Trends in Saudi FDA drug approvals and GMP inspections: an observational study
Published on 2023/10/18
Generics and Biosimilars Initiative Journal (GaBI Journal). 2023;12(3):76-86
Author byline as per print journal: Ali M Alhomaidan, PhD; Mohammed Abdulaziz Alageel, MSc; Turki Abdulaziz Alrafie, MSc; Hassan Mohammed Alqarni, MSc; Ibraheem Yahya Khbrani, MSc; Dalal J Alkhamis, MSc; Mohammed F Alkhalifah, MSc; Abdualmajeed S bin Jumaiah, MSc; Mohammed A Dahhas, PhD
|
Abstract: |
Submitted: 25 September 2023; Revised: 17 October 2023; Accepted: 17 October 2023; Published online first: 31 October 2023
Introduction
All pharmaceutical goods that are commercially available in Saudi Arabia are required by law to have a marketing authorization [1]. The frequency of new medicine approvals has dramatically increased over the last decade. In Saudi Arabia, 5,498 pharmaceutical items were registered in 2011. By 2020, this number rose to 10,424. Drug approvals and the variety of active ingredients on which these new medications are based have been steadily rising over time as novel technologies are developed and introduced, regulatory frameworks change, and merger and acquisition activities fuel more consolidation. It is anticipated that these trends will persist over the coming ten years.
The use of unsafe, adulterated, or ineffective medications can lead to therapeutic failure, disease exacerbation, drug resistance, and occasionally even death [2]. This is why it is necessary to regulate the use of all pharmaceuticals. Additionally, the introduction of any unsafe or ineffective medication erodes trust in healthcare institutions, medical experts, and pharmaceutical producers and suppliers. Neither consumers nor governments should waste money on cheap, inefficient medications. To successfully oversee the production, distribution, and use of medicines in order to safeguard and advance public health, governments must set up powerful national regulatory agencies. SFDA, which was founded under Council of Ministers Resolution No. 1 from 07/01/1424 H, is responsible for assuring the correctness, quality, safety, and efficacy of medicines in Saudi Arabia, as well as the regulation and supervision of manufacturing facilities, importation, and registration of these products [3].
The task of conducting an open, prompt examination of pharmaceutical products for quality, safety, and efficacy poses challenges for pharmaceutical regulatory authorities. To establish a benchmark against which the impact of change can be measured and to develop realistic improvement initiatives, their performance in addressing that challenge should be routinely evaluated against established international qualitative and quantitative standards and recognized best practices and procedures. This will enable agencies to appraise their own performance and, ultimately, ensure patients have quick access to advanced, safe, and beneficial medicines.
GMP inspections are one of the tasks carried out by SFDA. This paper will explore some of the most prevalent flaws identified during inspections. The goal of sharing the inspection non-conformities is to give the industry the opportunity to evaluate the deficiencies found and subsequently address them as part of a self-continuous improvement effort. A company must request Saudi Food and Drug Authority (SFDA) to perform a GMP inspection visit (based on SFDA’s GMP guideline) when submitting a new product for registration and changing the location of production. Furthermore, a routine GMP inspection may be performed for manufacturers based on inspection priority for local manufacturers or to investigate complaints or recall requests.
Method
All medications that SFDA approved were identified and examined for this investigation. We reviewed all items registered between 2011 and 2021. The overall number, class, and most prevalent deficiencies of the drugs licensed each year were evaluated. Information on new molecular entities authorized by SFDA, biologicals, and generic drugs were all covered in this review. Additionally, the deficiency data pattern from the GMP inspections between 2018 and 2021 were examined. Hundred per cent of inspection deficiencies for inspections conducted between 2018 and 2021 were represented by the sample that was taken. This study primarily focused on inspection issues relating to production, material management, validation, premises and equipment, quality management, quality control, and personnel.
Results
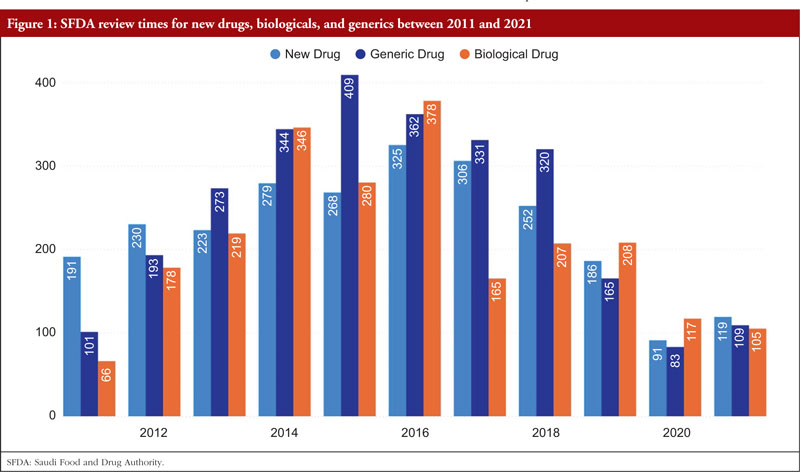
Reducing review timelines is a factor that helps pharmaceutical corporations achieve sales income more quickly while also allowing medications to reach patients more swiftly. The total number of days required by SFDA to examine new medications climbed from 191 days in 2011 to 325 days in 2016; however, it then rapidly decreased to 119 days in 2021. The total number of days required by SFDA to assess biologicals climbed from 66 in 2011 to 378 in 2016, and then fell to 105 in 2021. For all cycles of review, SFDA review times for generic pharmaceuticals grew from 101 days in 2011 to 409 days in 2015 and then decreased once again to 109 days by 2021,as shown Figure 1.
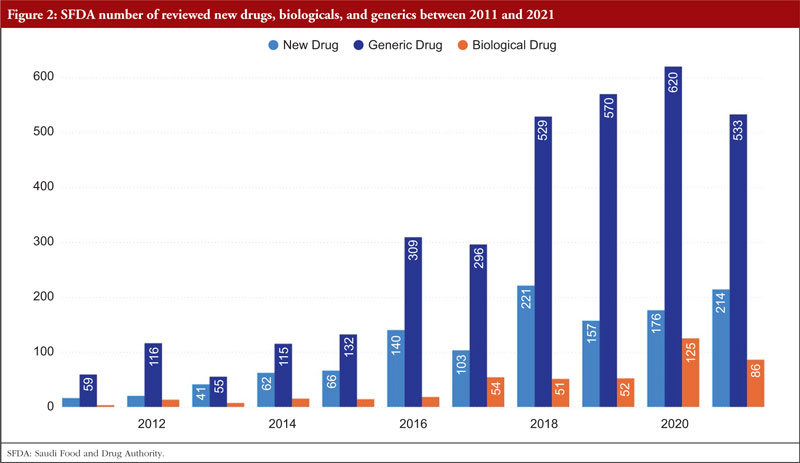
Over time, SFDA’s list of approved items has grown significantly. There were 214 new medications licensed in 2021 compared to 21 in 2011. With the number of generic pharmaceuticals approved annually rising from 59 in 2011 to 533 in 2021, the generic drugs pathway has been extensively employed. From 4 items in 2011 to 86 in 2021, the number of approved biologicals also grew, as shown Figure 2.
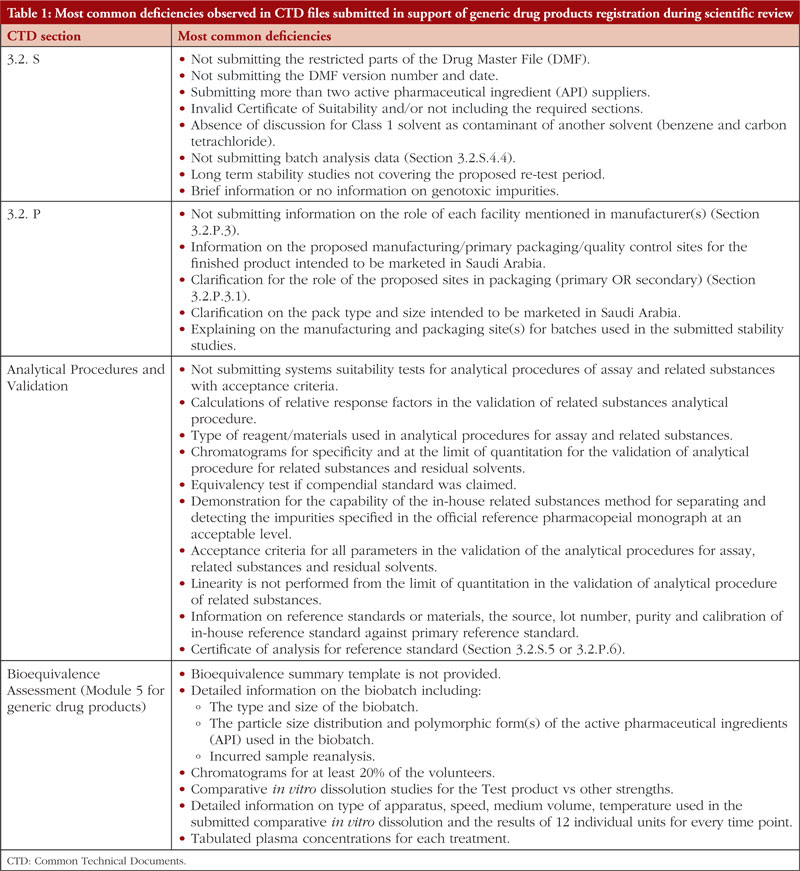
If SFDA did not approve a medicine during the first review cycle, it was mandated to send to the applicant an evaluation report outlining the shortcomings that the sponsor must correct. Here we list the most frequent deficiencies reported in the Common Technical Documents (CTD) files that were submitted in response to generic drug product registration during the scientific review of data as part of SFDA’s ongoing effort to streamline the review process and lower the number of deficiencies cited for the applications. These deficiencies are presented in Table 1. However, this list does not include every defect that has been found.
In total, 118 GMP inspections were conducted in 2018. Three producers of human-targeted pharmaceuticals and four veterinary medicine manufacturers were among the seven whose operations were suspended in 2018. Eighty-four of the 118 GMP inspections that were conducted in 2018 had major or critical defects. Twenty of the inspections with major/critical problems took place in Saudi Arabia, while 64 were performed abroad.
Twenty-three of the 124 GMP inspections conducted in 2019 were in Saudi Arabia, while 101were overseas. 2019 saw the suspension of eleven manufacturers. There were also 590 major deficiencies and 94 critical deficiencies identified in 2019.
2020 saw 43 GMP inspections, of which 12 were conducted abroad and 31 in Saudi Arabia. This reduction can be attributed to the COVID-19 pandemic. In total, 237 major flaws and 25 critical deficiencies were identified.
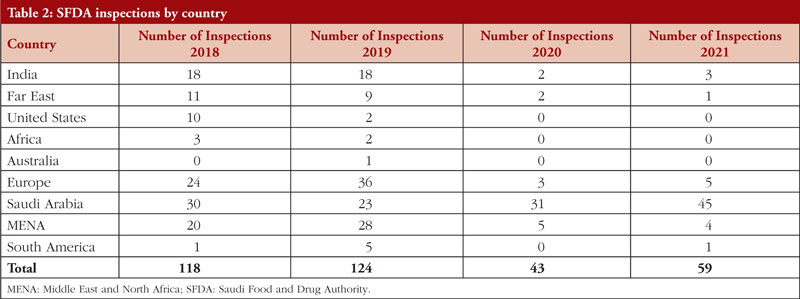
In 2021, 59 GMP inspections were conducted, with 14 being conducted abroad and 45 in Saudi Arabia (5 in Europe, 1 in China, 3 in India, 1 in South America, and 4 in the MENA region). There were 207 major deficiencies and 36 critical deficiencies. Between 2018 and 2021, SFDA performed 344 GMP inspections both within and outside of Saudi Arabia. The number of drug product inspections SFDA conducted between 2018 and 2021 is shown in Table 2 per nation.
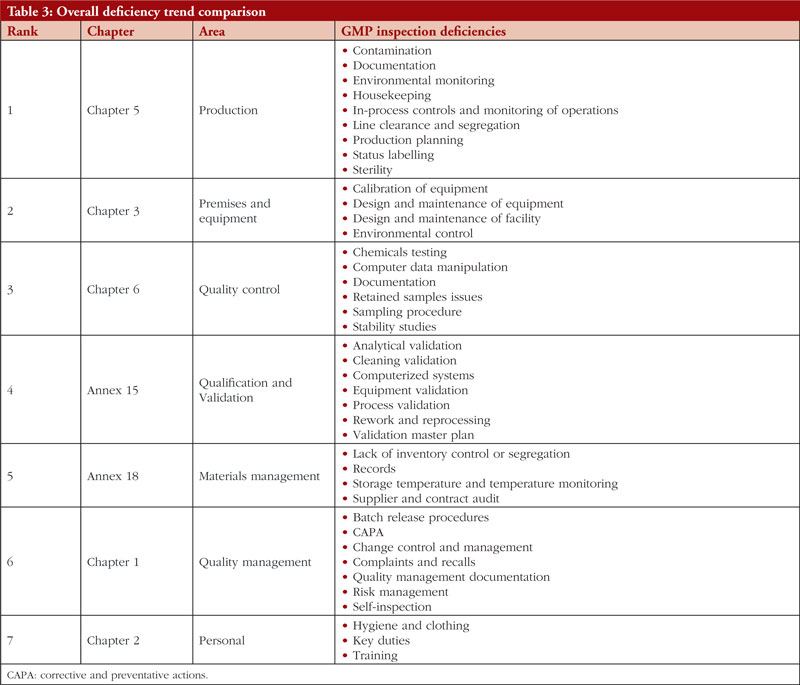
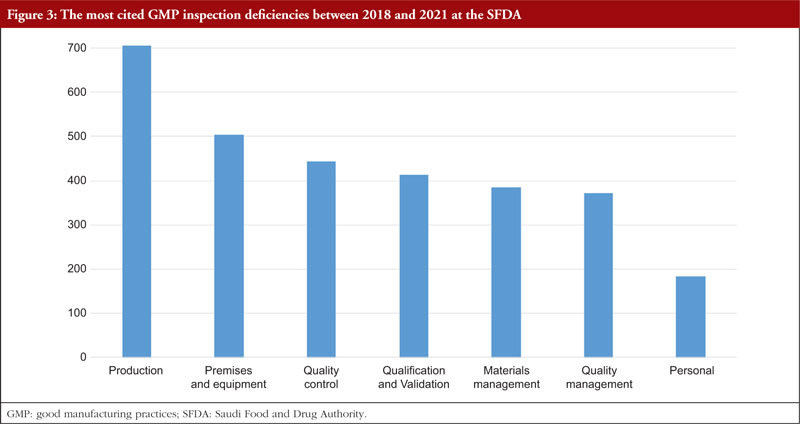
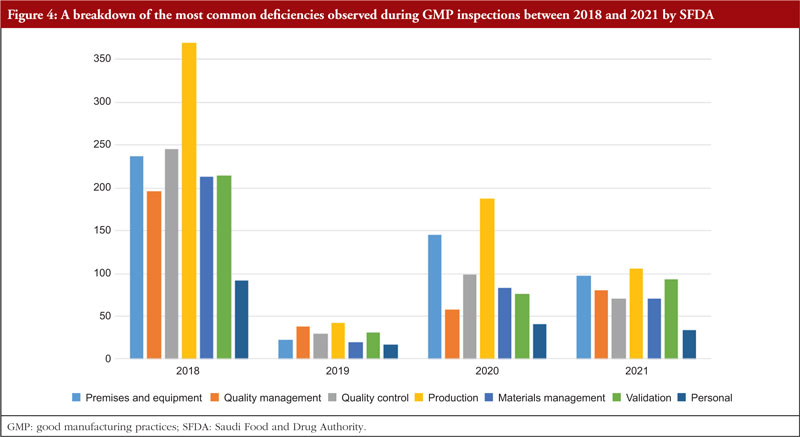
The trend is shown in Table 3, along with all classifications of deficiencies found in the top 7 chapters and annexes between 2018 and 2021. This data was derived from the GMP inspection reports. Between 2018 and 2021, the following, in order of frequency, are the most typical GMP flaws: Production, premises and equipment, quality control validation, materials management, quality management, and personnel. The most frequent flaws and a breakdown of them are shown in Figures 3 and 4.
Discussion
A crucial component of medication control is the registration of pharmaceuticals, commonly referred to as product licensing or marketing authorization. SFDA is required to license every drug that is sold, used, and distributed in Saudi Arabia. However, the quality and safety of products are assured by GMP inspection of the site, laboratory quality control testing and scientific examination of products prior to registration to ensure all marketed pharmaceutical goods fulfill the standards for safety, efficacy, and quality.
An effort is made to understand domestic and international trends related to drug evaluation as well as to obtain scientific knowledge. The most recent scientific discoveries serve as the foundation for evaluating pharmaceuticals. However, it is also important to take into account the context of the research studies that have been undertaken, their historical context, and previous choices regarding similar drugs. The decision on approval or rejection is based on a scientific and objective evaluation of the evidence supplied, taking into account the objective evaluation of benefits and hazards together with consideration of the patient’s perspective as well. Attempts are made to find the best solution for any issues that may arise during the new medication evaluation process by giving the applicant guidance and seeking understanding from many relevant parties after describing the cause and grounds for that specific concern. Efforts are invested in fostering mutual understanding with the applicant to support a smooth review, always keeping in mind the necessity to maintain open and honest communication at all times in order to secure a fair and unbiased stance.
SFDA approves over 900 medications annually. Many national regulatory bodies continue to confront difficulties as a result of resource limitations, despite their efforts to improve regulatory performance and work towards quicker clearance timeframes [2, 4]. The necessity for national regulatory agencies to use regulatory convergence initiatives, collaborative registration processes, and functional continental networks to carry out their regulatory mandates are brought on by growing workloads, developing technologies, and a lack of competence [5].
As can be observed in Table 1, this is by far the most active area in terms of common flaws and remarks made about applicants for generic drugs. The incidence of flaws illustrates how crucial the information regarding the controls suggested for the regular release and stability analysis of the drug product is. All suggested specifications (tests, procedures, and criteria) should be supported by solid scientific and regulatory reasoning, according to the applicants. This is not a complete list of the shortcomings in the sections on drug product release and stability, as was noted at the beginning of the study. However, the authors have made an effort to explain the fundamental causes of frequent deficiencies in stability testing and the control of therapeutic product. We want to explain why these flaws are mentioned and show how pharmaceutical development studies performed during the product’s original development could decrease the likelihood that these flaws become problematic.
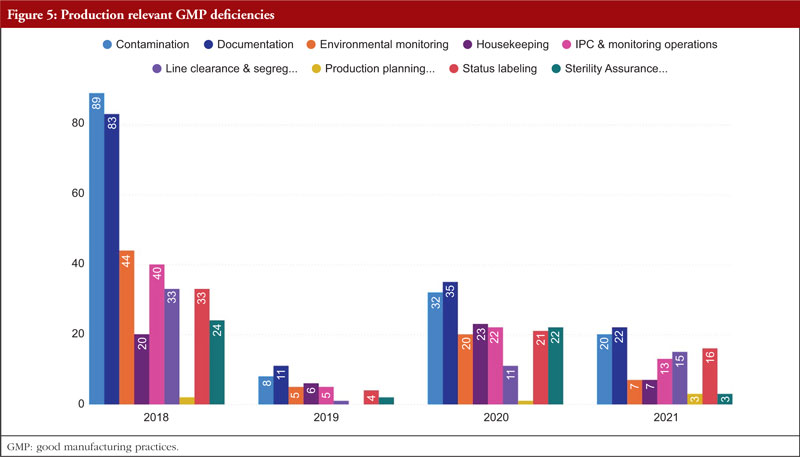
In regard to the faults found during the GMP inspection, we will begin by discussing Chapter 5, Production, as it is the source of the majority of the overall deficiencies, severe deficiencies, and major deficiencies. Chapter 5 is mentioned in almost 23% of all defects between 2018 and 2021. To produce high-quality products that satisfy regulatory criteria, production processes must adhere to GMP. Depending on the type of manufacturing undertaken, GMP for manufacturing operations includes criteria for the avoidance of cross-contamination during production, process validation, and environmental conditions. These GMP are required and should be supported by established and approved procedures and documentation for production activities [6]. Figure 5 shows production relevant GMP deficiencies.
During manufacture, sterile pharmaceuticals are more susceptible to contamination from particulate, pyrogenic, and microbiological sources. Special measures must be taken during the production of these products due to the health risk posed by using contaminated sterile supplies. All concerned personnel must possess the necessary knowledge, expertise, and training. Quality control is crucial, and production must adhere to rigorously established and approved preparation and sterilizing procedures. The environment in which aseptic procedures (such as equipment setup and filling) are carried out must be maintained in a controlled way and at an adequate quality in order to ensure product sterility. The handling of sterile materials before, during, and after the filling and closing, processes is an example of an activity that takes place in a crucial area [7].
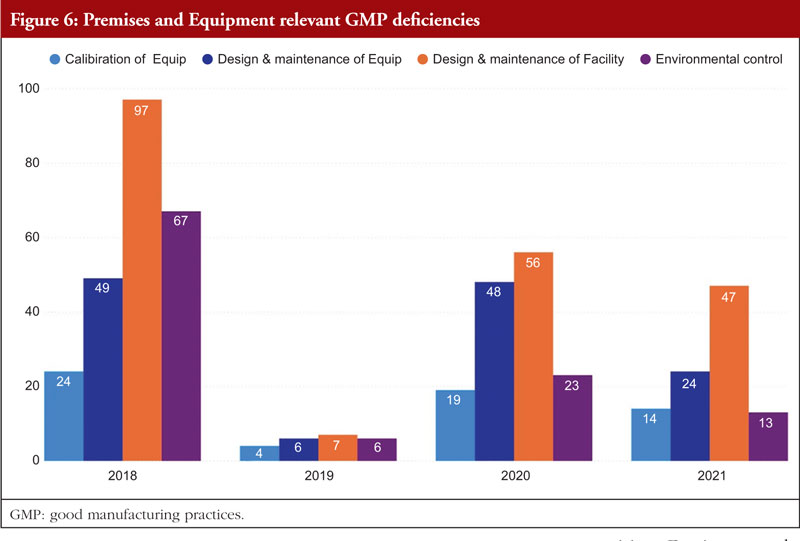
Chapter 3, Premises and Equipment, comes in second to Chapter 5 and is mentioned in almost 16% of all deficiencies observed between 2018 and 2021. The first requirement for ensuring that products are created safely is building design. Buildings must be of adequate size and designed in such a way to support these activities in order to allow cleaning, maintenance, and proper operations. In order to avoid cross-contamination between different sections, drug product containers, closures, labels, in-process materials, or drug products, manufacturers should have enough room for the orderly positioning of equipment and supplies. The unidirectional flow of parts, drug product containers, closures, labels, in-process materials, and drug products from minimally regulated regions to those with greater control should also be developed to reduce the risk of contamination. Each region of the production area that these materials pass through is under closer observation and supervision. For instance, the section where the bulk product is packed into containers will have more environmental controls than the loading dock at which raw materials and components are received [7]. Figure 6 shows Premises and Equipment relevant GMP deficiencies.
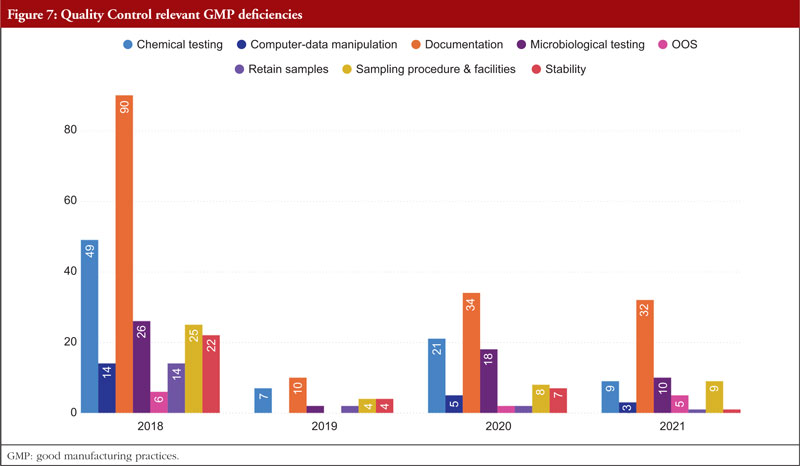
Third among the most often mentioned chapters and annexes between 2018 and 2021 is Chapter 6, Quality Control. The organization, documentation, and release procedures that guarantee that the required and appropriate tests are performed, and that materials or products are not released for sale or supply until their quality has been judged satisfactory are all concerned with quality control deficiencies. These deficiencies include sampling, specifications, testing, as well as organization, documentation, and release procedures. Quality control is not limited to laboratory procedures. It must be considered in all decisions that can have an impact on the product’s quality. It is thought that the successful operation of quality control depends on its independence from manufacturing. A quality control department should be present in every manufacturing site. The head of this department should have the necessary training, experience, and access to one or more control laboratories. It should be separate from other departments. To guarantee that all quality control procedures are successfully and consistently carried out, sufficient resources must be provided. Figure 7 shows Quality Control relevant GMP deficiencies.
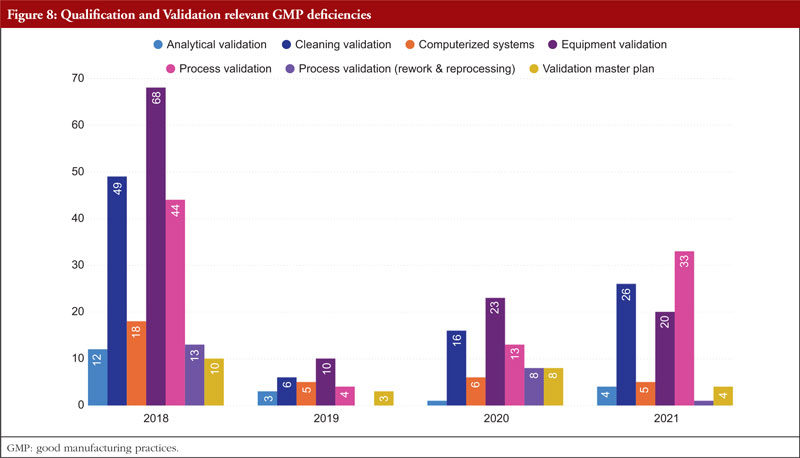
The next item on the list is Annex 15, Qualification and Validation. The validation of analytical results, cleaning validation, validation of computerized systems, validation of equipment, rework and reprocessing, and validation of processes are the most commonly referenced paragraphs. Any GMP facility, utility, piece of equipment, or process must be qualified and validated in the pharmaceutical industry. The goal of qualification and validation activities is to produce written proof that a facility’s utilities (such as water, gases, and air) and processes are created and run in accordance with SFDA’s GMP standards. Figure 8 shows Qualification and Validation relevant GMP deficiencies.
Appropriate certification and validation procedures must take place in order to guarantee that the facility, utilities, process, and equipment have been planned, constructed, deployed, and operated as intended. Design lays the groundwork for the overall success of a qualification and validation programme. For something to be built, installed, or manufactured, certain attributes must be present. These requirements are prescribed by specifications. The item being constructed, installed, or manufactured must meet specific requirements. You can categorize requirements as either user requirements or functional requirements. Specification and design requirements are focused on the characteristics that are essential to the quality of the final product and patient safety, and these criteria may be stated in the requirements and specification papers.
The manufacture and packaging of APIs and pharmaceutical goods must comply with regulations, which forbid both environmental and drug-related contamination. Equipment for washing and cleaning must be chosen and utilized with care to avoid becoming a source of contamination. The goal of the cleaning validation is to confirm the cleaning process’s efficacy in removing product residues, preservatives, degradation products, excipients, and/or cleaning agents, as well as in controlling any microbiological contamination. The manufacturer must also make sure that there is no possibility of cross-contamination of active components. To avoid impurities carrying over and building up, equipment needs to be cleaned at the proper intervals. The maintenance of cleanliness levels in the plant is partially ensured by a written cleaning process validation procedure [7].
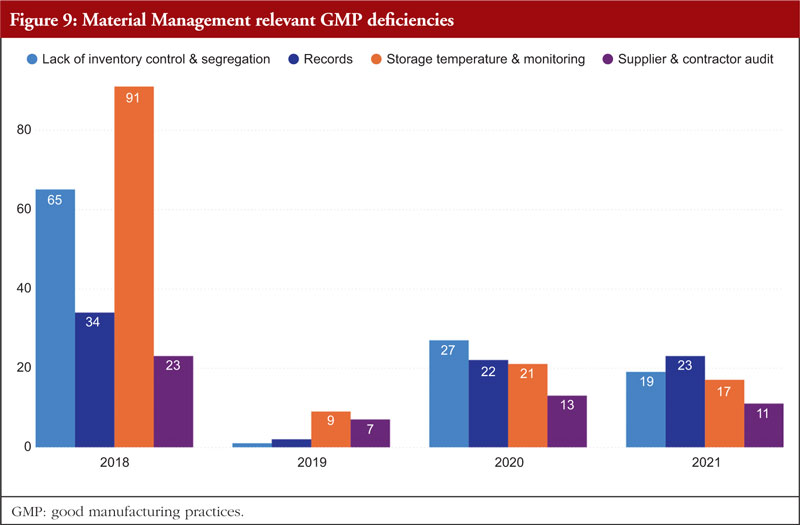
Material Management is next. The most frequently identified shortcomings include a lack of inventory control or segregation, problems with recordkeeping, problems with storage temperature and temperature monitoring, and audits of supplier contracts. Every document related to the shipping or delivery must be checked after receiving any material to verify accuracy and completeness. Excipients and active pharmaceutical components should only be sourced from recognized vendors for starting materials. Starting materials used in the production of APIs should be received in accordance with specific requirements in addition to those for starting materials used in medicinal products. It is best practice to confirm that incoming materials are correct, test them (if necessary), and then release them before combining them with current supplies. Figure 9 shows Material Management relevant GMP deficiencies.
Written sampling protocols that include the identities of the person(s) authorized to take samples, the techniques and tools to be used, the quantities to be taken, and any measures to be maintained to prevent contamination of the material or any degradation in its quality are preferable. All materials should have established sampling protocols and strategies that have been approved by the quality unit. Materials should be kept off the ground and placed in storage facilities with enough capacity to allow for orderly storage as well as to make cleaning and inspection easier. Any necessary label storage conditions, such as regulated temperature and humidity, should be maintained in storage locations. The monitoring equipment should be frequently examined and calibrated, and temperature monitoring records should be kept.
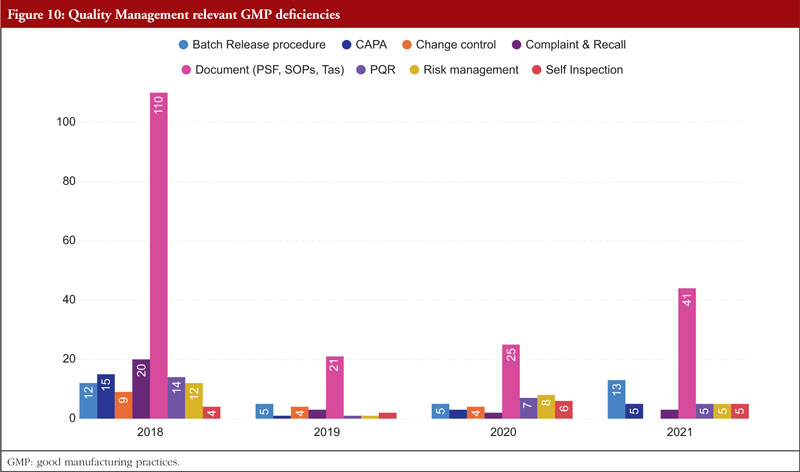
The next chapter is Chapter 1, Quality Management. The most frequently stated problems relate to batch release policies, CAPA, change control and management, complaints and recalls, quality management documentation, risk management, and self-inspection. Senior management is ultimately in charge of making sure a strong pharmaceutical quality management system is implemented that meets the quality goals. They are in charge of ensuring that the company’s duties, obligations, and powers are clearly stated, disseminated, and put into action. Figure 10 shows Quality Management relevant GMP deficiencies.
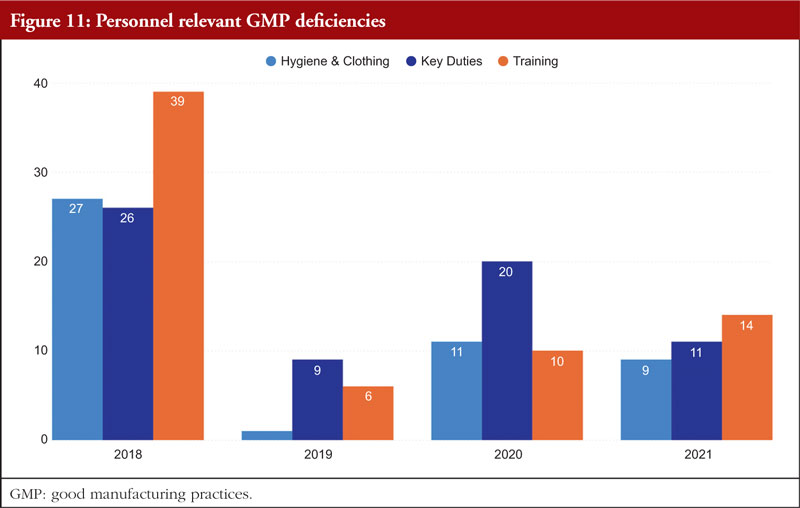
Chapter 2, Personnel, is the final chapter. Personnel must be properly educated, trained, and experienced in order to carry out their given responsibilities in accordance with GMP. A product may be declared adulterated by SFDA as a result of inadequate training. In such cases, the manufacturer would be prohibited from selling or distributing it. It is necessary to complete both GMP and specialized work task training. It is the responsibility of managers or supervisors to specify the training required for a certain position in a list of the necessary paperwork and educational programmes, such as a job curriculum. The curriculum can specify the scheduling requirements if frequent retraining is necessary; for example, as is the case with aseptic procedures. The training that must be completed before an employee can carry out a certain task should be specified in the curriculum, and the training completed should be formally recorded. Figure 11 shows Personnel relevant GMP deficiencies.
The provision of training should not be seen as a one-time event that occurs only when an employee is hired. When processes, batch records, and/or test methods are changed, or a task has not been completed recently, training pertaining to job tasks is required. Similar to this, when a worker’s position changes within an organization, his or her training, education, and experience should all be reviewed to ascertain what needs to be improved in order to accomplish the new duties. A current résumé or job history might be kept in the employee’s training file to show changes in work experience and education [7].
Conclusion
This paper examined the trends in SFDA medication approvals and GMP inspections from 2011 to 2020 and provided some insight into the most prevalent flaws in CTD dossiers that were examined for licensing purposes. The findings underline the significance of manufacturers leveraging such reports to pinpoint common sources of errors and strategize to prevent them in the future. By doing so, factories can enhance the quality and safety of their products while also maintaining compliance with regulatory standards. Furthermore, manufacturers can harness the insights gained from these generated reports to facilitate training for their employees. By analysing the documented errors and deficiencies, they can develop targeted training programmes to address these specific issues. This approach ensures that the workforce is equipped with the knowledge and skills necessary to avoid making similar mistakes in the future. By learning from past errors, manufacturers can create a culture of continuous improvement and maintain a proactive stance in preventing potential issues from arising. This integration of data-driven insights into training initiatives establishes a cycle of enhanced performance, minimized errors, and heightened product quality, ultimately contributing to the reputation and success of the company.
Disclaimer
The views expressed in this paper are those of the authors and do not necessarily reflect those of the SFDA or its stakeholders. Guaranteeing the accuracy and validity of the data is the sole responsibility of the research team.
Competing interests: The authors declare no competing interests for this work.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Ali M Alhomaidan, PhD
Mohammed Abdulaziz Alageel, MSc
Turki Abdulaziz Alrafie, MSc
Hassan Mohammed Alqarni, MSc
Ibraheem Yahya Khbrani, MSc
Dalal J Alkhamis, MSc
Mohammed F Alkhalifah, MSc
Abdualmajeed S bin Jumaiah, MSc
Mohammed A Dahhas, PhD
References
1. Saudi Food and Drug Authority. Royal Decree No. (M/108) dated 22/8/1441 AH Council of Ministers Resolution No. (534) dated 21/8/1441 AH. Law of Pharmaceutical Products, Pharmaceutical Establishments, and Herbal Preparations. 2020. [homepage on the Internet]. [cited 2023 Oct 17]. Available from: https://laws.boe.gov.sa/BoeLaws/Laws/LawDetails/3d191772-e60b-4925-b5e7-aba50097641f/1
2. Darrow JJ, Avorn J, Kesselheim AS. FDA approval and regulation of pharmaceuticals, 1983-2018. JAMA. 2020;323(2):164-76.
3. Saudi Food and Drug Authority. Royal Decree No. M/6 dated 25/1/1428 Cabinet Resolution No. 31 dated 1/24/1428, ‘The Law of The Saudi Food and Drug Authority’, 2007.
4. Tsiftsoglou AS, Ruiz S, Schneider CK. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27(3):203-11.
5. Auclair JR. Regulatory convergence for biologics through capacity building and training. Trends Biotechnol. 2019;37(1):5-9.
6. Saudi Food and Drug Authority. Guide to good manufacturing practice for medicinal products. 2022 [homepage on the Internet]. [cited 2023 Oct 17]. Available from: www.sfda.gov.sa/sites/default/files/2022-08/SFDAGMPGuideline2022
7. The Saudi Food and Drug Authority. Guide to good manufacturing prac-tice for medicinal products. 2023. [homepage on the Internet]. [cited 2023 Oct 17]. Available from: https://www.sfda.gov.sa/sites/default/files/2023-01/SFDA-GMP-Guideline%20_0.pdf
|
Author for correspondence: Ali M Alhomaidan, PhD, Senior Consultant on Research and Innovation, Saudi Food and Drug Authority, 4904 Northern Ring Branch Road, Riyadh, Saudi Arabia |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2023 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


