Health Canada’s perspective on the clinical development of biosimilars and related scientific and regulatory challenges
Published on 2015/02/06
Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):36-41.
|
Abstract: |
Submitted: 16 January 2015; Revised: 16 February 2015; Accepted: 20 February 2015; Published online first: 6 March 2015
Introduction
In the past decades, biologicals have had a profound impact on the overall health and quality of life of patients with complex diseases such as rheumatoid arthritis, diabetes and cancers. Unlike pharmaceuticals, biologicals are produced from living organisms, e.g. human, animal, using biotechnology. The continued advances in medical research and technology have driven novel scientific platform innovations resulting in the development of newer biologicals that have expanded the treatment options for patients. For example, antibody-drug conjugates, and antisense RNA interference-, cell- and gene-based therapies are at various stages of clinical development and some have received marketing authorization. Meanwhile, the great success of many biologicals, e.g. infliximab, rituximab, along with their recent or pending expiry of patent protection have opened the door to a distinct class of biologicals ‘biosimilars’ (subsequent entry biologics [SEBs] in Canada) with one growth hormone biosimilar [1, 2] and one monoclonal antibody (mAb) biosimilar [3] authorized, so far, in Canada. This new class of biologicals poses novel regulatory and scientific challenges due to its complexities. Nonetheless, in many countries, biosimilars benefit patients and the healthcare system, due to cost factors and the opportunity, therefore, to treat a larger number of patients who might otherwise not have access to such useful products. Thus, the objectives of this review intend to highlight the Canadian regulatory review process for biosimilars, and to discuss the regulatory and scientific issues associated with the clinical assessment of biosimilars, based on the current thinking in Health Canada (HC), guidance documents and the information available for each product.
Canadian regulatory review process for biologicals and biosimilars
In Canada, biologicals are regulated under Schedule D of the Food and Drugs Act and Division 4 of the Food and Drug Regulations. Because biologicals exhibit a number of properties that distinguish them from pharmaceuticals, the regulatory requirements for biological submissions differ from those for pharmaceuticals [4]. Guidance documents issued by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and adopted by HC for biologicals are generally applicable to biosimilars [5–12]. Many jurisdictions as well as the World Health Organization (WHO) have published specific guidance documents regarding the data requirements for the marketing authorization of biosimilars [13–18]. In Canada, biosimilars fall under the same provision as those for new drugs: Division 8 of the Food and Drug Regulations [13]. The biosimilar regulatory framework is based on the scientific and regulatory principles within the existing regulatory framework for biologicals. A biosimilar, like a new biological, must be filed as a new drug submission, see Table 1. The premise underlying a biosimilar submission is to demonstrate similarity to a previously authorized biological (reference biological drug [RBD]) marketed in Canada and relies, in part, on prior information regarding the efficacy and safety of the RBD. The demonstration of similarity is primarily deduced from side-by-side quality studies. The biosimilar development programme not only requires a full chemistry and manufacturing (C&M) data package as is expected for a standard new biological, but also an extensive structural and functional characterization between the biosimilar and the RBD chosen. These studies should be carried out using multiple orthogonal analytical methods, e.g. physicochemical and biological analyses, with high accuracy, sensitivity and specificity. The establishment of similarity at the analytical/functional level would form the basis for a reduced non-clinical and clinical package for biosimilars, see Table 1. Any differences in quality attributes should have no adverse impact upon safety or efficacy. The type and extent of the non-clinical and clinical data are dependent on the level of ‘residual uncertainty’ that remains based on the results of the quality studies. A fingerprint like analysis algorithm to compare the quality attributes may be used to leverage a more selective approach to subsequent clinical studies [16]. Overall, the establishment of biosimilarity is based on the totality-of-evidence. The authorization of a biosimilar does not imply that the RBD and the biosimilar are considered pharmaceutically and therapeutically equivalent from the regulatory perspective in Canada, since the drug substances of the biosimilar and the RBD are not identical [13]. Any declaration of therapeutic equivalence is not within the purview of the federal regulator, but is within the authority of each Province in Canada, as health care is within the authority of the provincial health authorities [4]. Once a biosimilar is authorized, it is regarded as a stand-alone biological: manufacturers do not have to compare it with the original reference product for post-market changes.
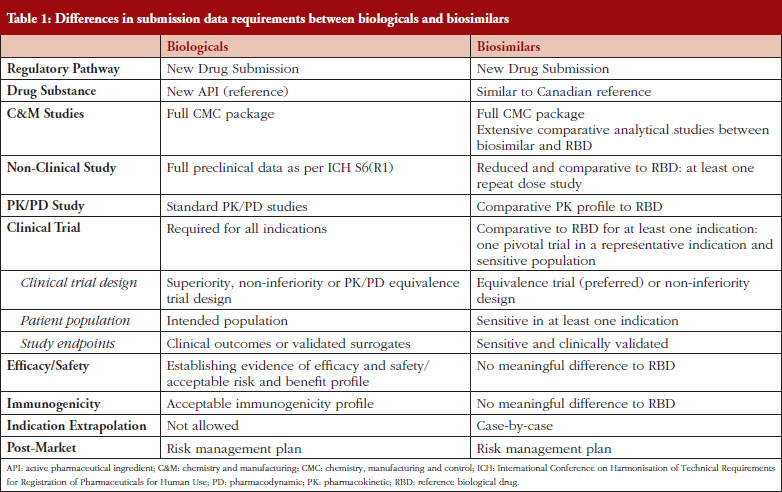
Special issues and considerations for the clinical assessment of biosimilars
The purpose of the clinical programme for a biosimilar is to resolve any residual uncertainties related to the similarity between the biosimilar and the RBD, and generally includes comparative pharmacokinetic (PK)/pharmacodynamic (PD), clinical safety/efficacy and immunogenicity studies. In principle, because a biosimilar is a biological, the clinical issues associated with biologicals also apply to biosimilars. However, since biosimilars usually follow a shortened clinical pathway and the clinical trials aim to exclude clinically meaningful differences in PK/PD, efficacy and safety, rather than establishing de novo risk/benefit, biosimilars are facing specific challenges and entail special considerations that are discussed below.
Choice of reference biological drug
One of the challenges associated with a global biosimilar development programme is the selection of the RBD. Clinical comparative studies should employ a suitable RBD. The Canadian biosimilar guidance document states that the RBD should be authorized for sale and marketed in Canada [13]. If multiple versions of an RBD are available on the market, it is preferable that the one licensed in Canada be used in the comparative studies. A non-Canadian RBD could be accepted if a rationale demonstrating its suitability as proxy for the version authorized in Canada is provided. The non-Canadian RBD should be marketed by the same innovator company or corporate entity that is approved to market the medicinal ingredient in the same dosage form in Canada [13]. Due to a global marketing strategy and in order to allow for a single development process, sponsors are using multiple versions of an RBD, e.g. American, Canadian and European versions made by the same manufacturer at the same or different manufacturing site(s), in clinical studies. To demonstrate that different versions of the RBD are virtually the same, sponsors would usually conduct three-way bridging studies including PK/PD studies between the different RBD versions and the biosimilar. In the selection of an RBD, it is important to note that the following products would not be considered suitable RBDs: i) different protein, e.g. granulocyte-macrophage colony-stimulating factor biological for granulocyte colony-stimulating factor biosimilar; ii) different protein modifications, e.g. non-pegylated biological for a pegylated biosimilar; iii) different amino acid sequences compared with the proposed biosimilar; or iv) protein made in a different expression system, e.g. animal-derived biological for a plant-derived biosimilar. HC considers that the absence of relevant animal-specific post-translational modifications, and/or post-translational modifications unique to plant expression systems, and their impact on immunogenicity, would make the development of biosimilars using plant-derived systems challenging. This would also increase uncertainties for decision-making due to limited regulatory experience and lack of sufficient information in the public domain [19].
Comparative PK/PD studies
PK/PD studies represent an essential part of the biosimilar clinical programme as they support biosimilarity (PK/PD comparability) between the biosimilar and the RBD. A similar PK/PD profile between the two products could alleviate some residual uncertainty and thus guide the extent of subsequent comparative clinical studies. PK/PD studies could help to monitor immunogenicity in clinical trials, e.g. via altered PK data, and could provide scientific evidence for extrapolation of indications. PK/PD data could also be used to compare different routes of administration or different strengths and formulations of a biosimilar. For example, if a biosimilar is proposed to be administered subcutaneously and intravenously, but only one route of administration is used in clinical studies, a bridging PK/PD study should be conducted to demonstrate that the two routes of administration are bioequivalent. Further, in a situation where a new strength or formulation is proposed for an intended subcutaneous (SC) or intramuscular (IM) route of administration, a bridging PK/PD study should be performed to show that the bioavailability between the different strengths or formulations, by the intended route of administration, is the same. If the intravenous (IV) route is proposed and used in the clinical studies, additional PK/PD data to compare the different strengths or formulations may not be required as the bioavailability is 100%. In cases where no studies are conducted via the SC route, it is unlikely that an indication using the SC route will be granted by extrapolation.
One key consideration for PK/PD studies is the use of a relevant patient population given that patient status, receptor internalization rate and expression of target receptor (density and subtypes) may affect the disposition and clearance of the biosimilar. In some cases, the use of healthy volunteers could be inappropriate as their PK/PD parameters may not be reflective of those observed in patients, due to differences in immune status. Also, target-mediated effects on PK cannot be fully assessed and a clinically relevant dose may induce a ceiling effect on healthy volunteers. Nonetheless, healthy volunteers may be pursued if justified, e.g. no efficacy and safety concerns. The most sensitive study design associated with comparative PK studies to detect potential differences between the biosimilar and the RBD is the single dose crossover design. However, this design could be limited by the properties of the biological such as a long half-life or by the formation of anti-drug antibody (ADA) that could impact the PK/PD profile. As well, patient population may require a continued dosing for ethical reasons. Alternatively, parallel or steady-state design could be considered. The criteria for comparative bioavailability as established in Canada for small molecules, should be generally followed [13]. However, they may not always apply to biologicals [20]. The criteria for PK comparability also differ between jurisdictions [21–23]. For instance, according to the HC guidance document for PK/PD studies, the 90% confidence interval (CI) of the relative mean area under the concentration (AUC) versus time curve to the time of the last quantifiable concentration (AUCT), as well as of the relative mean maximum concentration (Cmax) of the test (biosimilar) to the RBD should be within 80–125% inclusively [21]. At the same time, the US Food and Drug Administration (FDA) recommends applicants to provide the geometric means, arithmetic means, geometric mean ratios and 90% CI for AUC0-t, AUC0-inf, and Cmax [22]. For steady-state studies, not only the 90% CI of the relative mean area under the concentration versus time curve at steady-state over the dosing interval (AUCtau) and the ratio of the test to the reference (Cmax) at steady-state should be within 80–125% inclusively, but also the relative mean minimum concentration (Cmin) at steady-state of the test to the reference should not be less than 80% [21]. HC may accept potency correction if the measured drug content of the biosimilar and the RBD differ by 5% or more from each other. In such cases, the use of potency correction should be justified and predefined, and the applicable bioequivalence standards should be met on both potency-corrected and uncorrected data [21]. PD parameters could be investigated in the context of combined PK/PD studies or part of clinical trials. The required CI for PD parameters is usually set at 95%. The PD endpoints used should be considered as surrogate markers and be clinically validated. Otherwise, PD data would not provide strong support for biosimilarity. Note that for most biologicals, no suitable PD surrogates exist.
Equivalence versus non-inferiority design for efficacy/safety clinical studies
In line with the principle of demonstrating similarity, HC recommends that equivalence clinical trials for biosimilars be designed to show comparability to their respective RBDs. Based on the ICH definition, an equivalence trial is a trial designed to show that two interventions do not differ in either direction by more than a pre-specified insignificant margin [8]. A pre-specified and clinically acceptable equivalence margin (two-sided test) that is adequate to detect clinically meaningful differences between the biosimilar and the RBD should be selected. This equivalence margin should be based on available historical data for the RBD and should be smaller than the differences observed in superiority trials for the RBD [8–9]. Biosimilar guidelines prepared by various jurisdictions or WHO do not provide standard equivalence margins for most biologicals used to detect clinically meaningful differences in the targeted diseases. The equivalence margins should be considered on a case-by-case basis, and regulators may recommend a different margin than that proposed by sponsors. Therefore, sponsors of biosimilar products should consult with regulators prior to the initiation of pivotal trials to ensure that the selected equivalence margins are acceptable. In justified cases, a non-inferiority trial design could be acceptable such as a situation where the response rate for an RBD is very high, e.g. 90%, and superiority is unlikely to occur [24]. However, the use of non-inferiority trial requires superiority to be tested and if demonstrated, the product can no longer be considered as a biosimilar (at least in Canada). The demonstration of non-inferiority could also limit the extrapolation to other indications.
Sensitive patient population and study endpoints
Biosimilar clinical trials should be carried out in a ‘sensitive’ and homogeneous patient population. A ‘sensitive’ population is defined as one in whom potential differences between the biosimilar and the RBD are likely to be detected. For instance, metastatic breast cancer may not represent a sensitive population for trastuzumab biosimilar clinical trials because of the heterogeneous nature of metastatic disease, and the greater risk of immune impairment and of the development of secondary cancers [25]. Patients who have not received previous treatment, e.g. first-line therapy, are preferred compared with those who have been treated with different lines of therapy. Patients who previously received several or different lines of therapy are more heterogeneous and may be expected to respond differently, this may mask the detection of differences between the SEB and RBD. A patient population that received a drug as a monotherapy would also be considered to be a more sensitive population compared with one that was administered a concomitant medication, e.g. immunosuppressant, or had been treated with combination therapies. SEB sponsors are encouraged to consult HC with regard to the selection of a sensitive population prior to the initiation of a clinical trial.
Likewise, clinically relevant and sensitive study endpoints should be selected. These endpoints could be different from those traditionally used for the RBDs as they may not be considered as the most sensitive endpoints to detect differences in efficacy. If surrogate endpoints are used, they should correlate with clinical outcomes. The European Medicines Agency (EMA) states that for oncology trials, objective response rate (ORR) would be a more sensitive endpoint compared to progression-free survival (PFS) or overall survival (OS) since ORR would be less affected by patient- and disease-related factors [14]. It is also suggested that continuous outcomes, e.g. change in DAS28 over time, would be more sensitive than dichotomous outcomes, e.g. ACR20, in RA for determining clinical comparability [20]. However, regulatory bodies may not always agree upon the choice of study endpoints. Considering the case of RA, some may require ACR20, e.g. FDA [26], while others such as HC may prefer DAS28. For a biosimilar sponsor with a global development programme that is guided or required by various regulators to fulfil local regulatory or clinical practice requirements, it may be possible to pre-specify different primary study endpoints with the statistical power in the same trial to comply with various regulatory requirements. HC generally considers the commonly used surrogate markers or those defined for FDA as acceptable endpoints for clinical comparability studies. HC would not accept endpoints from an unauthorized indication in Canada for the purpose of establishing efficacy and safety comparability in pivotal clinical trials, as from a regulatory perspective this is to compare an unauthorized indication to other unknowns [27].
Immunogenicity and safety
Immunogenicity is an important aspect of biosimilar safety. The ability of biologicals to induce an immune response via the production of ADAs may result in unpredicted and unforeseen consequences that range from none to serious effects on PK, efficacy and/or safety [28]. For example, patients treated with recombinant erythropoietin developed pure red cell aplasia due to formation of neutralizing anti-epoetin antibodies (Abs) that cross-reacted with endogenous erythropoietin [29]. In a biosimilar clinical programme, immunogenicity should be evaluated in at least one clinical trial using the most sensitive population in which immune responses and adverse reactions are likely to occur. Immunogenicity testing should demonstrate that immunogenicity of the biosimilar is not increased compared to that of the RBD and that there is no change in terms of ADA concentration, titre and type between the two products. Manufacturing variations may lead to minor differences in structure, post-translational modifications, or impurities in a way that could shift the immunogenicity profile of a biosimilar and potentially affect safety [30]. Notably, glycosylation is of particular concern for mAb biosimilars since glycosylation on the Ab Fc domain could affect the activation of effector functions [31]. Immunogenicity testing should employ state-of-the-art assays that are capable of detecting difference in ADA response. Ideally, two assays should be used to validate the methodology, one using the biosimilar and the other using the RBD as the capture ligand. If only one assay is used, the biosimilar should be incorporated as the capture ligand. Neutralizing or cross-reacting Abs are the most concerning, and thus, any significant effect of these Abs on PK, efficacy and safety between the two products should be assessed.
Immunogenicity and safety, e.g. the nature, severity and frequency of adverse events, should be compared between the biosimilar and the RBD in clinical trials that enrolled a sufficient number of patients (> 100 patients) for an acceptable period of time (at least one year). Safety assessment should take into account the safety profile of the RBD throughout its life cycle to ensure that there is no new safety signal for the biosimilar. While safety data including immunogenicity are collected during the pre-market stage, it is unlikely that they are able to detect meaningful, less common (rare adverse events) or longer-term adverse drug reactions. For example, long-term safety concerns such as tumour progression or haematological malignancies have been associated with epoetin and filgrastim biosimilars, respectively [32]. Albeit analytical methods to detect ADAs are available, they remain limited and may not predict all biological properties. Therefore, a rigorous safety monitoring is required in the post-marketing setting.
Extrapolation of indications
Extrapolation to other indications for which the biosimilar has not been tested in clinical trials is appealing to many biosimilar manufacturers, but it represents a paramount concern. In Canada, an extensive and compelling comparability C&M in conjunction with reduced PK/PD and clinical studies may potentially permit extrapolation of the clinical data from the sensitive population to other indications for which the Canadian RBD is authorized at the time of filing [13]. For instance, HC granted all Canadian indications approved for the reference product to the first biosimilar somatropin, i.e. treatment of adult and children with growth hormone deficiency [1], while non-Canadian indications for the RBD were not granted, i.e. Prader-Willi syndrome, small for gestational age, Turner syndrome, and idiopathic short stature [33]. The decision to extrapolate could also be challenged by residual quality attributes and differences in the mechanism of action, pathophysiological mechanism of the disease, route of administration, posology, PK/PD profiles, concomitant therapies, clinical endpoints, study populations and/or safety/immunogenicity profiles between indications [4, 34]. Differences in clinical experience compared with the RBD may also preclude extrapolation [4]. HC’s view is that uncertainties related to the biosimilar which could result in potentially meaningful clinical effects need to be addressed prior to marketing authorization. HC, similar to many other regulatory agencies, endorses the concept of extrapolation, but each indication extrapolation has to be scientifically justified and will be considered on a case-by-case basis. Although decision-making is driven by scientific considerations, legal or public health frameworks and clinical practice vary in different countries. Indications that are still under patent or data protection in Canada would not be granted a Notice of Compliance until such protections expire or an allegation can justify immediate market entry that is either accepted by the innovator or upheld by the court. Consequently, as for any other therapeutic product, regulatory bodies may reach different decisions based on the same data package. For example, HC authorized the first mAb biosimilar infliximab for only a subset of indications and uses while other regulatory agencies granted many uses [3, 35–38].
Post-market
As for any new biological product, risk management plan should be in place for biosimilars. The post-marketing pharmacovigilance programme should be tailored to each biosimilar to address specific product-related issues. In some cases, a more extensive pharmacovigilance may be needed. Recently, the government of Canada has introduced Bill C-17 also known as Vanessa’s law which gives HC the authority to take full measures to strengthen safety oversight of drugs throughout their life cycle [39]. Hence, Bill C-17 fosters a robust Canadian regulatory system that will further contribute to the safety of biosimilars for Canadians.
Conclusion
The advent of biotechnology has enabled the development of biologicals that have revolutionized the prevention and treatment of multiple diseases. With the patent expiry of many biologicals, the arena of biosimilars is emerging, providing patients with wider access to biologicals. Unlike new biologicals, the clinical pathway of biosimilars is usually shortened and based on the demonstration of comparability with the innovator product (RBD). As such, the clinical development of biosimilars is associated with specific regulatory and scientific issues. Biosimilar clinical trials should be thoroughly assessed and scrutinized. Key concerns related to the choice of the RBD, trial design, safety and immunogenicity discussed in this review should form the basis of the clinical assessment. The comparative PK/PD and clinical studies should also be conducted in clinically relevant settings that are the most sensitive to detect potential differences between the two products, especially the use of sensitive population and study endpoints. A rigorous post-market safety and pharmacovigilance plan must be established to ensure the long-term safety of patients. Extrapolation of indications to conditions that have not been studied remains a challenge for biosimilars. Although the concept of extrapolation appears to be widely accepted, indications granted by each jurisdiction may vary for the same product. As the knowledge and experience with biosimilars increase, regulatory agencies may be able to overcome the scientific and regulatory hurdles and harmonize their decision-making at a global level.
Competing interest: None of the authors declare any competing interests.
Provenance and peer review: Commissioned; externally peer reviewed.
Authors
Ally Pen1, PhD
Agnes V Klein2, MD, DPH
Jian Wang1, MD, PhD
1Clinical Evaluation Division – Haematology/Oncology, Centre for the Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Products and Food Branch, Health Canada, Ottawa, Ontario, K1A 0K9, Canada
2Director, Centre for the Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Products and Food Branch, Health Canada, Ottawa, Ontario, K1A 0K9, Canada
References
1. Health Canada. Summary Basis of Decision (SBD) for Omnitrope. 14 September 2009 [homepage on the Internet]. 2009 Sep 23 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/sbd-smd/phase1-decision/drug-med/sbd_smd_2009_omnitrope_113380-eng.pdf
2. Klein AV. The first subsequent entry biologic authorized for market in Canada: the story of Ominitrope, a recombinant human growth hormone. Biologicals. 2011;39(5):278-81.
3. Health Canada. Drugs and Health Products. Summary Basis of Decision (SBD) for Inflectra. 11 March 2014 [homepage on the Internet]. 2015 Feb 16 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php
4. Klein AV, Wang J, Bedford P. Subsequent entry biologics (biosimilars) in Canada: approaches to interchangeability and the extrapolation of indications and uses. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):150-4. doi:10.5639/gabij.2014.0303.033
5. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Structure and content of clinical study reports E3. 30 November 1995 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E3/E3_Guideline.pdf
6. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Ethnic factors in the acceptability of foreign clinical data E5(R1). 5 February 1998 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E5_R1/Step4/E5_R1_Guideline.pdf
7. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Guideline for good clinical practice E6(R1). 10 June 1996 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf
8. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Statistical principles for clinical trials E9. 5 February 1998 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf
9. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Choice of control group and related issues in clinical trials statistical principles for clinical trials E10. 20 July 2000 [homepage on the Internet]. 2006 Mar 2 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E10/Step4/E10_Guideline.pdf
10. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Quality of biotechnological products: analysis of the expression construct in cells used for production of r-DNA derived protein products Q5B. 30 November 1995 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5B/Step4/Q5B_Guideline.pdf
11. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Specifications: test procedures and acceptance criteria for biotechnological/biological products Q6B. 10 March 1999 [homepage on the Internet]. 2006 Feb 28 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf
12. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised tripartite guideline. Preclinical safety evaluation of biotechnology-derived pharmaceuticals S6(R1). 12 June 2011 [cited 2015 Feb 16]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf
13. Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs). 5 March 2010 [homepage on the Internet]. [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guid es/seb-pbu/seb-pbu_2010-eng.php
14. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. 30 May 2012 [homepage on the Internet]. 2012 Jun 11 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
15. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005.22 February 2006 [homepage on the Internet]. 2006 Mar 8 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
16. U.S. Food and Drug Administration. Draft guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. May 2014 [homepage on the Internet]. 2014 May 9 [cited 2015 Feb 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidandes/UCM397017.pdf
17. World Health Organization. Guidelines on the quality, safety, and efficacy of biotherapeutic protein products prepared by recombinant DNA technology [homepage on the Internet]. 2014 Aug 20 [cited 2015 Feb 16]. Available from: http://www.who.int/biologicals/biotherapeutics/TRS_987_Annex4.pdf?ua=1
18. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 19-23 October 2009 [homepage on the Internet]. 2010 Jun 4 [cited 2015 Feb 16]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
19. Health Canada. Guidance document: Plant molecular farming (pmf) applications: plant-derived biologic drugs for human use. 8 May 2014 [homepage on the Internet]. 2014 May 7 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/pmf-mcv-eng.pdf
20. Kay J, Feagan BG, Guirguis MS, et al. Health Canada/BIOTECanada Summit on regulatory and clinical topics related to subsequent entry biologics (biosimilars), Ottawa, Canada, 14 May 2012. Biologicals. 2012;40(6):517-27.
21. Health Canada. Guidance document: comparative bioavailability standards: formulations used for systemic effects. 22 May 2012 [homepage on the Internet]. 2012 Mar 21[cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-eng.pdf
22. U.S. Food and Drug Administration. Guidance for Industry. Bioavailability and bioequivalence studies for orally administered drug products-general considerations. March 2003 [homepage on the Internet]. 2003 Feb 11 [cited 2015 Feb 16]. Available from: http://www.fda.gov/downloads/Drugs/Guidances/ucm070124.pdf
23. European Medicines Agency. Guideline on the investigation of bioequivalence. 20 January 2010 [homepage on the Internet]. 2010 Mar 10 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
24. Zhang N, Chi E. Statistical considerations for the development of biosimilar products. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):21-5. doi:10.5639/gabij.2014.0301.007
25. Cortes J, Curigliano G, Dieras V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat. 2014;144(2):233-39.
26. U.S. Food and Drug Administration. Draft Guidance for Industry. Rheumatoid arthritis: developing drug products for treatment. May 2013 [homepage on the Internet]. 2013 May 13 [2015 Feb 16]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM354468.pdf
27. Justice Laws Website. Food and Drug Regulations [homepage on the Internet]. 2015 Feb 18 [cited 2015 Feb 16]. Available from: http://laws-lois.justice.gc.ca/PDF/C.R.C.,_c._870.pdf
28. De Groot AS, Scott DW. Immunogenicity of protein therapeutic. Trends Immunol. 2007;28(11):482-90.
29. Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469-75.
30. Brinks V. Immunogenicity of biosimilar monoclonal antibodies. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):188-93. doi:10.5639/gabij.2013.0204.052
31. Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci. 2012;1253:170-80.
32. Bennett CL, Chen B, Hermanson T, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol. 2014;15(13):e594-e605.
33. European Medicines Agency. European public assessment report for Omnitrope – product information [homepage on the Internet]. 2008 Apr 14 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000607/WC500043695.pdf
34. Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation. J Clin Pharmacol. 2014 June 26. doi:10.1002/jcph.339. [Epub ahead of print]
35. European Medicines Agency. European public assessment report for remsima (infliximab) – product information [homepage on the Internet]. 2015 Jan 15 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf
36. European Medicines Agency. European public assessment report for inflectra (infliximab) – product information [homepage on the Internet]. 2014 Dec 10 [cited 2015 Feb 16]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf
37. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Japan [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Feb 16]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Japan
38. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in South Korea [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Feb 16]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-South-Korea
39. Health Canada. Protecting Canadians from Unsafe Drugs Act (Vanessa’s Law) Amendments to the Food and Drugs Act (Bill C-17). 6 November 2014 [homepage on the Internet]. 2015 Feb 16 [cited 2015 Feb 16]. Available from: http://www.hc-sc.gc.ca/dhp-mps/legislation/unsafedrugs-droguesdangereuses-eng.php
|
Author for correspondence: Jian Wang, MD, PhD, Chief, Clinical Evaluation Division – Haematology/Oncology, Centre for the Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Products and Food Branch, Health Canada, 200 Tunney’s Pasture Driveway, Ottawa, Ontario, K1A 0K9, Canada |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


