Immunogenicity of biosimilar monoclonal antibodies
Published on 2013/08/06
Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):188-93.
|
Abstract: |
Submitted: 5 July 2013; Revised: 5 September 2013; Accepted: 6 September 2013; Published online first: 20 September 2013
Introduction
The upsurge of antibody-induced pure red cell aplasia during erythropoetin alpha treatment more than a decade ago triggered the awareness that immunogenicity of therapeutic proteins can be a serious health risk [1]. Since then, multiple reports have shown that antibodies formed against therapeutic proteins such as factor VIII, interferon beta, and monoclonal antibodies (mAbs) can change pharmacokinetics, lower efficacy, and can lead to hypersensitivity reactions [2–5]. In fact, current belief is that all biotech-derived products can be immunogenic [6]. Therefore, appropriate immunogenicity testing is required for authorization of new therapeutic proteins on the US and EU drug market by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) respectively.
Since the development of so-called biosimilar products, the discussion on immunogenicity has intensified. Biosimilars are protein equivalents of generic chemical drugs that come onto the market after patent expiration of an originator therapeutic protein. However, the size and complexity of proteins make it virtually impossible to produce an exact copy of an originator product, hence the name ‘similar’. A major concern with biosimilars is that small (undetectable) differences compared with originator products might lead to unexpected immunogenicity in patients [7].
Up to now 16 biosimilars have been approved by EMA, including erythropoietin, growth hormone and granulocyte colony-stimulating factor products [8]. With the exception of a report on unexpected immunogenicity of the biosimilar erythropoietin alpha HX575 [9], these products seem to be of equal or perhaps better quality compared with the originator products [10].
Unfortunately, there is very limited clinical data on the immunogenicity of mAb biosimilars, since they have not yet entered the EU or US drug market.
Monoclonal antibodies are often considered a separate class of therapeutic proteins because of their increased complexity in structure and function compared to non-mAb proteins. Analysis of mAbs, including their physicochemical characterization, in vitro functioning, in vivo safety and immunogenicity, is therefore challenging. Several researchers have indicated that similarity testing between originator mAbs and biosimilar mAbs is more difficult than similarity testing between non-mAb originator and biosimilar products [11, 12]. EMA and FDA have acknowledged the difficulty in assessing similarity between mAb products, and in 2012 EMA published specific guidelines for similarity and immunogenicity testing of biosimilar mAbs [13, 14]. This difficulty in assessing similarity fuelled the discussion on immunogenicity, but now with a focus on mAb biosimilars. Again a major concern is whether small (undetectable) differences between originator mAb and biosimilar mAb might lead to unexpected immunogenicity in patients.
This review discusses similarity assessment between biosimilar mAbs and their originator products in terms of (i) product quality attributes; (ii) non-clinical, and (iii) clinical studies, with a focus on immunogenicity. It ends with a summary of the sparse clinical data on immunogenicity of mAb biosimilars.
How to assess similarity – quality attributes
Monoclonal antibodies are very big molecules with complex structures and functions that partly rely on their production processes. Detailed information on the production processes of originator products is not publically available, and production technologies are improving. Therefore, companies developing a biosimilar mAb will use distinct production conditions compared with the conditions used to produce the originator mAb. Differences in expression system, culture conditions, purification processes and primary packaging can evoke distinct product quality attributes such as differences in structure, post-translational modifications, biological activity and stability of the protein [15, 16]. As such, biosimilar mAb products should be tested for similarity of product quality attributes compared with the originator product before continuing with in vitro and in vivo comparability exercises.
First, a full physicochemical characterization of the mAb biosimilar is performed [17], and subsequently its characteristics are compared with the originator product. This last comparison is proof of biosimilarity at the quality level. Without going into detail on the exact methodology, it is important to mention that multiple orthogonal methods are used to study and compare the physicochemical characteristics of the mAbs. These characteristics include primary (amino acid) sequence, protein folding, truncations, post-translational modifications (glycosylation), the amount of mAb protein, the presence of degradation products and presence of aggregates [18–20]. The suitability of different excipients and primary packaging (vials, for example) will be tested, and the presence of host-cell impurities will be determined.
Regarding immunogenicity, several product quality attributes have been shown to be of importance [21]. First, the primary sequence can have an effect on immunogenicity. When the human immune system is exposed to a non-human protein, the protein will be recognized as foreign, and will induce an adaptive immune response. Early mAbs were of murine origin and caused an antibody response that greatly decreased their efficacy, most likely due to their foreignness [22]. After the development of recombinant DNA techniques, chimeric, humanized and fully human mAbs have been brought onto the market. Indeed, these mAbs are thought to be less immunogenic [23], however, it has been shown that even fully human mAbs such as adalimumab can be immunogenic [24].
The second quality attribute shown to affect immunogenicity of therapeutic proteins, including mAbs, is aggregation. [25–29]. Interestingly, not all aggregates are immunogenic, but rather a specific subset is. However, there is only limited data on the exact types of aggregates that are triggering immunogenicity, and on the amounts of aggregates needed to induce anti-drug antibodies. So, for now the aim is to lower aggregate content in the finalized product to a minimum. Why aggregates can induce immunogenicity is not exactly known, one hypothesis suggests that uptake of aggregates by antigen presenting cells and subsequent activation of the immune system is more efficient compared with uptake and activation of the immune system by protein monomers [30], while another hypothesis suggests that aggregates might mimic the structure of microorganisms [31].
Another quality attribute associated with immunogenicity of therapeutic proteins is host-cell impurities. Purification processes may be insufficient to completely eliminate impurities such as DNA, host-cell proteins, or endotoxins from the final product. As a result, therapeutic proteins might contain low levels of host-cell impurities. A study performed at FDA has shown that trace levels of lipopolysaccharide from E. coli and DNA impurities containing CpG motifs can activate the immune system and therefore increase immunogenicity of therapeutic proteins in mice [32].
Additionally, post-translational modifications such as glycosylation can have a significant impact on immunogenicity. Glycosylation is a common post-translational modification that can affect physicochemical properties of the protein such as solubility, folding, stability and biological activity. Glycosylation is species dependent, and is influenced by the cell line and culture conditions used during production processes [33]. Glycosylation of cetuximab is a well-known example of how glycan structures can affect immunogenicity. Cetuximab is a chimeric mouse–human IgG1 mAb used to treat colorectal cancer and squamous cell carcinoma of the head and neck, and contains galactose-a-1,3-galactose (a-Gal). This glycan can induce IgE responses within minutes of first exposure to cetuximab, leading to severe hypersensitivity reactions [34, 35]. a-Gal is not endogenously expressed by humans, but the cell line used to produce cetuximab (the murine Sp2/0 cell line) does give rise to this type of glycan. Possibilities to prevent a-Gal glycosylation are genetically modifying the gene encoding for a 1,3-galactosyltransferese, the enzyme involved in the synthesis of a-Gal in the Sp2/0 cell line, or changing to another expression system [21].
Last, the presence of metal and glass particles in the final product can cause immunogenicity. Spiking glass and metal particles into low immunogenic therapeutic proteins such as growth hormone and interferon beta has been shown to increase immunogenicity in mice [36, 37]. Additionally a clinical study showed that low amounts of tungsten present in the syringes of an erythropoietin biosimilar caused denaturation and aggregation of the protein and increased the incidence of immunogenicity [38].
Similarity – non-clinical studies
After determining similarity in product quality attributes between the originator and biosimilar mAb, subsequent preclinical in vitro and in vivo similarity assessment takes place. These tests are designed to optimally investigate and compare the complex biological activity of the multiple domains of the mAb and its biosimilar. These domains are the Fab region, which includes the variable domains, and which is involved in target interaction, and the Fc region, which is involved in antibody dependent cell mediated cytotoxicity and in complement dependent cytotoxicity. The Fc region, but sometimes also the Fab region, is glycosylated, and the type and size of glycan is important in effector function [39].
The EMA guideline on similar biological medicinal products containing monoclonal antibodies includes crucial steps in similarity testing—non-clinical and clinical issues are summarized below [14].
The first step describes in vitro testing. The guideline states that ‘because in vitro assays may be more specific and sensitive than studies in animals, these assays can be considered paramount in the non-clinical comparability exercise’. Comparative in vitro studies are thus considered very important, if not the most important when assessing preclinical similarity in biological functioning. In these tests, the originator and biosimilar mAb should show similar: (i) binding to target antigen(s); (ii) binding to representative isoforms of the three Fc gamma receptors; (iii) Fab-associated functions, e.g. neutralization of a soluble ligand, receptor activation or blockade; and (iv) Fc-associated functions, e.g. antibody dependent cell mediated cytotoxicity, complement dependent cytotoxicity and complement activation.
Importantly, the studies should be designed to detect minute differences between the biosimilar and in the original product in the relationship between protein concentration and biological activity, and should not just assess the response per se. Also, multiple batches should be included in similarity testing.
The second step describes the need for animal studies [14]. Important considerations when deciding if animal studies are necessary for similarity assessment are the presence of: (i) relevant quality attributes in the mAb biosimilar that have not been detected in the originator product, such as differences in post-translational modification; (ii) significantly different numbers of quality attributes than those present in the originator product; and (iii) relevant differences in formulation, e.g. use of excipients that are not commonly used for mAbs. If no concerns are identified, in vivo animal studies could be unnecessary, especially because suitable animal models are often unavailable. However, if concerns are identified, additional animal studies on pharmacokinetics, pharmacodynamics and/or safety might be necessary [14].
Nevertheless, in vivo similarity assessment between mAb originator and biosimilar in terms of pharmacokinetics, pharmacodynamics and safety (including immunogenicity) is often tested first in clinical studies. The following section describes how immunogenicity testing and comparison takes place during these clinical studies.
Similarity – clinical studies
There are multiple patient and treatment-related factors that can affect immunogenicity of therapeutic proteins during clinical studies [40]. These include the disease state of the patients, genetic factors, age, concomitant medication, duration and route of administration, and previous exposure to similar proteins. Additionally, the sampling schedule and the assay used to detect antibodies will influence immunogenicity assessment [40, 41]. Therefore, before starting a clinical study measuring immunogenicity, all these factors need to be taken into account, and an optimal study design should be chosen depending on the product.
Concerning immunogenicity testing of mAb, and their biosimilars, in particular, several critical factors should be considered in the EMA guidelines on (1) immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use, and (2) similar biological medicinal products containing monoclonal antibodies–non-clinical and clinical issues [13, 14].
First, well-validated assays to detect anti-drug antibodies should be in place, and these tests should be the same for the mAb biosimilar and originator product. Assays used to detect antibodies against mAb can be technically challenging. Often, conventional ELISAs and radio-immunoprecipitation assays involve the use of anti-immunoglobulin reagents to detect antibodies bound to the drug. However, in the case of anti-mAb antibody assays, these anti-immunoglobulin reagents will not only bind to the antibodies bound to the drug, but also the drug itself [42]. As a result, different assay approaches should be developed for mAbs. These can include the bridging format ELISA, electrochemiluminescence-based detection of anti-drug antibodies, or surface plasmon resonance-based assays. For these assays, the matrix interference due to serum or plasma components, interference due to the presence of the mAb, sensitivity and specificity should be assessed. If antibodies are detected, it is expected that the neutralizing capacity of the antibodies will be tested. The guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use suggests that competitive ligand binding assays may be the choice of assay [13].
Second, the best population to study immunogenicity should be chosen. That means that immunogenicity might be tested during a population pharmacokinetics study, in which sparse sampling and determination of drug concentration together with anti-drug antibody detection is acceptable. However, for some mAb, immunogenicity can be better detected in healthy volunteers, who develop a strong immune response after a single dose within a few days [14].
Third, the dose of the originator and biosimilar mAb used during clinical studies should be optimal for antibody assessment. The EMA guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues states that ‘some mAbs inhibit antibody formation when administered at high doses, and therefore studies conducted with low doses can be more sensitive to compare immunogenicity of the mAb biosimilar and original product’ [14].
The guidelines also stress that studies on immunogenicity are of particular importance when a different expression system is used to produce the biosimilar mAb compared with the originator product. Differences in post-translational modifications like glycosylation, could affect biological activity and immunogenicity. Therefore, they describe that ‘In some instances, IgE testing prior to clinical administration needs to be considered for patients if the mAb contains non-human carbohydrate structures, e.g. a-Gal, in order to prevent severe anaphylaxis’ [13].
An important consideration in developing biosimilar mAbs is that products with lower immunogenicity than the original products would not be excluded for market approval. However, higher immunogenicity than the originator mAb would question biosimilarity between the two products.
Furthermore, if the incidence of immunogenicity is expected to be rare and thus unlikely to be measured before market approval, an additional post-marketing study designed to detect subtle differences in immunogenicity may be requested.
MAb biosimilar products under development
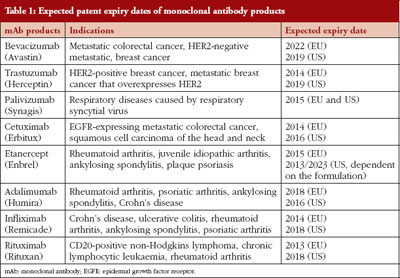
It is expected that eight mAb products will encounter patent expiration in the EU and US before 2020, see Table 1 [43, 44], opening the drug market for the development of biosimilar mAb products. Until now (August 2013), mAb biosimilars have not yet entered the EU or US markets, however, two products have received recommended approval from EMA’s Committee for Medicinal Products for Human Use (CHMP). Between 2003 and 2011, about 40% of all requests for scientific advice EMA received were for biosimilar mAbs [45], indicating the extensive interest in developing these products. This is not surprising as the monoclonal antibody products on the market are very profitable and can generate huge revenues. For example, global sales of Infliximab (Johnson & Johnson) are estimated at US$6.1 billion, and those of adalimumab (Abbott) are estimated at US$9.3 billion [45]. Because of the high number of biosimilar mAbs under development, only those with ongoing market approval, with ongoing clinical trials (clinicaltrials.gov), or with publically accessible clinical data are discussed.
Infliximab
Infliximab is a chimeric human–mouse antitumour necrosis factor alpha monoclonal antibody used for the treatment of autoimmune diseases such as rheumatoid arthritis, Crohn’s disease and ankylosing spondylitis. It is expected that the EU patent of Infliximab will expire in 2014, followed by the US patent in 2018, see Table 1.
In July 2012, the Korean FDA approved an Infliximab biosimilar (Remsima) for the Korean market, and in June 2013 two biosimilar Infliximab products (Remsima and Inflectra) received recommended approval from EMA’s CHMP for the EU market [46].
There are two clinical studies published on Remsima that could give an indication on the immunogenicity risk of this product. A phase III study by Yoo et al. performed in patients with active rheumatoid arthritis showed that besides similar efficacy and general safety, immunogenicity incidences are also similar between the originator and biosimilar product. For both products, an incidence of antibody positive patients of ~25% was found after 14 weeks of treatment and ~50% at 30 weeks of treatment [47]. The authors mention that the effect of antibody formation on treatment efficacy was tested, but they do not clearly show these results. Nevertheless, no significant difference in response was found between Infliximab and the biosimilar mAb. A phase I study by Park et al. in patients with active ankylosing spondylitis showed antibody incidences of 9.1% in patients treated with the Infliximab biosimilar and 11.0% in Infliximab treated patients at week 14. At week 30, patients treated with the Infliximab biosimilar displayed antibody incidences of 27.4%, and 22.5% of the patients treated with Infliximab were antibody positive [48]. In this study the authors do not report on the use of a bioassay (or similar) to check the antibodies for neutralizing activity, however, post hoc analysis revealed that antibody positive patients had a less robust response to the drug. No significant differences were found between the Infliximab biosimilar and Infliximab.
Etanercept
Etanercept is a fusion protein consisting of a portion of the human tumour necrosis factor receptor linked to the Fc portion of human IgG1, and is used to treat inflammatory conditions. It is expected that in 2015 the EU patent for etanercept will expire, and the US patents are expected to expire in 2013 and 2023, depending on the formulation, see Table 1.
Gu et al. have performed a study in which they showed comparable tolerability and pharmacokinetics in 19 healthy Korean men between etanercept and its biosimilar [49], however, no immunogenicity data has been published for this study. A similar study by Yi (2012) in 37 healthy volunteers also showed comparable tolerability and pharmacokinetics between etanercept and its biosimilar (HD203) [50]. It is unclear from these reports if the same biosimilar product was used.
Trastuzumab
Trastuzumab is a humanized mAb that interferes with the HER2/neu receptor, and is used to treat HER2 positive cancers, including HER2 breast cancer. Celltron, the Korea-based biosimilar company that launched the Infliximab biosimilar Remsima on the Korean market, is also pursuing the market approval of a trastuzumab biosimilar. On 6 June 2013, they applied for approval at the Korean Ministry of Food and Drug Safety (MFDS, formerly the Korea Food and Drug Administration). Clinical comparison trials have been executed for approval, however detailed data on immunogenicity are not yet publically available. In addition, Synthon has a global licensing agreement with Amgen and Watson Pharmaceuticals for the development of a biosimilar trastuzumab. A clinical phase I trial shows bioequivalence between Synthon’s trastuzumab biosimilar and the originator product, and they are now starting a phase III study with this biosimilar.
Rituximab
Rituximab is a humanized mAb used to treat CD20-positive non-Hodgkins lymphoma, chronic lymphocytic leukaemia and rheumatoid arthritis, see Table 1. Sandoz, Celltron and Biocad have ongoing clinical trials for similarity testing of their rituximab biosimilars (clinicaltrials.gov; search ‘biosimilar’). Sandoz is starting a phase III trial to compare the efficacy, safety and pharmacokinetics of their rituximab biosimilar (GP2013) to rituximab in patients with advanced stage follicular lymphoma (identifier: NCT01419665). Celltrion has an ongoing phase I study designed to demonstrate comparable pharmacokinetics between their biosimilar (CT-P10) and rituximab in patients with active rheumatoid arthritis (identifier: NCT01534884). In addition, Celltrion is starting a phase I study to provide initial evidence of safety, pharmacokinetics, pharmacodynamics, and efficacy of their rituximab biosimilar in patients with diffuse large B-cell lymphoma (CT-P10; identifier: NCT01534949). Lastly, Biocad is starting a phase III trial to prove that efficacy, safety and immunogenicity of their rituximab biosimilar (BCD-020) is equivalent to rituximab in patients with rheumatoid arthritis (identifier: NCT01759030). Unfortunately, no information on the outcome of these trials is available yet.
Conclusion
Immunogenicity is a major concern when developing biosimilar mAb. Despite the limited clinical data currently available, first reports show similar immunogenicity between infliximab and infliximab biosimilars. Although these data are encouraging, post-marketing surveillance of these products, and additional clinical studies, will be needed to obtain a good overview on immunogenicity of biosimilar mAb.
Disclosure of financial and competing interests: Dr Vera Brinks has received a grant from the Dutch Technology Foundation ‘STW’, grant number 11196, and has worked on projects that involve funding from Biogen Idec, Genzyme, Merck Serono, Sandoz, Shire and Vifor Pharma.
This manuscript received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Provenance and peer review: Commissioned, externally peer reviewed.
References
1. Casadevall N. Pure red cell aplasia and anti-erythropoietin antibodies in patients treated with epoetin. Nephrol Dial Transplant. 2003;18 Suppl 8: viii37-41.
2. Bartelds GM, Krieckaert CLM, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JWR, Dijkmans BAC, Aarden L, Wolbink GJ. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460-8.
3. Bertolotto A, Deisenhammer F, Gallo P, Sölberg Sørensen P. Immunogenicity of interferon beta: differences among products. J Neurol. 2004;251 Suppl 2: II15-24.
4. Atzeni F, Talotta R, Salaffi F, Cassinotti A, Varisco V, Battellino M, et al. Immunogenicity and autoimmunity during anti-TNF therapy. Autoimmun Rev. 2013;12(7):703-8.
5. Kaveri SV, Dasgupta S, Andre S, Navarrete AM, Repessé Y, Wootla B, et al. Factor VIII inhibitors: role of von Willebrand factor on the uptake of factor VIII by dendritic cells. Haemoph. Off. J World Fed Hemoph. 2007;13 Suppl 5: 61-4.
6. Schellekens H. The immunogenicity of therapeutic proteins. Discov Med. 2010;9(49):560-4.
7. Schellekens H. When biotech proteins go off-patent. Trends Biotechnol. 2004;22(8):406-10.
8. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Sep 5]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
9. Haag-Weber M, Eckardt KU, Hörl WH, Roger SD, Vetter A, Roth K. Safety, immunogenicity and efficacy of subcutaneous biosimilar epoetin-a (HX575) in non-dialysis patients with renal anemia: a multi-center, randomized, double-blind study. Clin Nephrol. 2012;77(1):8-17.
10. Brinks V, Hawe A, Basmeleh AHH, Joachin-Rodriguez L, Haselberg R, Somsen GW, et al. Quality of original and biosimilar epoetin products. Pharm Res. 2011;28(2):386-93.
11. Calvo B, Zuñiga L. Therapeutic monoclonal antibodies: strategies and challenges for biosimilars development. Curr Med Chem. 2012;19(26):4445-50.
12. Ebbers HC, van Meer PJ, Moors EH, Mantel-Teeuwisse AK, Leufkens HG, Schellekens H. Measures of biosimilarity in monoclonal antibodies in oncology: the case of bevacizumab. Drug Discov Today. 2013;18(17-18):872-9.
13. European Medicines Agency. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use [homepage on the Internet]. 2012 [cited 2013 Sep 5]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf
14. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies-non-clinical and clinical issues [homepage on the Internet]. 2012 [cited 2013 Sep 5]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
15. Kozlowski S, Swann P. Current and future issues in the manufacturing and development of monoclonal antibodies. Adv Drug Deliv Rev. 2006;58(5-6):707-22.
16. Tolbert WR, Rupp RG. Manufacture of pharmaceutical proteins from hybridomas and other cell substrates. Dev Biol Stand. 1989;70:49-56.
17. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1) [homepage on the Internet]. 2012 [2013 Sep 5]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf
18. Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11(7):527-40.
19. Kálmán-Szekeres Z, Olajos M, Ganzler K. Analytical aspects of biosimilarity issues of protein drugs. J Pharm Biomed Anal. 2012;69:185-95.
20. Zölls S, Tantipolphan R, Wiggenhorn M, Winter G, Jiskoot W, Friess W, et al. Particles in therapeutic protein formulations, Part 1: overview of analytical methods. J Pharm Sci. 2012;101(3):914-35.
21. Van Beers MMC, Bardor M. Minimizing immunogenicity of biopharmaceuticals by controlling critical quality attributes of proteins. Biotechnol J. 2012;7(12):1473-84.
22. Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100(2):354-87.
23. Clark M. Antibody humanization: a case of the ‘Emperor’s new clothes’? Immunol Today. 2000;21(8):397-402.
24. Van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):164-72.
25. Van Beers MM, Sauerborn M, Gilli F, Hermeling S, Brinks V, Schellekens H, et al. Hybrid transgenic immune tolerant mouse model for assessing the breaking of B cell tolerance by human interferon beta. J Immunol Methods. 2010;352(1-2):32-7.
26. Van Beers MM, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W. Aggregated recombinant human interferon beta induces antibodies but no memory in immune-tolerant transgenic mice. Pharm Res. 2010;27(9):1812-4.
27. Van Beers MM, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W. Oxidized and aggregated recombinant human interferon beta is immunogenic in human interferon beta transgenic mice. Pharm Res. 2011;28(10):2393-402.
28. Filipe V, Jiskoot W, Basmeleh AH, Halim A, Schellekens H, Filipe V. Immunogenicity of different stressed IgG monoclonal antibody formulations in immune tolerant transgenic mice. MAbs. 2012;4(6):740-52.
29. Hermeling S, Schellekens H, Maas C, Gebbink MFBG, Crommelin DJA, Jiskoot W. Antibody response to aggregated human interferon alpha2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci. 2006;95(5):1084-96.
30. Rosenberg AS. Effects of Protein Aggregates: An Immunologic Perspective. AAPS J. 2006;8(3):E501-7.
31. Sauerborn M, Brinks V, Jiskoot W, Schellekens H. Immunological Mechanism Underlying the Immune Response to Recombinant Human Protein Therapeutics. Trends Pharmacol Sci. 2010;31(2):53-9.
32. Verthelyi D, Wang V. Trace Levels of Innate Immune Response Modulating Impurities (IIRMIs) Synergize to Break Tolerance to Therapeutic Proteins. PloS One. 2010;5(12):e15252.
33. Brooks SA. Appropriate Glycosylation of Recombinant Proteins for Human Use: Implications of Choice of Expression System. Mol Biotechnol. 2004;28(3):241-55.
34. Chung CH, Mirakhur B, Chan E, Le Q-T, Berlin J, Morse M, et al. Cetuximab-induced Anaphylaxis and IgE Specific for Galactose-alpha-1,3-galactose. N. Engl J Med. 2008;358(11):1109-17.
35. Qian J, Liu T, Yang L, Daus A, Crowley R, Zhou Q. Structural Characterization of N-linked Oligosaccharides on Monoclonal Antibody Cetuximab by the Combination of Orthogonal Matrix-assisted Laser Desorption/ionization Hybrid Quadrupole-quadrupole Time-of-flight Tandem Mass Spectrometry and Sequential Enzymatic Digestion. Anal Biochem. 2007;364(1):8-8.
36. Van Beers MMC, Gilli F, Schellekens H, Randolph TW, Jiskoot W. Immunogenicity of Recombinant Human Interferon Beta Interacting with Particles of Glass, Metal, and Polystyrene. J Pharm Sci. 2012;101(1):187-99.
37. Fradkin AH, Carpenter JF, Randolph TW. Glass Particles as an Adjuvant: a Model for Adverse Immunogenicity of Therapeutic Proteins. J Pharm Sci. 2011;100(11):4953-64.
38. Seidl A, Hainzl O, Richter M, Fischer R, Böhm S, Deutel B, et al. Tungsten-induced Denaturation and Aggregation of Epoetin Alfa During Primary Packaging as a Cause of Immunogenicity. Pharm Res. 2012;29(6):1454-67.
39. Anthony RM, Wermeling F, Ravetch JV. Novel Roles for the IgG Fc Glycan. Ann N Y Acad Sci. 2012;1253:170-80.
40. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins [homepage on the Internet]. 2007 Dec 13 [cited 2013 Sep 5]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf
41. Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant. 2005;20 Suppl 6:vi3-9.
42. Aarden L, Ruuls SR, Wolbink G. Immunogenicity of anti-tumor necrosis factor antibodies-toward improved methods of anti-antibody measurement. Curr Opin Immunol. 2008;20(4):431-5.
43. GaBI Online – Generics and Biosimilars Initiative. US$67 billion worth of biosimilar patents expiring before 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Sep 5]. Available from: www.gabionline.net/Biosimilars/General/US-67-billion-worth-of-biosimilar-patents-expiring-before-2020
44. Drugbank [homepage on the Internet]. 2013 [cited 2013 Sept 5]. Available from: http://www.drugbank.ca
45. Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin J-H, Weise M, et al. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012;30(12):1179-85.
46. European Medicines Agency. European Medicines Agency recommends approval of first two monoclonal-antibody biosimilars [homepage on the Internet]. 2013 Jun 6 [cited 2013 Sep 5]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/06/news_detail_001837.jsp&mid=WC0b01ac058004d5c1. n.d.
47. Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013.
48. Park W, Hrycaj P, Jeka S, Kovalenko V, Lysenko G, Miranda P, et al. Randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605-12.
49. Gu N, Yi S, Kim T-E, Kim J, Shin S-G, Jang I-J, et al. Comparative pharmacokinetics and tolerability of branded etanercept (25 mg) and its biosimilar (25 mg): a randomized, open-label, single-dose, two-sequence, crossover study in healthy Korean male volunteers. Clin Ther. 2011;33(12):2029-37.
50. Yi S, Kim SE, Park M-K, Yoon SH, Cho J-Y, Lim KS, et al. Comparative pharmacokinetics of HD203, a biosimilar of etanercept, with marketed etanercept (Enbrel®): a double-blind, single-dose, crossover study in healthy volunteers. BioDrugs. 2012;26(3):177-84.
|
Author: Vera Brinks, MSc, PhD, Utrecht University, Department of Pharmaceutics, Utrecht Institute for Pharmaceutical Sciences, 99 Universiteitsweg, NL-3584 CG Utrecht, The Netherlands |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



An excellent birds eye view of the biosimilar therapeutic monoclonal antibody scenario. Application of in-silico tools to the research, development and further follow up to this area of medicine would be more intriguing, also probably time and wealth saving.
Dear Chandranand,
We very much appreciate your kind feedback.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office