Ocular emulsions and dry eye: a case study of a non-biological complex drug product delivered to a complex organ to treat a complex disease
Published on 2017/01/23
Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):13-23.
Author byline as per print journal: Anu Gore, PhD; Mayssa Attar, PhD; Chetan Pujara, PhD; Sesha Neervannan, PhD
|
Abstract: |
Submitted: 15 December 2016; Revised: 25 January 2017; Accepted: 27 January 2017; Published online first: 9 February 2017
Introduction
The classical generics approach based on showing pharmaceutical equivalence and bioequivalence has been the basis of the introduction of many safe and effective alternatives to innovative medicines [1–3]. This paradigm is based on the assumption that the molecular structure of the bioactive molecule is known and can be exactly reproduced and fully characterized. Typically, it is one well-defined molecule, the active pharmaceutical ingredient, embedded in an appropriate formulation. Regulatory experts from all over the globe, e.g. from the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the World Health Organization (WHO), have developed their guidance documents to assure equality in terms of quality, efficacy and safety between the innovator’s and various generic versions of these medicines.
In contrast to small molecule drugs, complex drugs such as biologicals are derived from living organisms – the generic versions of these are commonly referred to as biosimilars. Typically, for biosimilars, non-clinical and/or clinical studies are requested in addition to physicochemical (quality) analyses [4].
Then, there is the category of complex medicines that do not fall under the above definition of biologicals: the category of non-biological complex drugs (NBCDs) [5]. An NBCD is defined as a medicinal product, not being a biological medicine, and consists of different (closely related) structures that cannot be fully quantitated, characterized and/or described by (physico) chemical analytical tools alone. In addition, a product can be defined as an NBCD based on other complexities, such as complexity due to dosage form, complexity due to local delivery, and/or complexity of the disease that poses difficulties in establishing bioequivalence. The composition and quality of NBCD may be dependent on the manufacturing process and controls [6]. Therefore, with respect to assessment of similarity/equivalence for NBCD, new knowledge and policies need to be created. The challenges posed by the development of follow-on versions of NBCD that have been discussed in current literature include ‘families’ of liposomes, iron-carbohydrate (‘iron-sugar’) drugs and glatiramoids. It has been proposed that the same principles for the marketing authorization of copies of NBCD as for biosimilars be used: the need for animal and/or clinical data and the need to show similarity in quality, safety and efficacy [7].
In the current review paper, we discuss the category of ophthalmic emulsions as an NBCD. This classification is based on the unique challenges posed due to a combination of the complexities of the dosage form, the disease and the local delivery site. Ophthalmic emulsions are complex systems that are used to deliver poorly soluble drugs to the eye, a complex organ and, depending on the disease, offering potentially multiple target tissues. Further, ophthalmic dosage forms are locally acting, so pharmacokinetic (PK) bioequivalence is generally not possible or informative of delivery of efficacious levels. Recent FDA draft guidance, the June 2013 Draft Cyclosporine Bioequivalence (BE) Guidance (June 2013 Draft Guidance), on ophthalmic emulsions seems to acknowledge the complexity of ophthalmic emulsions – but there are still limitations in these to ensure bioequivalence of these generic emulsions. Using our experience with RESTASIS® ophthalmic emulsion and the recent FDA June 2013 Draft Guidance on cyclosporine ophthalmic emulsions (with its subsequent amendments) as a case study, we address areas where further understanding of locally acting ophthalmic emulsions is necessary to create scientifically robust guidance with respect to assessment of similarity/equivalence of ophthalmic emulsions.
The importance of safe and effective generic drugs
Generic drugs are intended to provide a lower cost option of safe and effective medicines thereby granting greater patient access to affordable healthcare [1]. A sound scientific basis and a robust regulatory process to demonstrate bioequivalence of the generic version of a branded drug are critical to ensuring the success of this option. In establishing the scientific basis for testing methods, the nature of the branded drug, the disease and site of action(s) must be considered. The challenges of developing a robust data package to support generics for locally acting drugs are widely recognized. In the past, FDA has generally recommended in vivo bioequivalence studies with clinical endpoints ‘because formulation differences for locally acting products may affect the availability of a drug at the site of action’. With that said, in an effort to encourage a path to bring forward safe and effective generic versions of ophthalmic medications, the regulators, including FDA, have made significant advances in categorizing appropriate paths for different drug options as presented below.
Aqueous solutions of topical ophthalmic medicines for the treatment of intraocular pressure present an opportunity to bring forward safe and effective generics. Absorption from a simple aqueous solution can be modelled in vitro. Secondly, the disease target tissue for the drug site of action in addition to the clinical endpoint of measurement of intraocular pressure are well established. However, with more complex formulations there have been challenges due to unique consideration for the eye as an organ. Several generic ophthalmic drugs unexpectedly have shown less efficacy than their reference listed drug (RLD) as well as clear drug-related toxicity, only after widespread clinical use. Some examples of past generic cases of inequivalence include diclofenac causing corneal melts [8], Ciprofloxacin suboptimal drug concentration [9], prednisolone acetate [10, 11], ketorolac and timolol gel forming solution [12, 13]. As such, differences in a generic drug product that are unknown, or known and deemed to be insignificant, could pose a significant hazard to the ophthalmic health of patients.
Overall, we learn from experience with drugs used to treat glaucoma that effective and safe ophthalmic generics are possible. However, great care must be taken depending on the drug, formulation and disease to be treated. Cyclosporine ophthalmic emulsion presents a multifaceted challenge that is presented as a case study in the section that follows.
Case study: RESTASIS® ophthalmic emulsion
RESTASIS® (cyclosporine ophthalmic emulsion) 0.05% contains cyclosporine (CsA), a topical immunomodulator with anti-inflammatory effects. Due to the poor aqueous solubility of CsA, it is formulated as an emulsion, containing castor oil, polysorbate 80, carbomer copolymer type A, purified water and sodium hydroxide to adjust pH. RESTASIS® ophthalmic emulsion is indicated to increase tear production in patients whose tear production is presumed to be suppressed due to ocular inflammation associated with keratoconjunctivitis sicca.
Complexity of disease
Keratoconjunctivitis sicca, colloquially known as dry eye disease, is among the leading causes of patient visits to ophthalmologists in the US. Twenty-three million Americans suffer from dry eye disease, which has two main causes: decreased secretion of tears by the lacrimal (tear-producing) glands, and loss of tears due to excess evaporation. Both causes lead to ocular discomfort, often described as a feeling of dryness, burning, a sandy/gritty sensation, or itchiness. Visual fatigue, sensitivity to light, and blurred vision are also characteristic of the disease. This is a serious disorder that, if left untreated or under-treated, progressively damages the ocular surface and may lead to vision loss. Dry eye disease is a disorder of the ‘tear film’, and ocular inflammation is known to play a major role in the symptoms and progression of the disease. Dry eye disease patients can suffer mild irritation (Level 1 severity). In patients with Level 2 to Level 4 severity scores, the symptoms are quite debilitating. If the condition in these cases is untreated or treated inadequately, e.g. only with an agent such as artificial tears, the disease will continue to progress, and may lead to severe eye damage and vision loss. Severe problems with untreated dry eye can also lead to corneal infection and scarring. Compared across different diseases, dry eye was found to cause degradation in quality of life that is on par with other severe disorders, such as class III/IV Angina. (AGN Citizens Petition/public response 2014).
Development of RESTASIS®
At the time Allergan initiated the RESTASIS® development programme in 1992, dry eye was a largely unmet medical need, predominantly affecting women. No therapeutic treatments were available, apart from the use of artificial tears and, for the most severe cases, blockage of the lacrimal drainage system with punctual plugs or cauterization.
Allergan’s investigators completed seminal work in the dry eye disease area, identifying the role of the T cell and chronic inflammation in the pathogenesis of dry eye disease, followed by application of cyclosporine (a drug previously used systemically to prevent transplant rejection) to target the disease locally [14–16]. The lipophilic nature of cyclosporine, however, made it extremely difficult to formulate an ocular-friendly preparation with appropriate bioavailability. The multiple target tissues of the ocular surface, e.g. cornea, conjunctiva, lacrimal glands; the composition of the tear film (not a simple salt solution), and the short retention time on the eye contributed many complex issues in creating an efficacious formulation. Various formulations were attempted with concentrations up to 2% cyclosporine; these were poorly tolerated and absorbed. Ultimately, Allergan successfully formulated RESTASIS® in its current form. An extensive clinical development programme was conducted with the emulsion formulation in its current form to demonstrate safety and efficacy and form the basis of the approval to market RESTASIS®.
Complexity of delivery to target tissues of the eye
Unlike other drug delivery routes, a topical ophthalmic formulation usually delivers the drug to the ocular tissues in a relatively short time frame of a few minutes. An eye-drop, irrespective of the instilled volume, is often eliminated rapidly within five minutes after administration, and only a small fraction (<3%) of the drug substance is delivered to the tear film and/or is absorbed and becomes bioavailable in ocular tissues. Further, normal human tear turnover acts to remove drug solution from the conjunctival cul-de-sac. Turnover may also be stimulated by many other factors including ocular irritation, formulation excipients and excipient quality, which may further enhance drug elimination.
A schematic of the various routes of drug delivery to ocular tissues is depicted in Figure 1. To achieve an effective and safe rate and extent of absorption, the drug product must accurately and differentially interact with each relevant tissue and the tear composition of a diseased eye that may change over the time course of the disease. In addition, the topical ophthalmic formulation must deliver and release its active ingredient to relevant ocular tissues in a timeframe of mere minutes (a product engineering challenge). Because ocular drug availability is extremely low, there is little or no margin for error.
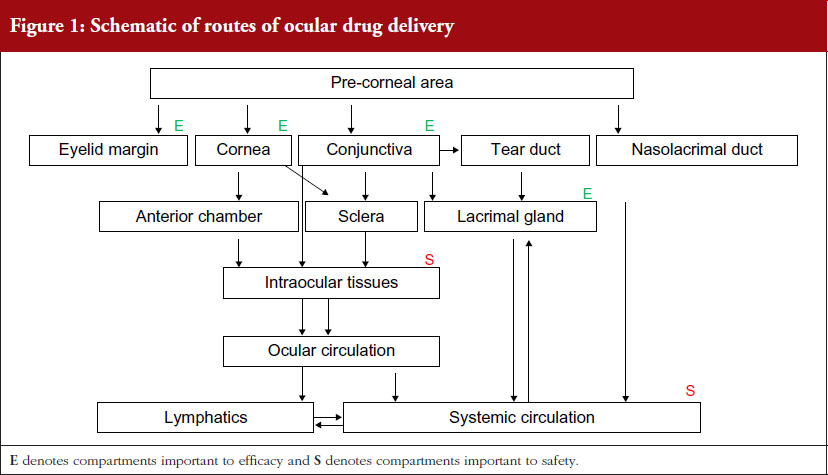
Bioavailability of topically applied cyclosporine is a result of complex differential rate processes and precorneal film dynamics that adjust continually toward equilibrium:
- Precorneal clearance of the applied dose, e.g. due to blinking and lacrimation
- Tear film drug concentration time curve, i.e. amount of cyclosporine in the tears
- Tissue permeability
- Post-tissue clearance
Delivery to the relevant ocular tissues was one of RESTASIS®’s core innovations. A complex emulsion was required to deliver the drug appropriately to the tissues. The specific target organs of RESTASIS® are understood to be the external tissues of the eye. But the required amount of drug in individual eye tissues, e.g. the lacrimal gland, conjunctiva, or epithelial layers of the cornea, to obtain relief of symptoms is not known. Nonetheless, the RESTASIS® emulsion has been shown through clinical studies to achieve the requisite tissue concentrations and time course for safety and effectiveness.
Complexity of the dosage form
Due to the poor aqueous solubility of CsA, it was formulated as an oil-in-water ophthalmic emulsion formulation in RESTASIS®. An emulsion is a dispersion of two or more immiscible liquids, stabilized by a surfactant or emulsifier coating droplets and preventing coalescence by reducing interfacial tension or creating a physical repulsion between the droplets. Although emulsions are often depicted schematically as drug containing oil droplets coated with a surfactant/emulsifier and dispersed in an aqueous phase, they are fundamentally complex dosage forms. The emulsion components can distribute themselves in various phases depending on their physicochemical properties as well as the process of manufacture of the emulsion. For example, the surfactant can partition into the water phase to form micelles in addition to acting as an emulsifier to stabilize oil droplets. The oil droplets may form globules with a range of sizes.
The drug dissolved in the oil can partition into the other phases, such as the water phase, micellar phase, microemulsion phase, or at the oil/water interface. The drug is thus expected to be present in the product in several locations including:
- in true solution in water
- in micellar equilibrium in the aqueous phase (in both the micellar core and in the surfactant palisade layers)
- in the oil droplets
- in the surfactant monolayer of the oil/water interface
- associated with viscosity agents.
The portion of the drug in each of these phases depends on not only the physicochemical properties of the drug and the emulsion composition, but is also impacted by the manufacturing process for the emulsion. This has been demonstrated for phospholipid emulsions [17, 18], and it has also been shown that process changes can impact the distribution of drugs in these phases [19].
The schematic showing the complexity of RESTASIS® emulsion is depicted in Figure 2.
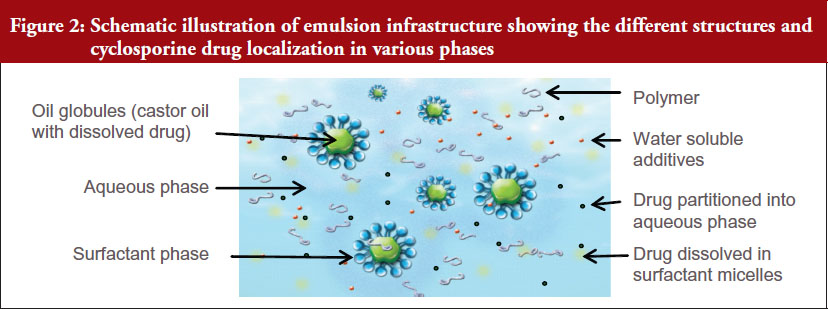
In RESTASIS® emulsion, castor oil (oil phase) is dispersed in the water phase using polysorbate 80 as the surface-active agent. In addition, the water phase contains a secondary emulsifier and viscosity agent (carbomer polymer), which provides further stability to the emulsion droplets over the product shelf life. Polysorbate 80 is a strong surfactant with a high hydrophilic/lipophilic balance value of 15 and low critical micelle concentration (CMC) of 0.014 mM or 0.0018% w/v. The level of polysorbate 80 contained in RESTASIS® is several times above its CMC, and it is expected that some portion of the surfactant will be present in the aqueous phase. This portion of polysorbate 80 is capable of solubilizing both cyclosporine as well as the drug-oil mixture by formation of micelles or microemulsion. Cyclosporine A can be present in the various phases including large and small oil globules, castor oil/polysorbate 80 interface, micellar phase, and solution phase. Further, the distribution of cyclosporine is dependent on the qualitative and quantitative composition of the formulation, grades of excipients used and the process of manufacture.
The significance of the drug distribution in various components or phases of the emulsion and its impact on efficacy and safety of the product is not fully understood. Thermodynamics dictate, however, that the drug localized in different phases may preferentially partition into different ocular tissues depending on their lipophilic or hydrophilic characteristics. For example, the drug in the aqueous phase shows greater affinity towards tissues such as the cornea or conjunctiva, and the oil compartment shows greater affinity towards lipid tissues such as the eyelid margin containing the meibomian glands. Thus, the rate and extent of distribution in these tissues can be affected by the amount of drug in the different phases of the emulsion.
Complexity due to interaction with the ocular surface
As mentioned above, the oil globules in RESTASIS® are stabilized by surfactant and secondary emulsifier and viscosity agent (carbomer polymer) which is salt sensitive. When a drop of this emulsion is applied to the eye, the salt sensitive polymer rapidly loses viscosity releasing the oil droplets and micelles on the ocular surface. The dilution and action of salt is expected to further break apart the surfactant-stabilized globules and micelles. The oil released migrates to lipid layer and coalesces to form larger droplets while surfactant associates with lipids and mucins in the tear film. These processes are schematically depicted in Figures 3A and 3B. The release of CsA on the ocular surface would be determined by rate and extent of these processes as well as the portion of CsA in each of these phases as they interact with the ocular surface.

Development pathway for generics for RESTASIS® – FDA June 2013 Draft Guidance for Cyclosporine ophthalmic emulsions
As discussed in the previous section, RESTASIS® is a formulation of a poorly water soluble drug requiring a complex delivery system – which is indicated to treat a complex disease – that requires delivery of the drug to multiple target tissues in a complex organ (eye). This complexity would make it difficult to demonstrate bioequivalence with traditional in vitro Q1, Q2 and Q3 methodologies alone, and FDA has, therefore, traditionally not allowed such a pathway. Further, RESTASIS® is intended for local delivery, and clinical testing on RESTASIS® has shown that there are negligible systemic levels of CsA – hence PK bioequivalence is not possible. FDA has historically recommended in vivo studies with clinical endpoints to demonstrate bioequivalence for complex drugs/formulations for some locally acting topical drug products. For these products, measuring the bioavailability of the active moiety or active ingredient in blood, plasma, and/or urine (and thus assessing bioequivalence of the proposed generic drug product to the RLD) does not adequately represent the safety and efficacy at the site of action because the drug is minimally absorbed into systemic circulation. Due to the complexity of RESTASIS® described in sections above, before June 2013, FDA therefore required in vivo studies with clinical endpoints to show bioequivalence to RESTASIS®.
In June 2013, FDA published a draft product-specific bioequivalence recommendation for cyclosporine ophthalmic emulsion, i.e. the June 2013 Draft Guidance, that included both in vivo and in vitro options for developing a generic of RESTASIS® . The June 2013 Draft Guidance states that a proposed abbreviated new drug application (ANDA) product may establish bioequivalence using the in vitro methodology if: (a) the proposed generic drug formulation is qualitatively (Q1) and quantitatively (Q2) the same as the RLD; (b) the proposed generic drug formulation demonstrates physiochemical properties similar to the RLD (Q3); and (c) the proposed generic drug formulation demonstrates in vitro drug release rates of cyclosporine comparable to those of the RLD. The physicochemical properties that were included in this June 2013 Draft Guidance were globule size distribution, viscosity, pH, zeta potential, osmolality, surface tension, with population bioequivalence analysis recommended only for globule size distribution.
FDA explained the scientific rationale behind the in vitro option in their response to Allergan’s citizen petition (ref. Docket no. FDA-2014-P-0304). FDA explained that it considers comparative clinical endpoint studies to be relatively insensitive at detecting the manufacturing and formulation variables, which have the greatest potential to affect the bioavailability of topical ophthalmic products. In particular, in vivo clinical endpoint studies, which measure formulation differences indirectly rather than directly, may be limited by confounding variables, such as different severities of disease and variability in the definition of the instrument used to measure efficacy, among other issues. FDA provided the rationale for selection of the six in vitro properties to be measured as each of these physicochemical properties has the potential to affect ocular bioavailability by changing the drug’s absorption, clearance, and permeation, as well as the product’s stability. Some of the properties, such as viscosity and pH, are also important for ensuring patient comfort. The in vitro release testing requested was to provide confirmation that a proposed generic drug product has a comparable release rate to that of the RLD, which can help ensure that the proposed generic drug product will deliver cyclosporine to the ocular tissues for absorption in a manner comparable to that of the RLD. In summary, FDA believed that a proposed cyclosporine ophthalmic emulsion formulation that meets the three recommended criteria outlined above – Q1/Q2 sameness, Q3 sameness, and an acceptable comparative in vitro release rate – should become available at the site of action at a rate and to an extent that is not significantly different from that of the RLD, thus meeting the requirement for demonstrating bioequivalence. However, FDA also recognized that if research later uncovers additional properties that may influence bioavailability, FDA would review the evidence to determine whether the June 2013 Draft Cyclosporine BE Guidance should be revised to include those properties. Whether the data and information in a particular ANDA are sufficient to demonstrate bioequivalence is an issue to be determined during review of a proposed generic drug product’s marketing application. (Ref. FDA response to Allergan’s Citizen’s petition Docket no. FDA-2014-P-0304).
Since the publication of the first draft guidance in June 2013, FDA has revised the guidance on two subsequent occasions. The first revision was in February 2016 and the second revision was in October 2016. These revisions have improved, in many regards, the scientific rigour of analyses as compared to the 2013 guidance. For example, the February 2016 revised draft guidance acknowledges that the active drug in cyclosporine ophthalmic emulsion is distributed across different phases of the emulsion. As a consequence, any proposed generic drug product developer must apply multiple, complementary methods, under controlled, repetitive test parameters, to accurately characterize not only globule content and size, but also distribution across the emulsion phases, using the RESTASIS® as the reference product. The October 2016 revision removes the requirement of utilizing complementary globule size measurement methods, but provides additional details regarding the statistical metric that are preferred to assess the difference, e.g. in terms of distance, between the shapes of distribution profiles. Further, the October 2016 revision to the June 2013 Draft Guidance adds the requirement that no changes, e.g. source, grade; should be made to the structure forming excipient or solubilizing excipient in the product for commercial batches unless adequate supporting data and risk assessment are provided to demonstrate that the changes will not affect the product performance and quality. There are still several gaps in the recommended testing before accepting a generic drug product as bioequivalent based on only in vitro data – these are further discussed in the later sections of this review paper.
Allergan investigations regarding suitability of the June 2013 Draft Guidance to demonstrate bioequivalence of a generic cyclosporine emulsion
To evaluate if the in vitro characterization techniques listed in FDA’s June 2013 Draft Guidance were sufficient to discriminate between non-bioequivalent emulsions, Allergan conducted extensive experimental investigations. Allergan developed a series of different methods to make emulsions similar to RESTASIS® by making selected modifications such as excipient grades, processing times, homogenization methods, sterilization techniques and/or processing temperature. This yielded nine test emulsions that were Q1/Q2 to RESTASIS® with respect to ingredients and composition. Each of these test formulations was compared against RESTASIS® using the six physicochemical properties listed in the June 2013 Draft Guidance (globule size, pH, viscosity, zeta potential, surface tension and osmolality) to evaluate if they would meet the Q3 portion of the guidance. In addition to the six physicochemical properties listed in the guidance, Allergan conducted additional testing on these products, which included drug distribution in various emulsion phases, rheology, globule size measurements with a variety of different techniques and the effect of dilution with tears on these physicochemical parameters. Further, several non-clinical assessments were conducted comparing these test emulsions with RESTASIS®.
The June 2013 Draft Guidance also required that acceptable comparative in vitro drug release rate tests of cyclosporine from the test and RLD formulations to be demonstrated. Allergan did not conduct in vitro drug release testing on these emulsions as currently no in vitro release test exists for RESTASIS®, and none has been established or validated for an ophthalmic emulsion. Common methods proposed in the literature [20–22] for testing in vitro drug release for disperse systems, such as sample and separate technique, membrane diffusion technique (dialysis sac or diffusion cells), and continuous flow-through technique are not applicable to RESTASIS® due to a variety of reasons, such as: (a) complex nature of the emulsion; (b) poor solubility of cyclosporine in relevant dissolution media; (c) difficulties in separating dissolved/released cyclosporine from micellar or oil solubilized portion; (d) similarity of molecular weights of cyclosporine with other excipients in the formulation; and (e) lack of evidence to show correlation of in vitro drug release testing to clinical performance as it cannot simulate the complex drug release and delivery to receiving compartments. In a publication [23] by Rahman et al., 2014, the researchers were unable to use in vitro drug diffusion to discriminate cyclosporine emulsions manufactured utilizing different processes – even when these emulsions showed significant differences in other parameters, such as globule size and viscosity. FDA has acknowledged the difficulties of developing suitable drug release tests for ophthalmic products in various workshops on in vitro/dissolution testing of novel/special dosage forms [24]. Further, over the last couple of years FDA has sponsored more than 20 grants for research on in vitro release characterization and drug delivery modelling for complex or locally acting drugs – nine of these grants focused on ophthalmic products in 2016 [25, 26]. Based on the current state of this science, it is not possible to utilize available methods as a tool to compare quality or bioequivalence of the emulsions.
In the following sections, results from 10 emulsions are presented, including RESTASIS® and nine test emulsions. All emulsions are only identified with numbers (or letters) and details of excipient grades, processing times, homogenization methods, sterilization techniques and/or processing temperature are not included for proprietary reasons.
1. Physicochemical characterization of emulsions manufactured with modified processes
The results for the six physicochemical properties (globule size distribution, viscosity, osmolality, pH, zeta potential and surface tension) measured for the test formulations and compared to RESTASIS® showed that several of these formulations would meet the requirements of the June 2013 Draft Guidance with respect to those six properties. For example, the guidance recommended that the globule size distribution of the emulsions should meet 95% CI established by comparing to three lots of RESTASIS® – but the method to be used for this globule size measurement was not specified. Globule size measurement methodology is highly technique-dependent and can be selected or modified to yield findings of similarity for dissimilar emulsions. Allergan used multiple methods to characterize globule size and found that five out of the 10 emulsions tested by Allergan (Emulsions 2, 4, 8, 9 and 10) would have globule size distribution that would be considered equivalent to RESTASIS® according to the globule size distribution criteria stated in the June 2013 Draft Guidance, using at least one method. For the other five physicochemical properties, no guidance was provided on comparing the results of a generic drug formulation to RESTASIS®. Allergan found that nine of the 10 emulsions would be considered equivalent to RESTASIS® with respect to these five physicochemical properties. (Note: although RESTASIS® is included as one of the 10 emulsions evaluated, it is not identified for proprietary reasons).
To summarize the results of the physicochemical properties:
- All emulsions showed comparable results to RESTASIS®with respect to pH, Osmolality, zeta potential and surface tension
- Nine of the 10 emulsions show comparable results to RESTASIS®for viscosity
- Five of the 10 emulsions met the globule size criteria as described in the June 2013 Draft Guidance by a minimum of one measurement method (either static light scattering or dynamic light scattering)
- In vitro drug release was not tested for any of the 10 emulsions due to lack of a suitable method
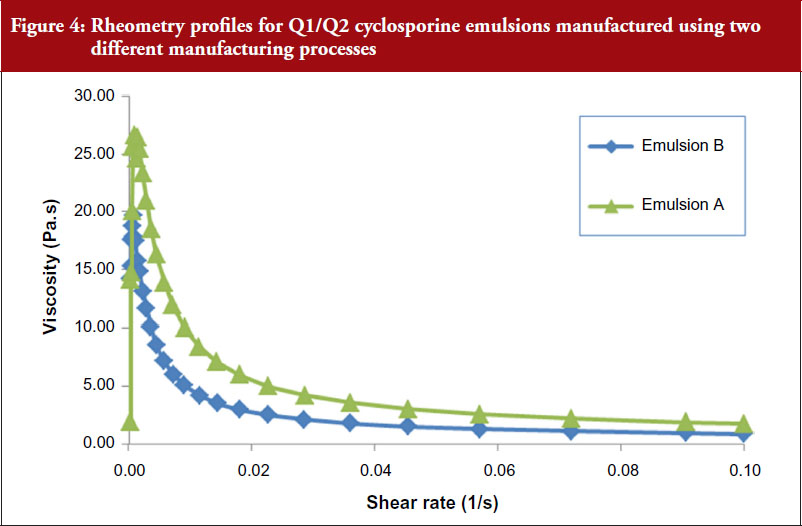
Although the four test emulsions plus RESTASIS® met the requirements of the June 2013 Draft Guidance with respect to the six physicochemical properties listed, differences were observed between those four test emulsions and RESTASIS® in other tests. For example, emulsions that appeared to meet globule size requirements by commonly used methods, such as dynamic or static light scattering were found to have quite distinct size distribution profiles when measured undiluted using Field Flow Fractionation. Similarly, emulsions with similar viscosity values measured at single point of shear were found to be distinctly different when their rheology profiles were compared as shown in Figure 4.
Another property of these emulsions that was measured for these test emulsions although it was not specified in the June 2013 Draft Guidance, was the distribution of cyclosporine in various emulsion phases. Distribution of cyclosporine within an emulsion can be affected by the manufacturing process and excipient grade. Differential distribution within an emulsion formulation may impact drug delivery to target tissues [27].
To understand the effect of manufacturing process and excipient grade on the distribution of cyclosporine in the aqueous and oil phases of the emulsions, nine emulsions were centrifuged to obtain two phases – a cream layer and a clear/translucent phase. The concentration of cyclosporine and castor oil in the clear/translucent phase solubilized in polysorbate 80 micelles or present as microemulsion was measured, see Figure 5.
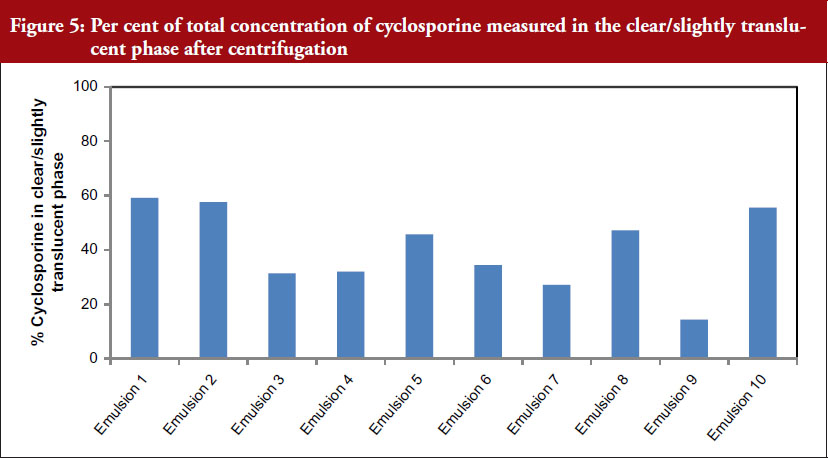
These data indicate that the manufacturing process appears to impact the distribution of cyclosporine in the emulsion. The per cent cyclosporine measured in the clear translucent phase was approximately 60% to 14%. However, Emulsions 2, 4, 8, 9 and 10 meet Q1/Q2 criteria and the physicochemical parameter measures as described in the guidance.
The significance of the drug distribution in various components or phases of the emulsion and its impact on efficacy and safety of the product is not fully understood. To better understand the impact of this differential distribution with the emulsion, these nine emulsion formulations were applied to the surface of human corneal epithelial cells and penetration into and across these cells was measured, see Figure 6.
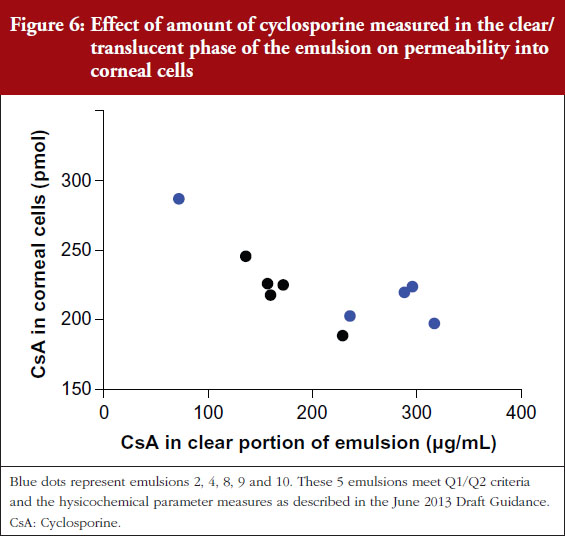
These data illustrate that permeability into human corneal epithelial cells increased as the proportion of drug in the clear/translucent portion of the emulsion decreased.
Together this indicates that modifications to the manufacturing process and/or excipient grade that appear to affect the distribution of cyclosporine within the emulsion may also affect the rate and extent of absorption into a key target site.
These data can be explained when considering the interaction at the formulation-drug-tissue interface. Specifically, thermodynamics dictate that the drug localized in different phases may preferentially target partitioning into different ocular tissues depending on their lipophilic or hydrophilic characteristics. For example, the drug in the aqueous phase shows greater affinity towards tissues such as the cornea or conjunctiva, and the oil compartment shows greater affinity towards lipid tissues such as the eyelid margin containing the meibomian glands. Thus, the rate and extent of distribution in these tissues may be affected by the amount of drug in the different phases of the emulsion as presented in Figure 7 below [27].
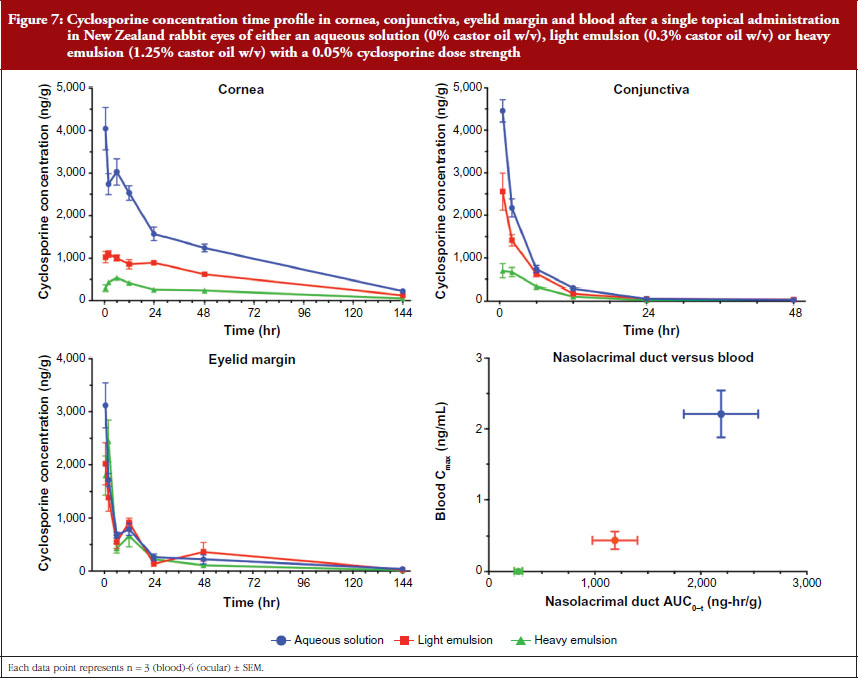
The complexity of these processes likely increases in a diseased eye and further research is required to elucidate how the time course of disease pathology and treatment affects the rate and extent of cyclosporine absorption.
2. Kinetics at the ocular surface and consequences to safety and efficacy
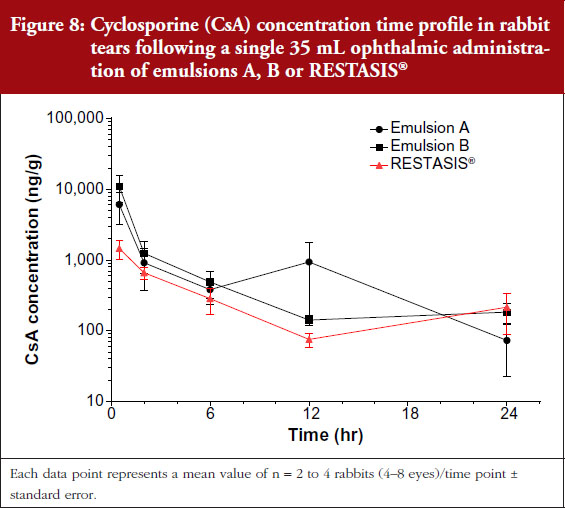
Two of the test emulsions that meet Q1/Q2 criteria and the physicochemical parameter measures as described in the June 2013 Draft Guidance were administered to New Zealand white rabbits. These two emulsions were labelled A, and B for in vivo study purposes. Tear pharmacokinetics were characterized following a single 35 μL administration and ocular tolerability (ocular discomfort and hyperemia) were characterized following multiple daily ophthalmic doses for seven days. Emulsions A and B were compared to RESTASIS®.
No anatomical barriers exist in drug partitioning from the formulation into the tear film; yet clear differences were measured in rabbit tear pharmacokinetics for these emulsions, see Figure 8.
Close examination of tear data at the 0.5 hour time point, demonstrate that emulsions A and B tended to achieve higher cyclosporine concentrations with a wider range of variability as compared to RESTASIS®, see Figure 9.
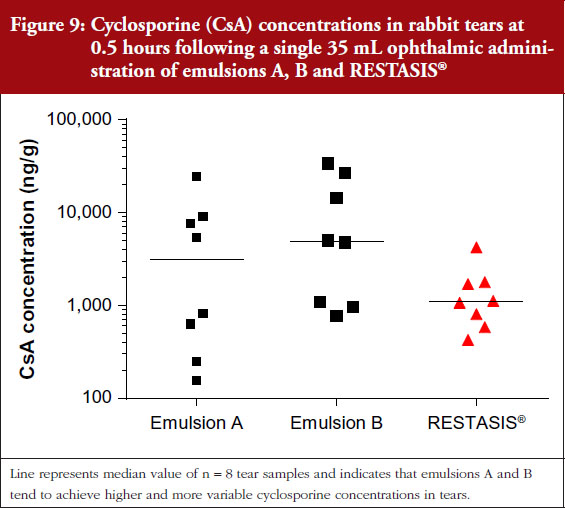
These higher cyclosporine tear concentrations are thought to contribute to the increased severity of ocular discomfort and conjunctival hyperemia observed in rabbits administered emulsions A and B as compared to RESTASIS®.
These data demonstrate that modifications to the manufacturing process and/or excipient grade may affect the extent of cyclosporine distributing from the emulsion into the tear film, which may lead to increased discomfort.
For ophthalmic emulsions, further complexity arises as we consider the changes in these physicochemical parameters when the drops are applied to the ocular surface. When a drop of the emulsion is administered to the ocular surface, it is expected to interact with the tear fluid and release the drug on the ocular surface. This interaction and drug release depends on emulsion characteristics as well as the conditions on the ocular surface. Dilution of the emulsions with normal saline (to represent ocular surface mixing with tear fluid) shows that these physicochemical parameters change significantly – with the magnitude of change being different for the various emulsions. RESTASIS® composition contains carbomer which is a salt sensitive polymer. It is expected that when this emulsion is administered to the ocular surface there will be a reduction in viscosity and destabilization of the emulsion as the emulsion gets progressively diluted with tear fluid – allowing for rapid release of the drug on the ocular surface. The kinetics and extent of this destabilization are expected to impact drug absorption and distribution to the ocular tissues. An example of this is the distribution of cyclosporine in the various phases of the emulsion. For RESTASIS® on dilution with normal saline, higher amounts of cyclosporine partition into the clear phase versus other emulsions. For example, no change in partition is observed for emulsion 9 while a large increase in the partitioning to the clear/translucent phase is observed for emulsion 4, see Figure 10. Together these observations further highlight the multifaceted complexities of these emulsions.
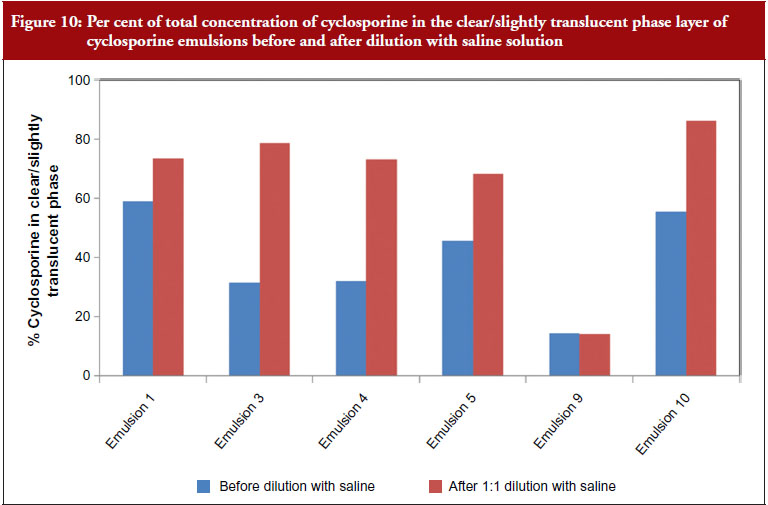
The results of these studies show that the physicochemical tests recommended by the June 2013 Draft Guidance were not sufficient to fully characterize the emulsion properties and their impact on delivery of the drug to the eye. The results of these investigations show that some of the emulsions, despite their apparent similarities based on the macro properties (globule size, pH, viscosity, zeta potential, surface tension and osmolality) can result in products that may not necessarily be bioequivalent to RESTASIS®.
FDA Revised Draft Guidance for Cyclosporine ophthalmic emulsions
The results of the investigations conducted by Allergan were provided to FDA for their consideration. Although FDA did not agree with all of Allergan’s recommendations, they did consider the information to revise the draft guidance on at least two occasions. The first revision occurred in February 2016, while the second revision occurred in October 2016. Both the revisions to the draft guidance have improved, in many regards, the scientific rigour of analyses as compared to the 2013 guidance. It is noteworthy that none of the emulsions 1–10 above met the criteria of the revised guidances. However, several key gaps still remain for the recommended testing – which are important to address prior to accepting a generic drug product as bioequivalent based on only in vitro data.
The February 2016 Revised Draft Guidance acknowledges that the active drug in cyclosporine ophthalmic emulsion is distributed across different phases of the emulsion. As a consequence, any proposed generic drug product developer must apply multiple, complementary methods, under controlled, repetitive test conditions, to accurately characterize not only globule content and size, but also distribution across the emulsion phases, using RESTASIS® as the reference product. FDA has acknowledged recently that none of the common methods known or proposed in the literature for testing in vitro drug release for disperse systems is a good fit for cyclosporine ophthalmic emulsions and that the burden is on ANDA applicants to develop a suitable in vitro method for measuring drug release. The only requirement for in vitro release methodology in the February 2016 Revised Draft Guidance is that the method should discriminate the effect of production process variability on the test formulation. This requirement may not provide sufficient guidance as many in vitro release methods could ‘discriminate’ extreme types of ‘process variability’, which produce emulsions with globule sizes and other parameters varying by several orders of magnitude; but may not be able to discriminate emulsions with more subtle differences that may be observed during development and validation of emulsions. Additional requirements are needed such that the in vitro method should be able to discriminate the effect of globule size distribution variability and the effect of differences in drug distribution in the various emulsion phases. Further, due to the unique constraints of the site of delivery, the method also needs to be capable of addressing the short duration of dose on ocular surface.
The February 2016 Revised Draft Guidance also adds more rigorous requirements for other measurements such as globule size by requiring complementary globule size analytical methods, acknowledging that ‘more than one size characterization method may be necessary to accurately detect the entire globule size distribution’. Further guidance on methods and validation criteria that would be used to determine suitability of these methods would be beneficial to potential ANDA applicants. For the other physicochemical parameters where population bioequivalence (PBE) is not required, it would be useful to clarify the statistical analysis that should be used to determine sameness of the emulsions.
The October 2016 revision to the draft guidance expands on the in vitro requirements in two major categories – globule size distribution measurements and the importance of the grade of the functional excipients in addition to the Q1/Q2 requirement. This revision removes the requirement for complementary globule size measurement methods, and continues to recommend Dynamic Light Scattering methods to obtain size distribution profiles using intensity-weighted histograms. It is acknowledged that the conventional population bioequivalence (PBE) based on just D50 and SPAN may not be sufficient to demonstrate bioequivalence. The preference of using statistical metrics to assess the difference in the globule size distribution of a generic drug product to RESTASIS® is discussed, and a suggested approach is the earth mover’s distance (EMD) method. In many aspects, this is a more rigorous approach for comparing size distributions of test emulsions with the reference product, but there is still a gap in that the selection of number of samples and replicates is left up to the applicant. In prior guidances for other emulsion products, FDA has provided the required number of minimum datasets to be used to account for variability and ensure suitable power for PBE analysis. For example, the draft guidance related to difluprednate ophthalmic emulsion states: ‘The applicants should provide no less than 10 datasets from three batches each of the Test and Reference products to be used in the PBE analysis’. Similar requirements for the cyclosporine ophthalmic emulsion guidance are required to ensure that generic drug product manufacturers employ the required level of rigour in demonstrating similarity of test product to the reference product. Further, the EMD method is a statistical measure that is very sensitive to data grouping, but no guidance has been provided for this critical attribute. Minimum requirements for this attribute should take into account the minimum number of groups used to classify size data that encompass the entire distribution profile. This clarification is necessary to ensure that the discriminatory power of this method is not compromised.
Other major gaps that continue to be carried forward into the October 2016 revision are the limitations of the in vitro release testing methodology, since no changes were made in this requirement since the February 2016 version. These have been discussed in earlier sections and are mainly due to lack of appropriate methodology given the current state of scientific understanding in this field. Given the complexity of the disease and its ocular distribution, the very short time that the drug is in contact with the target tissues, and the complex multiphase formulation, currently there is no valid in vitro release test that would be predictive of clinical performance.
While the updates to the draft guidance for cyclosporine emulsions seem to be acknowledging the complexity of ophthalmic emulsions with expectations of additional physicochemical characterization to show equivalence, a clear link to in vivo performance is still missing and the robustness of the characterization methods have not been addressed. Therefore, the following must be considered for Q1/Q2 cyclosporine emulsions to demonstrate equivalence to RESTASIS®:
- In vitro drug release methods that can be linked to in vivo performance
- Robust emulsion physicochemical characterization methods that provide meaningful information on impact to in vivo performance
Conclusion
Ophthalmic emulsions are complex systems that are used to deliver poorly soluble drugs to the eye, a complex organ with potentially multiple target tissues, to treat complex diseases. The case study of RESTASIS® described here discusses the category of ophthalmic emulsions as NBCD and describes the unique challenges posed due to complexity of the dosage form and the delivery site. The clinical performance of a complex emulsion such as RESTASIS® may depend on how it is formulated, manufactured, its mode of action, and its condition of use. The clinical performance of a complex drug product with localized delivery is expected to be a function of its physicochemical properties, but the current state of science in this area is not sufficient to demonstrate adequacy of the selected properties. Based on the totality of evidence for RESTASIS®, additional research is warranted to establish the relationship of these in vitro methods to clinical performance.
Competing interests: All authors are employees of Allergan plc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Anu Gore1, PhD
Mayssa Attar2, PhD
Chetan Pujara1, PhD
Sesha Neervannan1, PhD
1Department of Pharmaceutical Development, Research and Development, Allergan plc
2Department of Non-Clinical and Translational Sciences, Research and Development, Allergan plc
References
1. U.S. Food and Drug Administration. What are generic drugs? 19 June 2015 [homepage on the Internet]. [cited 2017 Jan 25]. Available from: http://www.fda.gov/Drugs/ResourcesForYou/Consumers/BuyingUsingMedicineSafely/UnderstandingGenericDrugs/ucm144456.htm
2. U.S. Food and Drug Administration. CFR – Code of Federal Regulations Title 21. US Food and Drug Administration, 21 Sept. 2016 [homepage on the Internet]. [cited 2017 Jan 25]. Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=320&showFR=1&subpartNode=21:5.0.1.1.8.1
3. Schellekens H, Klinger E, Mühlenbach S, Brin JF, Storm G, Crommelin DJ. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
4. Crommelin DJ, de Vlieger JS, Weinstein V, Mühlebach S, Shah VP, Schellekens H. Different pharmaceutical products need similar terminology. AAPS J. 2014;16(1):11-4.
5. Crommelin DJA, de Vlieger JSB, editors. Non-biological complex drugs: the science and the regulatory landscape (AAPS Advances in the Pharmaceutical Sciences Series) 2015th edition. AAPS/Springer; 2015.
6. TI Pharma. Non Biological Complex Drugs Working Group (NBCD). [homepage on the Internet]. [cited 2017 Jan 25]. Available from: http://www.tipharma.com/pharmaceutical-research-projects/completed-projects/nonbiologicalcomplexdrugsworkinggroupnbcd.html
7. Schellekens H, Stegemann S, Weinstein V, de Vlieger JSB, Flühmann B, Mühlebach S, Gaspar R, Shah VP, Crommelin DJ. How to regulate non-biological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2014;16(1):15-21.
8. Flach AJ. Corneal melts associated with topically applied nonsteroidal anti- inflammatory drugs. Trans Am Ophthalmol Soc. 2001;99:205-10.
9. Weir RE, Zaidi FH, Charteris DG, Bunce C, Soltani M, Lovering AM. Variability in the content of Indian generic ciprofloxacin eye drops. Br J Ophthalmol. 2005; 89(9):1094-6.
10. Fiscella RG, Jensen M, Van Dyck G. Generic prednisolone suspension substitution. Arch Ophthalmol. 1998;116(5):703.
11. Roberts CW, Nelson PL. Comparative analysis of prednisolone acetate suspensions. J Ocul Pharmacol Ther. 2007;23(2):182-7.
12. Fiscella RG, Gaynes BI, Jensen M. Equivalence of generic and brand-name ophthalmic products. Am J Health Syst Pharm. 2001;58(7):616-7.
13. Garbe D. True extent of NSAID problems now becoming clearer. Ocul Surg News. 2000;2:43-44.
14. Stern ME, Beuerman RW, Fox RI, Gao J, Mircheff AK, Pflugfelder SC. A unified theory of the role of the ocular surface in dry eye. Adv Exp Med Biol. 1998;438:643-51.
15. Stern ME, Schaumburg CS, Dana R, Calonge M, Niederkorn JY, Pflufelder SC. Autoimmunity at the ocular surface: pathogenesis and regulation. Mucosal Immunol. 2010;3(5):425-42.
16. Barabino S, Chen Y, Chauhan S, Dana R. Ocular surface immunity: homeostatic mechanisms and their disruption in dry eye disease. Prog Retinal Eye Res. 2012;31(3):271-85.
17. Benita S, Levy MY. Submicron emulsions as colloidal drug carriers for intravenous administration: comprehensive physicochemical characterization. J Pharm Sci. 1993;82(11);1069-79.
18. Sila-On W, Vardhanabhuti N, Ongpipattanakul B, Kulvanich P. The influence of physicochemical properties of preservative compounds on their distribution into various phases of oil in water submicron emulsion. PDA J Pharm Sci Technol. 2006;60(3);172-81.
19. Sila-on W, Vardhanabhuti N, Ongpipattanakul B, Kulvanich P. Influence of incorporation methods on partitioning behaviour of lipophilic drugs into various phases of a parenteral lipid emulsion. AAPS PharmSciTech. 2008;9(2);684-92.
20. Brown CK, Friedel HD, Barker AR, Buhse LF, Keitel S, Cecll TL, et al. FIP/AAPS joint workshop report: dissolution/in vitro release testing of novel/special dosage forms. AAPS PharmSciTech. 2011;12(2):782-94.
21. Liu Z, Zhang X, Wu H, Li J, Shu L, Liu R, Li L, Li N. Preparation and evaluation of solid lipid nanoparticles of baicalin for ocular drug delivery system in vitro and in vivo. Drug Dev Ind Pharm. 2011;37(4):475-81.
22. Ammar OH, Salama HA, Ghorab M, Mahmoud AA. Nanoemulsion as a potential ophthalmic delivery system for dorzolamide hydrochloride. AAPS PharmSciTech. 2009;10(3):808-19.
23. Rahman Z, Xu X, Katragadda U, Krishnaiah YS, Yu L, Khan MA. Quality by design approach for understanding the critical quality attributes of cyclosporine ophthalmic emulsion. Mol Pharma. 2014;11(3):787-99.
24. Siewert M, Dressman J, Brown C, Shah VP. FIP/AAPS guidelines for dissolution/in vitro release testing of novel/special dosage forms. Dissolution Technologies. 2003;10(1):6-15.
25. U.S. Food and Drug Administration. Lionberger R. GDUFA Regulatory Science Update. 20 May 2016 [homepage on the Internet]. [cited 2017 Jan 25]. Available from: http://www.fda.gov/downloads/ForIndustry/UserFees/GenericDrugUserFees/UCM506176.pdf
26. Choi S. Alternative approaches to demonstrate bioequivalence of ophthalmic products and the role of regulatory science. AAPS Workshop on locally acting drug products: bioequivalence challenges and opportunities. 12-13 Nov 2016. AAPS Workshop on Locally Acting Drug Products: Bioequivalence Challenges and Opportunities. Nov. 12–13, 2016.
27. Attar M, Graham R, Borbridge L, Neervanan S. The rate and extent of cyclosporine absorption is formulation and tissue dependent following topical ophthalmic administration. ARVO Annual Meeting 2014. Program Number: 457 Poster Board Number: D0076.
|
Author for correspondence: Mayssa Attar, PhD, Department of Non-Clinical and Translational Sciences, Research and Development, Allergan plc, 2525 Dupont Drive, RD-2B Irvine, CA 92612, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


