Biosimilars in Italy: what do real-world data reveal?
Published on 2017/06/12
Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(3):114-9.
Author byline as per print journal: Assistant Professor Gianluca Trifirò, MD, PhD; Ylenia Ingrasciotta, MSc; Ilaria Marcianò, MSc; Armando A Genazzani, DPhil, MD
|
Abstract: |
Submitted: 18 April 2017; Revised: 18 July 2017; Accepted: 19 July 2017; Published online first: 1 August 2017
Introduction
In recent years, national health services (NHSs) across the globe have been faced with ever increasing expenditures due to the marketing of high-cost innovative drugs. In Italy, the amount spent on pharmaceuticals by public hospitals almost doubled from Euros 2,124 million in 2011 to Euros 4,095 million in 2015 [1]. This trend is unlikely to stop as there is an increasing human life expectancy (from 1970–2010, the average life expectancy increased at an annual average rate of 0.21 for males and 0.17 years for females [2]), and the constant development of highly innovative therapies such as biological drugs.
Biological drugs contain one or more active substances, produced or extracted from biological systems or via biotechnological procedures [3, 4]. Biological drugs include hormones, growth factors, interleukins, interferons, vaccines, monoclonal antibodies and fusion proteins. They have led to a dramatic change in the management of several high burden diseases in different therapeutic areas, such as rheumatology (e.g. rheumatoid arthritis and ankylosing spondylitis), dermatology (e.g. psoriasis), gastroenterology (e.g. ulcerative colitis and Crohn’s disease), and oncology. These drugs selectively target specific proteins which play a key role in the disease progression or the tumour growth [5], thus offering substantial benefits in terms of response rate and quality of life [6].
With these therapeutic innovations, NHSs of western countries face new challenges. They must find a balance between economic sustainability and ensuring market access to innovative medicines. Research and development also needs to be rewarded financially. In order to ensure the sustainability of health systems where payers require the value of innovation to be demonstrated, strict cost containment strategies must be adopted [7].
When chemically synthesized drugs were considered, the introduction of generics represented a great opportunity for NHSs to achieve relevant savings. In the US in 2015, around 90% of all prescriptions were for generic drug products [7]. In European countries in the same year, the generics market share was more heterogeneous. It ranged from around 5% in Greece, to 70% in The Netherlands [8]. However, when biological drugs were considered, there are more hurdles to overcome to get access to markets. This is mainly due to the fact that biologicals are highly reliant on their production process, which can lead to subtle differences in the product quality. Biosimilars are biological drugs containing a version of the active substance of an already approved original biological drug (reference product, whose patent has expired) and, due to the subtle differences, are required to demonstrate their biosimilarity to the corresponding reference product, in terms of: quality characteristics, biological activity, safety and efficacy, based on a comprehensive comparability exercise [4, 9].
In general, biosimilars are considered as comparable, effective and safe therapeutic alternatives to their reference products [10]. They can provide around 20–30% purchase cost reduction in comparison to the reference product [11]. Such cost reductions can reach significantly higher percentages (69% in 2015) where a higher uptake of biosimilars occurs as demonstrated in the case of infliximab in Norway [12]. In cases such as these, tendering is usually the preferred purchasing tool of health services [13]. Overall, this means that biosimilars may represent a valid opportunity for additional cost savings.
Biosimilars in Italy: an overview of dispensing and monitoring
In 2015, the 30 most costly molecules to Italy’s hospitals included drugs that did not have biosimilars available yet, i.e. adalimumab, and drugs whose corresponding biosimilar is already on the European market, i.e. etanercept, insulin glargine, somatropin, epoetin alfa. Biosimilar uptake in Italy was relatively low when compared to countries such as Norway. Uptake was also widely variable and varied for different drugs. For example, it varied from 0.3% for biosimilar follitropin-alfa, to 31.2% for biosimilars of erythropoiesis-stimulating agents (ESAs), in terms of defined daily dose (DDD) per 1,000 inhabitants of the total of the therapeutic class. However, an increasing trend in biosimilar consumption was observed in comparison to the previous year (+49.0% for biosimilar ESA, +21.5% for biosimilar somatropin and +16.5% for biosimilar granulocyte colony-stimulating factors [G-CSFs]) [14].
In the preliminary ‘Italian report on Drug use, 2016’ (January to September) a specific indicator to monitor the appropriateness of biosimilar ESA use was developed, highlighting an increasing number of naïve patients starting ESA therapy with a biosimilar (+20.2%, compared to the same period of the previous year) [1]. This indicator reflects the recommendation from the Italian Medicines Agency (Agenzia Italiana del Farmaco, AIFA), which clearly outlines that biosimilars are the preferred therapeutic alternative to be used to ensure economic savings, especially in naïve patients (both never-treated and previously exposed patients with an adequately long wash-out period), as no scientific or regulatory reasons justify the use of a more expensive product in these patients [4]. However, the AIFA recommendations also state that, whenever a patient is successfully treated with a biological drug, and this is substituted with another drug (even a cheaper one), the corresponding responsibility is that of the individual prescribing physician [4]. In practice, this discourages substitution. Automatic substitutions of reference products and biosimilars by healthcare professionals other than the prescribing physician are not allowed.
The Italian health service is run at the regional level. This allows the adoption of different loco-regional health interventions to influence the uptake of the most cost-effective molecule (usually the biosimilar over the reference product), but no systematic monitoring of biosimilars in clinical practice has been put in place at a national or regional level. Even though some biosimilars are subjected to AIFA monitoring registries, the corresponding data have never been shared, nor has any study been conducted using such registries. However, in some Italian regions specific web-based prescription monitoring systems for biologicals have been set up but no results have been published to date.
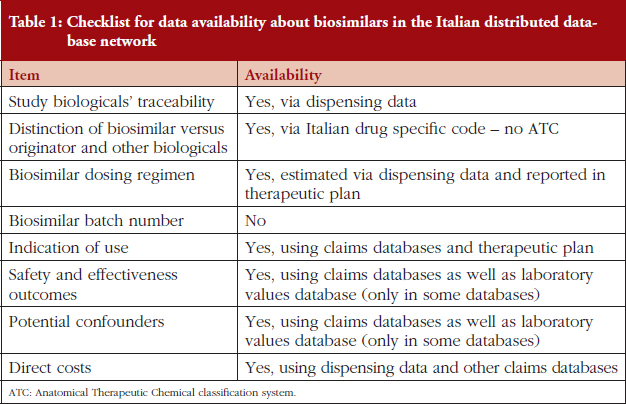
In this complex context, the drug-utilization studies may represent the first step of post-marketing assessment of biosimilars in real-world settings. The ideal data source for such assessments should allow:
a) Tracing biological drugs for which biosimilars are marketed.
b) Distinction between biosimilars and originators (both reference products and biological drugs still covered by patent), as the Anatomical Therapeutic Chemical classification system identifies the specific molecule but does not distinguish between the medicinal products and the drug-specific name is therefore necessary.
c) Plus: identification of information about biosimilar dosing regimen, batch number, indication of use, effectiveness outcome as investigated in pre-marketing randomized clinical trials (RCTs), safety outcomes as reported in risk management plans, potential confounders for effectiveness and outcome analysis, direct costs of the biosimilars for economic evaluations.
The Italian administrative healthcare databases are a valid source to explore the treatment pattern of biosimilars in different geographic areas over time, as well as the short- and long-term comparative effectiveness and safety of biosimilars versus originators in naïve users and switchers, together with the economic impact of biosimilars. Several observational studies have been conducted throughout Italy over time to explore the pattern of use of the available biosimilars and corresponding originators, as well as their comparative effectiveness [15–19].
This paper provides an overview of the available real-world data on the pattern of biosimilar use and the comparative effectiveness of the biosimilars and their corresponding originator biological drugs. The data were collected in Italy outside the constraints of traditional clinical trials.
Real-world data in Italy
The Italian Ministry of Health recently funded the 4-year project entitled ‘Assessment of short- and long-term risk-benefit profiles of biologicals through healthcare database network in Italy’ (RF-2010-2320172). This aimed to create a network of administrative healthcare databases from different Italian local health units (LHUs) and regions, to evaluate the pattern of use and the comparative safety and effectiveness of biological drugs for which biosimilars were available on the Italian market when the project was approved, i.e. ESAs, G-CSFs, somatropin. Six centres participated in the project: Caserta, Treviso and Palermo LHUs; and Tuscany, Umbria and Lazio regions, covering a total population of 14,133,687 persons (around 25% of the whole Italian population), see Figure 1. Various healthcare claims databases, linkable through unique and anonymous patients’ identifiers, were made available from the participating centres: demographic and mortality registries, emergency department visits and hospital discharge diagnosis, drug dispensing, healthcare service payment exemptions, outpatient diagnostic tests and specialists’ visits and, in some cases, also laboratory findings, see Table 1. From these sources, more than 65,000 ESA users, more than 30,000 G-CSF users, and almost 7,000 somatropin users in the years of 2009–2014 were retrieved.

In addition, two different population-based drug-utilization studies analysed the switching patterns between biosimilars and originator ESAs, using the administrative healthcare databases from the Sicilian LHU of Messina in the period January 2010 to May 2011 [20], and those from the Umbria region in the period July 2011 to December 2014 [19], respectively. One retrospective study analysed data from 11 Italian dialysis centres, from 2011 to 2014, to evaluate the effect of switching from originators to biosimilar ESAs [21].
Loco-regional healthcare interventions in Italy and biosimilar uptake
To date, two drug-utilization studies have been published in the context of the Italian Ministry for Health project (RF-2010-2320172). These were aimed at evaluating the prescribing patterns of ESAs and G-CSFs (both biosimilars and originators) in the participating Italian centres where different health policy interventions promoting biosimilar use were implemented [14–16]. Considering the uptake of biosimilars, Ingrasciotta Y et al. highlighted an increasing trend in biosimilar ESA use in all centres considered from 2009 to 2013 (overall, +33.6%) [15]. Heterogeneity in uptake was observed in the different centres, with a greater increase being observed in Treviso (up to 45.0% of biosimilar ESA users on total ESA users) than in the Caserta LHU (from 7.5% to 22.9%) during the study years. Similarly, an increase in the proportion of biosimilar G-CSF users over time was described by Marcianò I et al. Here, there were 0.2% of G-CSF users in 2009 and 66.2% in 2014, together with a notable difference between individual centres [16]. Ongoing analyses from the same project documented a similar trend for somatropin in the same study years. However, the biosimilar of this drug accounts for a much lower proportion of the total somatropin use (8.0%).
Potential reasons for such variability in biosimilar uptake may be ascribed to the different healthcare policy interventions promoting biosimilar use adopted at a loco-regional level. In some centres, naïve users must be dispensed a biosimilar; in others, clinicians are requested to justify any prescription of drugs other than the cheapest and are otherwise charged with the cost of the prescribed drug; and in others, achievement of predefined thresholds of biosimilar consumption assures economic incentives. These policies, as well as different tender procedures for the purchase of originators and biosimilars by public hospitals, the scepticism surrounding biosimilars, and the differences in patients’ access to biological drugs, may all have influenced the uptake of biosimilars in regions across Italy.
Switching patterns in Italy: biosimilars and originators
As mentioned previously, AIFA does not allow automatic substitution of a prescribed biological drug with another one, even if cheaper [4], probably because the safety of the substitution of biosimilars and originators is not supported by robust studies [22]. However, switching between different active pharmaceutical ingredients belonging to the same therapeutic class appears to be quite common in clinical practice. This generally concerns switching from one originator to another.
The analysis of the switching patterns of the Italian project (RF-2010-2320172) revealed that, during the first year of treatment, switching was frequent. This was both among different G-CSFs (20.3% of naïve G-CSF users [16]) and among different ESAs (17.0% of naïve ESA users [15]). In general, switching occurred most frequently from biosimilars to reference products, from other ESAs still covered by patent to biosimilars, and between originators in both patients affected by chronic kidney disease (CKD) (17.0%) and those with chemotherapy-induced anaemia (17.4%), see Figure 2. These results support previous data from a drug-utilization study in southern Italy [20] where switching between ESAs was also very frequent (21.8% of incident users, during a 1.5 years observation period). Among switchers, 27.0% switched more than once and switching back accounted for 20.7%. Furthermore, the most frequent switches occurred towards the reference product (32.9%). An additional real-world study on ESAs, using administrative healthcare databases from the Umbria region, documented that 12.2% of incident ESA users having at least two prescriptions of ESAs, experienced a switch during a 48-month observation period. The switching practice equally affected all ESAs and occurred most frequently between originators. The probability of switching increased with the duration of therapy, from 15% of incident users switching within the first year of treatment, up to 25% within two years [19].

Switching practices concern all biological drug users equally, not only those using biosimilars [15, 16, 19, 20]. This suggests that it is not influenced by the drug cost or by evidence provided by the comparability exercise. Furthermore, a switch between different biological drugs of the same therapeutic class is not associated with adverse events. Ebbers H et al. [23] reviewed data from clinical trials and pharmacovigilance databases, identifying almost 60 studies that evaluated the safety of switching between different ESAs or G-CSFs or somatropin (regardless of whether biosimilars or originators). Here, no safety issues were described following switching to and from different biological drugs (even though the studies were too short to evaluate long-term safety) [23].
Comparative effectiveness of biosimilar versus originator in an Italian post-marketing setting
According to AIFA, ESAs are prescribed to patients affected by chronic kidney disease (CKD) with haemoglobin (Hb) levels lower than 11 g/dL, and in the case of chemotherapy-induced anaemia with Hb levels lower than 10 g/dL. In both conditions, haemoglobinaemia ranges between 11–12 g/dL, avoiding an increase higher than 2 g/dL over a 4-week period [24].
To date, four pre-marketing clinical trials have been conducted on patients with CKD-related anaemia, and no differences in achieving the target Hb value after 12, 24, 28 or 56 weeks of treatment were found between biosimilar and reference product [25–28]. Furthermore, a meta-analysis of RCTs, comparing the efficacy and safety of ESAs in CKD patients, highlighted a lack of evidence suggesting the superiority of any ESA formulation [29]. Only one randomized study was conducted to evaluate the efficacy and safety of 12-week treatment with biosimilar epoetin alfa in chemotherapy-induced anaemia due to solid tumours [30]. On average, less than 500 patients were enrolled in each pre-marketing RCT on biosimilar ESAs.
With respect to post-marketing assessment, several observational studies have proven the effectiveness of epoetin zeta, darbepoetin alfa, and biosimilar epoetin alfa, using real-world data but they have not been compared to originators [31–33]. Only one European multicentre retrospective study compared the real-life clinical effectiveness and safety of biosimilar epoetin alfa versus darbepoetin alfa in patients with chemotherapy-induced anaemia, showing no differences [34].
In the context of the Italian project (RF-2010-2320172), a population-based study was conducted to evaluate and compare the effects of biosimilar and originator ESAs on haemoglobinaemia in CKD and cancer patients between 2009 and 2014. This used the administrative database from Treviso LHU which also included haemoglobinaemia values [17]. In this study, incident ESA users with at least one Hb measurement taken within the month prior to the first ESA dispensing, i.e. Index Date [ID], defined as the baseline Hb value, and another measurement between the second and the third month after ID (follow-up Hb value) were considered. This allowed for the identification of more than 1,000 incident users. Considering the effects of ESAs on Hb values during the follow-up period, no statistically significant differences were found for biosimilars versus reference product/other ESAs in patients being treated for either indications of use, when considering mean baseline Hb value and follow-up Hb value. Based on the difference between follow-up and baseline Hb values, i.e. delta Hb (ΔHb), ESA users were classified as non-responders (ΔHb ≤ 0 g/dL), responders (0 ‹ ΔHb ≤ 2 g/dL) and high responders (ΔHb › 2 g/dL). Around 15–20% of ESA users were non-responders in both CKD and cancer. Considering the distribution of responders and high responders in both indications of use, no differences across types of ESA users were found. The strength of ESA therapy, defined as the total number of dispensed DDD during the follow-up, divided by the days of follow-up, was investigated stratifying by type of administered ESA, i.e. reference product, biosimilar and other ESAs still covered by patent. No statistically significant difference in the strength of treatment between different types of ESAs was found in either CKD or cancer patients. The difference related to the higher doses used in chemotherapy-induced anaemia was in line with the dosing regimens described in the Summary of Product Characteristics.
In terms of responsiveness to the drug, no statistically significant difference between biosimilars (Binocrit, Abseamed, Retacrit) and reference product (Eprex)/ESAs covered by patent (Aranesp, Nespo, Neorecormon, Eporatio, Mircera) was found, despite a comparable prescribed cumulative dose of drug during the observation period. This finding is in contrast to the results reported in a recently published research letter [21] where Minutolo R et al. analysed data from 11 Italian dialysis centres between 2011–2014. Here, the goal was to evaluate the effect of switching from originator to biosimilar ESAs when treating anaemia. The authors concluded that switching from originator to biosimilar ESAs may require higher doses of biosimilar ESA to maintain Hb levels (so called ‘dosing penalty’) [21]. Several factors may explain the differences observed by Ingrasciotta [17] and Minutolo [21]: a) Ingrasciotta Y et al. investigated all naïve users of different ESAs while Minutolo R et al. analysed only those who switched from originators to biosimilars. As renal functionality declines progressively, thus requiring higher doses of all ESAs to achieve target Hb over time, it may be more appropriate to investigate all possible switches between biosimilars and reference product/other ESAs covered by patent and vice versa; b) Ingrasciotta Y et al. conducted a population-based study, thus using real-world data, while Minutolo R et al. conducted a multicentre study where patients received stable originator ESA doses, thus preventing the generalization of the results to the whole spectrum of the haemodialysis population.
Despite the AIFA recommendations surrounding the specific Hb values to start ESA treatment, 14–24% of CKD patients had a baseline Hb value higher than 11 g/dL. In addition, 22–38% of cancer patients started ESA treatment having a baseline Hb value higher than 10 g/dL, regardless of the type of ESA dispensed at ID. More worryingly, 33–54% of CKD patients and 26–39% of cancer patients reached a follow-up Hb value higher than 12 g/dL [17]. Given the widely known negative effects of high Hb values [35], these data may reflect an inappropriate use of ESA thus warranting further investigation.
Conclusions and future assessment of biosimilars in Italy
The Italian administrative healthcare databases and their distributed database network have provided real-world data about biosimilar patterns of use and the comparative efficacy of ESAs. In the future, post-marketing assessment of biosimilars will be required and this will need: a) data supporting the long-term safety and effectiveness of first generation biosimilars versus originators; b) further investigation and evaluation of the clinical effects of switching between originators and biosimilars, biosimilars and originators, and between different originators; c) the risk–benefit profile of the most recently marketed and more complex biosimilars, i.e. monoclonal antibodies.
In light of the growing number of biosimilars set to reach the market in the future across therapeutic areas, an international post-marketing monitoring system for biosimilars, based on combination of multiple healthcare databases from several countries, may provide useful data. The secondary use of healthcare administrative databases represents a great opportunity for post-marketing assessment of biosimilars and biological drugs in general.
This paper reported a heterogeneous uptake of first generation biosimilars over time and across regions of Italy. This is likely due to the specific loco-regional healthcare policy interventions, and confirms comparable effectiveness of the different types of ESAs in clinical practice, used for treatment of both cancer and CKD patients. However, a better monitoring of biosimilar prescribing is necessary to ensure that savings can be made without putting patients at risk.
Competing interests: Assistant Professor Gianluca Trifirò declares his participation to advisory boards on biosimilars, organized by Sandoz and Hospira; furthermore, he coordinates studies funded by pharmaceutical companies to his department. Ilaria Marcianò, Ylenia Ingrasciotta and Dr Armando Genazzani declare that they have no conflicts of interest.
Provenance and peer review: Commissioned; externally peer reviewed.
Authors
Assistant Professor Gianluca Trifirò1,2, MD, PhD
Ylenia Ingrasciotta2, MSc
Ilaria Marcianò1, MSc
Armando Genazzani3, DPhil, MD
1Unit of Clinical Pharmacology, Azienda Ospedaliera Universitaria, Farmacologo Clinico, Policlinico ‘G Martino’, Dipartimento di Medicina, Clinica e Sperimentale, 1 Via Consolare Valeria, IT-98125 Messina, Italy
2Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina, 1 Via Consolare Valeria, IT-98125 Messina, Italy
3Department of Pharmaceutical Sciences, University of Piemonte Orientale, 6 Via Bovio, IT-28100 Novara, Italy
References
1. Italian Medicines Agency. L’uso dei farmaci in Italia – Rapporto OsMed Gennaio-Settembre 2016 [homepage on the Internet]. [cited 2017 Jul 18]. Available from: http://www.aifa.gov.it/sites/default/files/Rapporto_OsMed_gennaio_settembre_2016.pdf
2. Timonin S, Shkolnikov VM, Jasilionis D, Grigoriev P, Jdanov DA, Leon DA. Disparities in length of life across developed countries: measuring and decomposing changes over time within and between country groups. Popul Health Metr. 2016;14:29.
3. European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products) [homepage on the Internet]. 2012 [cited 2017 Jul 18]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
4. Italian Medicines Agency. Position Paper sui Farmaci Biosimilari [homepage on the Internet]. 2013 [cited 2017 Jul 18]. Available from: http://www.agenziafarmaco.gov.it/sites/default/files/AIFA_POSITION_PAPER_FARMACI_BIOSIMILARI.pdf
5. Dos Reis C, Teixo R, Mendes F, Cruz RS. Biosimilar medicines – review. Int J Risk Saf Med. 2016;28(1):45-60.
6. Mach JP. Introduction to monoclonal antibodies. Cancer Immun. 2012;12:11.
7. Chin WW. A delicate balance – pharmaceutical innovation and access. N Engl J Med. 2015;373(19):1799-801.
8. IMS Health. The role of generic medicines in sustaining healthcare systems: a European perspective [homepage on the Internet]. 2015 [cited 2017 Jul 18]. Available from: https://www.imshealth.com/files/web/IMSH Institute/Healthcare Briefs/IIHI_Generics_Healthcare_Brief.pdf
9. European Medicines Agency. Guideline on similar biological medicinal products [homepage on the Internet]. CHMP/437/04 Rev 1. 23 October 2014 [cited 2017 Jul 18]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf
10. Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111-7.
11. Genazzani AA, Biggio G, Caputi AP, Del Tacca M, Drago F, Fantozzi R, et al. Biosimilar drugs: concerns and opportunities. BioDrugs. 2007;21(6):351-6.
12. Asbjørn M. Norway, biosimilars in different funding systems. What works? Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(2):90-2. doi:10.5639/gabij.2015.0402.018
13. Curto S, Ghislandi S, van de Vooren K, Duranti S, Garattini L. Regional tenders on biosimilars in Italy: an empirical analysis of awarded prices. Health Policy. 2014;116(2-3):182-7.
14. Italian Medicines Agency. L’uso dei farmaci in Italia – Rapporto OsMed 2015 [homepage on the Internet]. [cited 2017 Jul 18]. Available from: http://www.agenziafarmaco.gov.it/sites/default/files/Rapporto_OsMed_2015_AIFA.pdf
15. Ingrasciotta Y, Giorgianni F, Bolcato J, Chinellato A, Pirolo R, Tari DU, et al. How much are biosimilars used in clinical practice? A retrospective Italian population-based study of erythropoiesis-stimulating agents in the years 2009–2013. BioDrugs. 2015;29(4):275-84.
16. Marcianò I, Ingrasciotta Y, Giorgianni F, Bolcato J, Chinellato A, Pirolo R, et al. How did the introduction of biosimilar filgrastim influence the prescribing pattern of granulocyte colony-stimulating factors? Results from a multicentre, population-based study, from five italian centres in the years 2009–2014. BioDrugs. 2016;30(4):295-306.
17. Ingrasciotta Y, Giorgianni F, Marcianò I, Bolcato J, Pirolo R, Chinellato A, et al. Comparative effectiveness of biosimilar, reference product and other erythropoiesis-stimulating agents (ESAs) still covered by patent in chronic kidney disease and cancer patients: an Italian population-based study. Mantovani LG, editor. PLoS One. 2016 May 17;11(5):e0155805.
18. Trotta F, Belleudi V, Fusco D, Amato L, Mecozzi A, Mayer F, et al. Comparative effectiveness and safety of erythropoiesis-stimulating agents (biosimilars vs originators) in clinical practice: a population-based cohort study in Italy. BMJ Open. 2017;7(3):e011637.
19. D’Amore C, Da Cas R, Rossi M, Traversa G. Switching between epoetins: a practice in support of biosimilar use. BioDrugs. 2016;30(1):27-32.
20. Loiacono C, Sgroi C, Coppolino S, Cannata A, Ferrara R, Arcoraci V, et al. How much are biosimilars used in southern Italy?: a retrospective analysis of epoetin utilization in the local health unit of Messina in the years 2010–2011. BioDrugs. 2012;26(2):113-20.
21. Minutolo R, Borzumati M, Sposini S, Abaterusso C, Carraro G, Santoboni A, et al. Dosing penalty of erythropoiesis-stimulating agents after switching from originator to biosimilar preparations in stable hemodialysis patients. Am J Kidney Dis. 2016;68(1):170-2.
22. Pani L, Montilla S, Pimpinella G, Bertini Malgarini R. Biosimilars: the paradox of sharing the same pharmacological action without full chemical identity. Expert Opin Biol Ther. 2013;13(10):1343-6.
23. Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12(11):1473-85.
24. Italian Medicines Agency. Determinazione AIFA. Aggiornamento del Piano terapeutico AIFA per prescrizione SSN di Eritropoietine (ex Nota 12) [homepage on the Internet]. [cited 2017 Jul 18]. Available from: http://www.agenziafarmaco.gov.it/sites/default/files/2010-07-29_determina_aggiornamento_template_nota_12_g.u._75_del_31-03-2009.pdf
25. Wizemann V, Rutkowski B, Baldamus C, Scigalla P, Koytchev R, Epoetin Zeta Study Group. Comparison of the therapeutic effects of epoetin zeta to epoetin alfa in the maintenance phase of renal anaemia treatment. Curr Med Res Opin. 2008 Mar 18;24(3):625-37.
26. Krivoshiev S, Todorov V V, Manitius J, Czekalski S, Scigalla P, Koytchev R, et al. Comparison of the therapeutic effects of epoetin zeta and epoetin alpha in the correction of renal anaemia. Curr Med Res Opin. 2008;24(5):1407-15.
27. Krivoshiev S, Wizemann V, Czekalski S, Schiller A, Pljesa S, Wolf-Pflugmann M, et al. Therapeutic equivalence of epoetin zeta and alfa, administered subcutaneously, for maintenance treatment of renal anemia. Adv Ther. 2010;27(2):105-17.
28. Haag-Weber M, Vetter A, Thyroff-Friesinger U, INJ-Study Group. Therapeutic equivalence, long-term efficacy and safety of HX575 in the treatment of anemia in chronic renal failure patients receiving hemodialysis. Clin Nephrol. 2009;72(5):380-90.
29. Palmer SC, Saglimbene V, Mavridis D, Salanti G, Craig JC, Tonelli M, et al. Erythropoiesis-stimulating agents for anaemia in adults with chronic kidney disease: a network meta-analysis. Cochrane Database Syst Rev. 2014 Dec 8;(12):CD010590.
30. Weigang-Köhler K, Vetter A, Thyroff-Friesinger U. HX575, recombinant human epoetin alfa, for the treatment of chemotherapy-associated symptomatic anaemia in patients with solid tumours. Onkologie. 2009;32(4):168-74.
31. Michallet M, Luporsi E, Soubeyran P, Amar NA, Boulanger V, Carreiro M, et al. BiOsimilaRs in the management of anaemia secondary to chemotherapy in HaEmatology and Oncology: results of the ORHEO observational study. BMC Cancer. 2014;14(1):503.
32. Bustos A, Álvarez R, Aramburo PM, Carabantes F, Díaz N, Florián J, et al. Evaluation of clinical use and effectiveness of darbepoetin alfa in cancer patients with chemotherapy-induced anemia. Curr Med Res Opin. 2012;28(1):57-67.
33. Kerkhofs L, Boschetti G, Lugini A, Stanculeanu D-L, Palomo AG. Use of biosimilar epoetin to increase hemoglobin levels in patients with chemotherapy-induced anemia: real-life clinical experience. Future Oncol. 2012;8(6):751-6.
34. Rodriguez Garzotto A, Cortijo Casacajares S, Pernaut C, Ruiz Ares GJ, Otero Blas I, Heine O, et al. Erythropoiesis-stimulating agents for the treatment of chemotherapy-induced anemia: comparisons from real-world clinical experience. J Blood Med. 2014;5:43-8.
35. KDIGO Clinical practice guideline for anemia in chronic kidney disease. Kidney Int. Suppl. 2012;2(4):Aug (2).
|
Author for correspondence: Ylenia Ingrasciotta, Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina, 1 Via Consolare Valeria, IT-98125 Messina, Italy |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


