nov 29]. Available from: https://www.drugs.com/pro/januvia.html#s-34089-3
20. Fediuk DJ, Matschke K, Liang Y, Pelletier KB, Wei H, Shi H, et al. Bioequivalence of ertugliflozin/sitagliptin fixed-dose combination tablets and coadministration of respective strengths of individual components. Clin Pharmacol Drug Dev. 2019;8:884-94.
21. Migoya EM, Miller JL, Gutierrez M, Zheng W, Johnson-Levonas AO, Liu Q, et al. Bioequivalence of sitagliptin/metformin fixed-dose combination tablets and concomitant administration of sitagliptin and metformin in healthy adult subjects: a randomized, open-label, crossover study. Clin Drug Investig. 2010;30(12):855-66.
22. Loh GOK, Wong EYL, Tan YTF, Lee YL, Pang LH, Chin MC, et al. Simple and rapid LC-MS/MS method for determination of sitagliptin in human plasma and application to bioequivalence study. J Chromatogr B Analyt Technol Biomed Life Sci. 2020;1159:122337.
23. Reddy S, Ahmed I, Ahmad I, Mukhopadhyay A, Thangam S. Development and validation of a method for simultaneous estimation of metformin and sitagliptin in human plasma by LC–MS-MS and its application in a bioequivalence study. J Chromatogr Sci. 2015;53(9):1549-56.
24. Burugula L, Mullangi R, Pilli NR, Makula A, Lodagala DS, Kandhagatla R. Simultaneous determination of sitagliptin and simvastatin in human plasma by LC-MS/MS and its application to a human pharmacokinetic study. Biomed Chromatogr. 2013;27(1):80-7.
25. Thao NNN, Hieu NN, Tho DCMV, Loan TTT, Tuan ND. Development, validation, and application for simultaneous assay of metformin and sitagliptin in human plasma by liquid chromatography? Syst Rev Pharm. 2020;11(2):6-13.
26. Shi P, Liu X, Li T, Sun F-Fei, Liu Y-Ping, Liu S-Qin, et al. Pharmacokinetics and bioequivalence of sitagliptin phosphate/metformin hydrochloride tablets in healthy chinese subjects: a randomized, open-label, crossover study. Drugs R D. 2022;22(1):15-23.
27. National Pharmaceutical Regulatory Agency (NPRA). Ministry of Health Malaysia. ASEAN guidelines: the conduct of bioequivalence studies. 2015 [homepage on the Internet]. [cited 2022 Nov 29]. Available from: https://www.npra.gov.my/images/reg-info/BE/BE_Guideline_FinalMarch2015_endorsed_22PPWG.pdf
|
Author for correspondence: Chuei Wuei Leong, PhD, Formulation and R & D Technologies, Duopharma Innovation Sdn Bhd, No. 2, Jalan Saudagar U1/16, Zon Perindustrian Hicom Glenmarie, Seksyen U1, Shah Alam 40150, Selangor, Daryl Ehsan, Malaysia |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2023 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


