Glossary of key terms
Published: 2013/01/01
Last update: 13 February 2015
Confusion may sometimes surround terms used in the fields of generics and biosimilars. This has been recognized as a problem by EMA, who has expressed the need to propose a more precise definition for biosimilars due to problems arising from imprecise usage of the terms in the scientific literature and elsewhere [1].
The source of some of this confusion is due to authorities in various regions of the world defining terms differently and other instances are due to a misunderstanding of the actual nature, characteristics, and method of research and manufacture of these biological products.
In an attempt to prevent any such confusion for GaBI Online readers, here is a glossary of the relevant terms for biosimilars and generics as used in GaBI Online.
ANDA (Abbreviated New Drug Application)
A pathway to marketing approval in the US for generics that requires a reduced amount of data to be submitted to the FDA.
An Abbreviated New Drug Application (ANDA) contains data which when submitted to the FDA’s Center for Drug Evaluation and Research, Office of Generic Drugs, provides for the review and ultimate approval of a generic drug product. Once approved, an applicant may manufacture and market the generic drug product to provide a safe, effective, low cost alternative to the American public.
Active substance
Active ingredient or molecule that goes into a specific medicine and provides this medicine with properties for treating or preventing one or several specific disease(s).
Adverse event
EMA Definition
Any untoward medical occurrence in a patient or clinical trial subject administered a medicinal product and which does not necessarily have a causal relationship with this treatment.
An adverse event can therefore be any unfavourable and unintended sign (e.g. an abnormal laboratory finding), symptom or disease temporally associated with the use of a medicinal product, whether or not considered related to the medicinal product.
Adverse reaction
EMA Definition
A response to a medicinal product, which is noxious and unintended.
Response in this context means that a causal relationship between a medicinal product and an adverse event is at least a reasonable possibility.
Adverse reactions may arise from use of the product within or outside the terms of the marketing authorization or from occupational exposure. Conditions of use outside the marketing authorization include off-label use, overdose, misuse, abuse and medication errors.
Amino acid
Building blocks of proteins. There are 20 common amino acids found in proteins.
Anaemia
A condition that is due to a reduced number of red blood cells or reduced amounts of haemoglobin within them. This results in reduced oxygen-carrying capacity and reduced aerobic activity in blood cells. The symptoms include marked tiredness. Anaemia often occurs after chemotherapy.
Anaphylaxis
An acute and severe allergic reaction in humans.
Antibody (pl: antibodies)
Antibodies (also known as immunoglobulins, abbreviated to Ig) are proteins found in blood or other bodily fluids. They are produced by humans and animals in response to the presence of a specific antigen such as micro-organism, e.g. virus, bacterium. They are used by the immune system to identify and neutralize foreign objects, such as bacteria and viruses by binding to an antigen in a manner that leads to immunological responses such as death of bacteria, elimination of foreign proteins, etc. Usually antibodies are very useful and allow individuals to deal with current and subsequent infections. They can become a problem in some circumstances, e.g. autoimmune disease.
Antigen
A substance, often a protein, which stimulates the production of antibodies. Examples can include pollen grains, dust, bacteria or viruses.
Aplasia
Refers to a type of anaemia affecting the precursors to red blood cells but not to white blood cells.
Assay
Technique for measuring a biochemical or immunological response.
Autoimmune disease
A disease caused by the body producing an excessive immune response against its own tissues. Thereby, the immune system ceases to recognize one or more of the body’s normal constituents as ‘self’ and will create auto-antibodies that attack its own cells, tissues, and/or organs. Inflammation and tissue damage are common symptoms of autoimmune diseases.
Autoimmunity
A condition in which the body mounts an immune response against one of its own organs or tissues.
Automatic substitution
The practice by which a product other than the one specified on the prescriptions is dispensed to the patient, without the prior informed consent of the treating physician. A variation of substitution is practised in some countries, where, if the physician prescribes by International Nonproprietary Name (INN), the pharmacist may decide to dispense any product with the same active ingredient.
Bacteria
Very small, simple living organisms. Their living processes can be ‘hijacked’ by genetic engineering technology and used to manufacture biotechnology medicines.
Bioassay
Determination of the effectiveness of a compound by measuring its effect on animals, tissues or organisms in comparison with a standard preparation. Bioassays using animals or organs are now rarely used to measure potency of biologicals.
Bioavailability
EMA Definition
The rate and extent to which the active substance or active moiety is absorbed from a pharmaceutical form and becomes available at the site of action.
FDA Definition
The rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. For drug products that are not intended to be absorbed into the bloodstream, bioavailability may be assessed by measurements intended to reflect the rate and extent to which the active ingredient or active moiety becomes available at the site of action.
Bioequivalence
EMA Definition
Two medicinal products are bioequivalent if they are pharmaceutically equivalent or pharmaceutical alternatives and if their bioavailabilities after administration in the same molar dose are similar to such degree that their effects, with respect to both efficacy and safety, will be essentially the same.
Other Explanation
The absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of action when administered at the same molar dose under similar conditions in an appropriately designed study.
Bioequivalent drug products
FDA Definition
Pharmaceutical equivalent or alternative products that display comparable bioavailability when studied under similar experimental conditions.
Biological
See biological medicinal product
Biological medicinal product
EMA Definition
A biological medicinal product is a medicinal product whose active substance is made by or derived from a living organism.
Other Explanation
A medicinal product or a vaccine that consists of, or has been produced by the use of, living organisms such as cells or tissues. Often recombinant DNA (a form of DNA that does not exist naturally and which combines DNA sequences that would not normally occur together in order to establish new functions) forms the basis for biotechnologically manufactured products. Examples include therapeutic proteins such as antibodies, insulin or interleukins; but also vaccines, nucleic acids or tissues and cells.
Biologicals have large, complex, inherently diverse molecular structures that are not easily identified or characterized and many are manufactured using biotechnology. Biological products often represent the cutting edge of biomedical research and are sometimes the most effective way to prevent or treat a disease.
Biotechnology medicines are derived through three main sources: yeast cells, bacterial cells and mammalian cells. These host cells are genetically modified and are allowed to multiply in a process referred to as fermentation. Human protein, i.e. the active ingredient of the future medicine, is either produced and contained within the host cells or secreted into a nutrient solution. The cells and their human protein products are then removed, separated, purified and processed into a formulated medicine.
Biopharmaceuticals
Medicines made, or derived, from living organisms using biotechnology. See also biological medicine.
Biosimilar
See biosimilar medicinal product
Biosimilar medicinal product
EMA Definition
A biosimilar is a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product) in the European Economic Area (EEA). Similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy based on a comprehensive comparability exercise needs to be established.
Biological medicines are medicines that are made by or derived from a biological source, such as a bacterium or yeast. They can consist of relatively small molecules such as human insulin or erythropoietin or complex molecules such as monoclonal antibodies.
The active substance of a biosimilar and its reference medicine is essentially the same biological substance, though there may be minor differences due to their complex nature and production methods. Like the reference medicine, the biosimilar has a degree of natural variability. When approved, its variability and any differences between it and its reference medicine will have been shown not to affect safety or effectiveness.
An authorized biosimilar is generally used at the same dose to treat the same conditions. If there are specific precautions to be considered when taking the reference medicine, the same will generally apply to the biosimilar.
Biosimilars can only be authorized for use once the period of data exclusivity on the original ‘reference’ biological medicine has expired. In general, this means that the biological reference medicine must have been authorized for at least 10 years before a similar biological medicine can be made available by another company.
FDA Definition
A biosimilar is a biological product that is highly similar to a US-licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product.
Other Explanation
Biosimilar medicines are follow-on versions of original biological medicines. Biosimilars can be developed during the period in which the originator product is protected by patent exclusivity, but they can only be marketed after the patent protecting the originator product has expired.
Biosimilar medicines are independently developed to have the same mechanism of action as the original biological medicines, and are designed to treat the same diseases as the innovator’s product.
Biosimilarity
Property of a medicine to show similarity and lack of significant differences in terms of quality, efficacy and safety to a reference biological medicine to which it has been compared.
Biotechnology medicine
See biological medicinal product.
Biotechnology
A broad term generally used to describe the use of biology in industrial processes such as agriculture, brewing and baking. Recently, the word has come to refer more to the production of genetically modified organisms or the manufacture of products, notably medicines, from genetically modified organisms. Technology that manipulates living organisms so that they produce certain specific proteins including hormones or monoclonal antibodies; or any technological application that uses biological systems, living organisms, or derivatives thereof, to make or modify products or processes for specific use.
Cell culture
The process by which cells may be grown outside the body under controlled conditions.
Characterization
Tests to determine the properties of a molecule or active substance, e.g. molecular size/weight, chemical structure, purity. These tests are also called physiochemical characterisation.
Chemotherapy
The application of chemicals (drugs) to control the growth of cells that form a cancer.
Clinical study or trial
Study with the objective of determining how a (new) medicine is handled by, and affects, humans. Clinical studies or trials are conducted in healthy volunteers or in patients. Clinical studies routinely involve the use of a control group of patients that is given an inactive substance (placebo) that looks like the test product. Pivotal clinical studies involving a larger group of patients provide evidence on whether the medicine can be considered both safe and effective in a real clinical setting. There are four types of clinical studies:
- Phase I clinical study or trial
Study with the objective of determining how a medicine is handled by, and affects, humans, and of helping to predict the initial dosage range for the medicine and to test the safety profile. Although such studies are often conducted in a small cohort of healthy volunteers, phase I studies in patients are also possible in some situations. - Phase II clinical study or trial
Study of a small number of patients with the objectives of proving the efficacy concept of a medicine and of collecting data to establish the correct dose for that medicine. Phase II studies are not formally required for the development of biosimilar medicines as efficacy and dose are already established for the reference product. - Phase III clinical study or trial
Study involving a larger group of patients, which aims to provide definitive evidence on whether the medicine can be considered both safe and effective in a real clinical setting for drug registration. - Phase IV clinical study or trial
Study with the objective of collecting post-marketing surveillance data to track safety over time.
Comparability
The scientific evaluation of a comparison of two medicinal products to determine equivalence and any detectable differences at the level of quality, efficacy and safety.
Compulsory licence
A compulsory licence allows for a generic drug to be brought onto the market while the originator is still under patent protection under certain humanitarian conditions. For example, the 2005 Indian Patents Act allows for issuing of a compulsory licence in cases including those where:
a) the reasonable requirements of the public with respect to the patented invention have not been satisfied or;
b) it is not available to the public at a reasonably affordable price and;
c) the patent is not being worked.
The idea behind compulsory licensing is to provide low cost medicines and increase access to medicines for poorer people in a public health crisis.
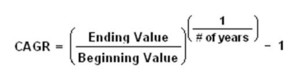
Compound annual growth rate (CAGR)
The compound annual growth rate (CAGR) is the year-over-year growth rate of an investment over a specified period of time.
The CAGR is calculated by taking the nth root of the total percentage growth rate, where n is the number of years in the period being considered.
This can be written as follows:
Defined Daily Dose (DDD)
The defined daily dose (DDD) is a statistical measure of drug consumption, defined by the World Health Organization (WHO). It is used to standardize the comparison of drug usage between different drugs or between different healthcare environments. The DDD is not to be confused with the therapeutic dose or with the dose actually prescribed by a physician for an individual patient.
The WHO’s definition is: ‘The DDD is the assumed average maintenance dose per day for a drug used for its main indication in adults.’
Diabetes
There are several forms of diabetes. It is a common condition in which the amount of glucose (sugar) in the blood is too high because the body is unable to use it properly. Normally, a hormone called insulin removes excess glucose from the blood.
DNA
Acronym for deoxyribonucleic acid. A molecule of DNA consists of a long chain of deoxyribose, a 5-carbon sugar, and phosphate groups with the bases adenine, thymine, cytosine and guanine. DNA contains the genetic code that controls the production of proteins in living things.
EMA
The European Medicines Agency (EMA) is responsible for approving all medicines before they are made available to doctors and patients in the 28 Member States of the European Union.
Enzyme
A protein catalyst that facilitates specific chemical or metabolic reactions necessary for cell growth and reproduction.
Erythropoietin (epoetin – EPO)
A hormone released from the kidneys and the liver in response to low oxygen concentrations in the blood. It controls the rate of red blood cell production.
Essentially similar products
EMA Definition
A medicinal product is essentially similar to an original product where it satisfies the criteria of having the same qualitative and quantitative composition in terms of active substances, of having the same pharmaceutical form, and of being bioequivalent unless it is apparent in the light of scientific knowledge that it differs from the original product as regards safety and efficacy.
By extension, it is generally considered that for immediate release products the concept of essentially similar also applies to different oral forms (tablets and capsules) with the same active substance.
EudraVigilance
Data processing network and management system for reporting and evaluating suspected adverse reactions during the development and following the marketing authorization of medicinal products in the European Economic Area (EEA).
Extrapolation
Extending the findings from one set of conditions to another, such as extending and applying the safety and efficacy data from clinical studies regarding one indication (medical condition) to another indication or extending data from clinical studies in one population (e.g. adults) to another, e.g. children.
Extrapolation concerns the extrapolation of four different aspects; efficacy, safety, immunogenicity and interchangeability. It may concern the indication, population or both.
FDA
The US Food and Drug Administration (FDA). This agency is responsible for approving all medicines before they are made available to doctors and patients in the United States.
Fermentation
Chemical reactions induced by living organisms (or enzymes derived from living organisms) to produce raw material for pharmaceutical products.
Formulation
The recipe and presentation of a medicine.
Generic medicinal product
EMA Definition
A medicinal product which has the same qualitative and quantitative composition in active substances and the same pharmaceutical form as the reference medicinal product, and whose bioequivalence with the reference medicinal product has been demonstrated by appropriate bioavailability studies. (Reg. 726/2004, Art 10, 2b) Generic ‘copies’ can only be marketed after the originator’s patent protection and/or marketing exclusivity has expired.
Other Explanation
A drug product that has the same composition in active substance(s) and the same pharmaceutical form as the originator reference medicine, as well as the same dosage form, strength, route of administration, quality and performance characteristics and intended use, and whose bioequivalence with the originator reference medicine, i.e. the same behaviour in the body, has been demonstrated by appropriate bioequivalence studies. A generic medicine may be made by a different company after patent expiry of the originator product.
Generics
See generic medicinal product.
Genetic disorder
A condition that results from a defective gene or chromosome.
Genetic engineering
A technology used to alter the genetic material of living cells in order to make them capable of producing new substances or performing new functions.
Glycosylation
The type and length of any sugar or carbohydrate groups attached to a given molecule.
Haemoglobin
The protein found in the blood of most vertebrates and some invertebrates that carries oxygen and small amounts of carbon dioxide.
Harvesting
Separation of raw biological material from cell culture.
Identification
The action of designating or identifying something.
Immune response
The immune response is the way the body recognizes and defends itself by producing antibodies against micro-organisms, viruses and substances recognized as foreign and potentially harmful to the body.
Immune system
The collection of mechanisms – cells, biological substances such as antibodies and cellular activities – within the body that work together to provide resistance to disease by identifying and killing pathogens, e.g. viruses and bacteria, and tumour cells.
Immunogenic/Immunogenicity
The capability of a specific substance to cause an immune reaction, i.e. induce the production of antibodies in the human body. The biological response to such a substance is termed an immune response or reaction.
INN (International Nonproprietary Name)
EMA Definition
The INN identifies pharmaceutical substances or active pharmaceutical ingredients. Each INN is a unique name that is globally recognized and is public property. A nonproprietary name is also known as a generic name.
Other Explanation
Scientific or generic name of an active substance. The World Health Organization (WHO) in Geneva allocates INNs for new active substances. The INN is a unique and universally accessible name used to identify each pharmaceutical substance or active pharmaceutical ingredient. For generic and biosimilar medicines cross-referring to originator products, it is the regulatory authority that decides whether the INN of the active substance as submitted for the generic or the biosimilar medicine is scientifically acceptable.
Insulin
A hormone of vertebrates and invertebrates produced in the pancreas by the islets of Langerhans that promotes the conversion of glucose to glycogen and which regulates the amount of glucose in the blood. The lack of insulin causes a form of diabetes.
Interchangeability
Refers to the medical/pharmaceutical practice of switching one medicine for another that is equivalent, in a given clinical setting. A product is considered to be interchangeable if it can be administered or dispensed instead of another clinically equivalence product without significant risk of an adverse health outcome.
Interchangeability can be at the population level, meaning both products can be used for treatment of the same condition in the same population. It can also be on an individual level, meaning that in an individual patient, the products can be alternated or switched. Interchangeability at the individual level is a condition for substitution.
Interferon
A class of proteins important in the immune response. There are three major types of interferon: alpha (leukocyte), beta (fibroblast) and gamma (immune). Interferon inhibits viral infections and may have anti-cancer properties.
In vitro
In vitro refers to biological or chemical studies carried out in the test tube (in vitro is Latin for ‘within the glass’) rather than in living systems.
In vivo
In vivo refers to biological or chemical studies carried out on whole, living organisms usually animals including humans and plants (in vivo is Latin for ‘within the living’), as opposed to a partial or dead organism or those done in vitro.
Line extension
A variation of an existing product. The variation can be a new formulation of an existing product or a new modification of an existing molecular entity. Line extensions are used by companies to extend patent protection of a brand-name drug.
Marketing authorization
The permission granted by a regulatory authority to a company to market a medicinal product in accordance with the conditions described on the label, following the company’s submission of required documentation and data relating to testing and clinical trials of the product.
Molecular
Of a molecule. Relating to or consisting of molecules.
Molecule
A group of atoms bonded together, representing the smallest fundamental unit of a chemical compound that can take part in a chemical reaction. Molecules are made up of one or more atoms. If they contain more than one atom, the atoms can be the same (an oxygen molecule has two oxygen atoms) or different (a water molecule has two hydrogen atoms and one oxygen atom). Biological molecules, such as proteins, can be made up of many thousands of atoms.
Monoclonal antibodies
Monospecific antibodies that are produced by a single clone of immune cells. They have become an important tool in molecular biology and medicine, and are the basis of many biopharmaceuticals.
Non-biological complex drug (NBCD)
A non-biological complex drug (NBCD) is defined as a medicinal product, not being a biological medicine, where the active substance is not a homo-molecular structure, but consists of different (closely related) structures that can’t be fully quantitated, characterized and/or described by (physico-)chemical analytical tools. The composition, quality and in vivo performance of NBCDs are dependent on the manufacturing process and controls.
Examples of NBCDs include liposomes, iron-carbohydrate (‘iron-sugar’) drugs and glatiramoids.
Neurodegenerative
A term used to describe diseases such as Parkinson’s disease and Alzheimer’s disease, which cause elements of the nervous system to deteriorate.
Nucleic acid
A macromolecule, i.e. a very large molecule, composed of chains of monomeric (having a single component) nucleotides, which are molecules that, when joined together, make up the structural units of RNA and DNA. In biochemistry, these molecules carry genetic information or form structures within cells.
Organism
A living thing. The term includes anything that has DNA, from bacteria to vertebrates.
Originator company
Company that was first to develop and produce a specific medicine (biological or pharmaceutical).
Originator medicinal product
A medicine that has been developed and produced by an originator company and that has been approved by the national regulatory authorities or the European Commission on the basis of a full registration dossier.
Patent
A patent is a legal mechanism granted by a State (national government) which allows the discoverer of a medicine the exclusive right to make and sell the product for a set period of time in order to recover the development costs, in exchange for public disclosure of their invention. Typically, however, a patent application must include one or more claims defining the invention, which must be new, non-obvious, and useful or industrially applicable.
Pharmaceuticals
Conventional or traditional chemical medicines.
Pharmaceutical alternatives
EMA Definition
Medicinal products are pharmaceutical alternatives if they contain the same active moiety but differ in chemical form (salt, ester, etc.) of that moiety or in the dosage form or strength.
FDA Definition
Drug products are considered pharmaceutical alternatives if they contain the same therapeutic moiety, but are different salts, esters or complexes of that moiety, or are different dosage forms or strengths (e.g. tetracycline hydrochloride, 250 mg capsules vs. tetracycline phosphate complex, 250 mg capsules; quinidine sulfate, 200 mg tablets vs. quinidine sulfate, 200 mg capsules).
Data are generally not available for the FDA to make the determination of tablet to capsule bioequivalence. Different dosage forms and strengths within a product line by a single manufacturer are thus pharmaceutical alternatives, as are extended-release products when compared with immediate- or standard-release formulations of the same active ingredient.
Pharmaceutical equivalent
EMA Definition
Medicinal products are pharmaceutically equivalent if they contain the same amount of the same active substance(s) in the same dosage forms that meet the same or comparable standards. Pharmaceutical equivalence does not necessarily imply bioequivalence as differences in the excipients and/or the manufacturing process can lead to faster or slower dissolution and/or absorption.
FDA Definition
To be considered pharmaceutical equivalents, two drugs must contain the same active ingredient(s), have the same dosage form and route of administration and have identical strength or concentration. Pharmaceutically equivalent drugs may differ, however, in shape, scoring configuration, release mechanisms, packaging, excipients (including colours, flavours, preservatives), expiration time and labelling (within certain limits). Generics are an example of pharmaceutical equivalents.
Other Explanation
Pharmaceutical equivalence implies the same amount of the same active substance(s), in the same dosage form, for the same route of administration and meeting the same or comparable standards.
Pharmacodynamic tests or studies
Study of the actions and effects of a medicine on living systems over a period of time.
Pharmacovigilance
Science and activities relating to the detection, assessment, understanding and prevention of any adverse effects of medicinal products placed on the market. These are safety control procedures to which medicines are subject before, during and after their approval by regulatory authorities.
Pharmcokinetic tests or studies
Studies to determine how medicines are absorbed, distributed metabolized and eliminated by the body.
Physiochemical characterization
Tests to determine the properties of a molecule or active substance, e.g. molecular size/weight, chemical structure, purity.
Polypeptides
Molecules made up of chains of amino acids, which may be pharmacologically active in the human body. They contain fewer amino acids, and hence have lower molecular weights than proteins.
Post-Authorization Safety Study
Any study with an authorized medicinal product conducted with the aim of identifying, characterizing or quantifying a safety hazard, confirming the safety profile of the medicinal product, or of measuring the effectiveness of risk management measures after their approval by regulatory authorities.
Proteins
Large molecules made of amino acids arranged in chains, e.g. erythropoietin.
Purification
Processes used to remove impurities (foreign or undesired materials) from a medicinal product.
Reference product
The original product to which a biosimilar or generic drug refers in its application for marketing approval.
RNA
Acronym for ribonucleic acid. RNA is made up of a long chain of components called nucleotides. Each nucleotide consists of a nucleobase, a ribose sugar and a phosphate group. The sequence of nucleotides allows RNA to encode genetic information that directs the synthesis of proteins.
Second-generation biological
Improved versions of the originator medicine, which represent explicitly different products (different active ingredient, longer acting, less frequent dosing, better efficacy, higher patient convenience), are called second-generation biologicals. Unlike first-generation biologicals, these are still protected by patents.
Similar biological medicinal product
See biosimilar medicinal product.
Substitution
The practice of dispensing one medicine instead of another equivalent and interchangeable medicine in any given patient at the pharmacy level without consulting the prescriber.
The FDA believes that products classified as therapeutically equivalent can be substituted with the full expectation that the substituted product will produce the same clinical effect and safety profile as the prescribed product.
There is no ‘substitutability determination’ at the EU level
Switching
Decision by the treating physician to exchange one medicine for another medicine with the same therapeutic intent (e.g. from originator to generic/biosimilar orvice versa) in a patient during the course of treatment.
In hospitals, the decision to switch a medicine is made by a multidisciplinary team including the clinical community (therapeutic/formulary committee).
Therapeutic equivalent
EMA Definition
A medicinal product is therapeutically equivalent with another product if it contains the same active substance or therapeutic moiety and, clinically, shows the same efficacy and safety as that product, whose efficacy and safety has been established.
FDA Definition
Drug products are considered to be therapeutic equivalents only if they are pharmaceutical equivalents and if they can be expected to have the same clinical effect and safety profile when administered to patients under the conditions specified in the labelling.
Other Explanation
A drug that has essentially the same effect, in the treatment of a disease or condition, as one or more other drugs. A drug that is a therapeutic equivalent may or may not be chemically equivalent, bioequivalent or generically equivalent.
Therapeutic equivalence is considered demonstrated if the 90% confidence intervals of the ratios for the log AUC0-tand Cmax between the two preparations lie in the range 80-125%.
Drugs classified as therapeutically equivalent can be interchanged with the full expectation that the substituted product will produce the same clinical effect and safety profile as the prescribed product.
Traceability
The ability to trace each individual unit of a medicinal product from the source to its final destination and vice versa.
Vaccine
A biological preparation, which is used to establish or improve immunity to a particular disease.
Acronyms – List of Abbreviations
EC – European Commission
EMA – European Medicines Agency
EU – European Union
FDA – US Food and Drug Administration
IP – Intellectual property
LMWH – Low molecular weight heparins
MAA – Market Authorization Approval
WHO – World Health Organization
Related articles
WHO definitions of generics
WHO definitions of biosimilars
Health Canada definitions of generics and biosimilars
FDA definitions of generics and biosimilars
EMA definitions of generics and biosimilars
Controversial nomenclature for new biosimilars
Reference
1. Weise M, Bielsky M, De Smet K, Ehmann F, Ekman N, Narayanan G, et al. Biosimilars—why terminology matters. Nat Biotechnol. 2011(8); 29:690-3.
Source: www.gabionline.net



Biosimilars have gained tremendous interest in recent years. However, there are many challenges associated with their development that include batch to batch variability, complexity of biologic formulations and variable results from clinical trials. These become a cause for concern for both the FDA and industry as they must agree upon a standard guidance to prove the similarity between the reference and a test product. The big questions still remain unanswered. How similar is similar enough? How far are we from having a biosimilar product in the US market?