PMDA update: the current situation and future directions
Published on 2012/08/20
Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1):41-4.
|
Abstract: |
Submitted: 10 August 2012; Revised: 18 October 2012; Accepted: 8 March 2013; Published online first: 11 March 2013
Introduction
The Pharmaceuticals and Medical Devices Agency (PMDA) is Japan’s drug regulatory agency whose mission is to protect the public health by assuring safety, efficacy and quality of pharmaceuticals and medical devices. This paper summarizes the structure and activities of the PMDA, including the setting up of a new Office of Standards and Guidelines Development, the Science Board and the Office of Review Innovation.
Recent changes at PMDA have enabled the organisation to shorten the review time for both priority- and standard-review products, and decrease the ‘drug lag’, or time difference between Japan and the US for new drug approval. The latter has been achieved mostly through speeding up the post-application review process. Efforts are also continuing to speed up the application process such as by offering advice to start-ups and academics with promising candidate medical products on how to gather the data necessary for a complete application.
The requirements for approval application of biosimilars are summarized in the guidelines on the ‘Quality, safety and efficacy assurance of biosimilars’ issued by the country’s Ministry of Health, Labour and Welfare (MHLW). Two biosimilars have been approved in Japan by 2009.
With regard to safety, PMDA is expanding its source of adverse drug reactions (ADR) information by utilizing electronic databases on insurance claims and medical records. PMDA also operates a Japanese language email messaging system for healthcare professionals carrying information on the safety of medical products.
Bearing in mind the globalization of development, manufacture, distribution and consumption of medical products, PMDA is taking steps to enhance its international activities, including cooperation across Asia and international harmonization of regulatory standards, according to its international vision.
Organization
PMDA has 678 regular staff as of 1 April 2012, having increased the number of reviewers and safety information analysts in order to better fulfill its mission. The agency plans to have more than 700 employees in fiscal year 2012, about three times as many as those employed when PMDA was established in 2004.
Recent organisational changes include the establishment of the Office of Standards and Guidelines Development in July 2011, the Science Board and the Office of Review Innovation in May 2012. The Office of Standards and Guidelines Development coordinates activities across the offices related to international standards such as the International Organization for Standardization (ISO), regulatory guidelines, orphan drugs and paediatric drugs. It also deals with development of the Japanese Pharmacopoeia.
PMDA established its Science Board as a high-level body to provide PMDA reviewers with scientific advice on assessing the products of advanced technologies and science, in close collaboration with leading academic researchers. The Board consists of external experts who are active in the frontlines of medical, dental, pharmaceutical, and engineering research [1].
In order to liaise well with the Science Board and guide its related offices, PMDA also established the Office of Review Innovation, led by one of the three Executive Directors of PMDA.
Drug lag
Current status
As shown in Table 1, PMDA has shortened the review time for both priority review products and standard review products. Table 2 shows how PMDA has also decreased the ‘drug lag’, or time difference in drug approval between Japan and the USA. This is the sum of the following two components. First, the ‘application lag’ for a particular year is defined as the median value of the time difference between the receipt of the applications at the US Food and Drug Administration (FDA) and PMDA, for the drugs approved in Japan within the year. Second, the ‘post-application (in-review) lag’ is defined as the median value of the difference between the times taken for review in the US and Japan for the drugs approved in Japan within the year.
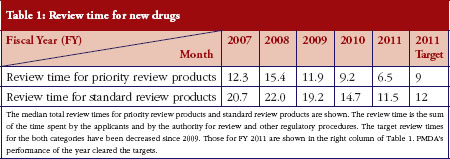
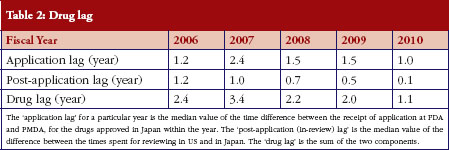
Table 2 also shows that while the post-application time lag is decreasing smoothly, the application lag has not significantly decreased, and remains as Japan’s next challenge.
Efforts to diminish the drug application time lag
PMDA’s efforts to reduce the time lag for drug applications include the enhancement of the regulatory science and the consultation service, which provide valuable support to drug development and facilitate regulatory submission. Currently, PMDA offers more than 30 different kinds of consultation. A few examples are touched upon here. Under the category ‘prior assessment consultation’, started by PMDA in 2009, new drug application (NDA) data are reviewed before formal submission, as they become available in portions. Under the category ‘pharmacogenomics biomarker consultation’, PMDA’s cross-sectional team consults on qualification of specific biomarkers. The consultation does not deal with particular products, but with the values of proposed biomarkers for regulatory decision-making, which helps in advancing regulatory science in that the established biomarkers will serve as new tools for drug development and review.
In July 2011, PMDA started a new category, the ‘pharmaceutical affairs consultation on R & D strategy’, intended to help start-ups that and academics who have promising candidate medical products. These small enterprises and researchers often lack the know-how for overcoming many of the obstacles to regulatory approval. PMDA experts provide information on which non-clinical and clinical studies are required for regulatory submission. The new service, whose focus is on the early stages of drug development and medical devices, is also expected to support innovation generally.
Generic drugs and biosimilars
The share of generic drugs in the Japanese prescription drug market was 23% by volume and 10% of sales for the fiscal year 2011. There appears to be sizeable room for further growth in the generics market. MHLW has been taking measures to enhance the use of generic drugs to alleviate the financial burden on patients and the national treasury, with the goal of increasing the generics market share to 30% by volume [2].
PMDA’s Office of OTC/Generic Drugs is responsible for reviewing generic drug applications. Table 3 summarizes the number of generic drugs approved and the median time taken for review and other regulatory procedures (not including the time spent by applicants) between fiscal years 2007 to 2011. PMDA has already achieved the target review time for generic drugs of 10 months, published in its mid-term plan for 2009 to 2013 [3].
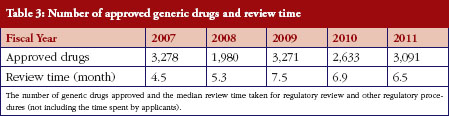
To assist with the approval of generic drugs, PMDA added the following two items to its consultation service menu in October 2011: ‘consultation on bioequivalence tests for generics’, and ‘consultation on quality requirements for generic drugs’.
In the current Japanese regulation, biosimilars are treated differently from chemical generics. PMDA’s Office of Biologics I is reviewing applications for follow-on biologics in cooperation with the Offices of New Drugs I to V. Japan’s regulations on biosimilars are still relatively young and PMDA reviewers are making daily efforts to achieve best practice.
MHLW published the ‘Guidelines for the quality, safety and efficacy assurance of biosimilars’ (Notification 0304007) on 4 March 2009 [4], which prescribes the general requirements and recommendations for applicants to (a) establish an independent manufacturing process; (b) identify the product’s quality characteristics in detail, and (c) demonstrate the product’s comparability to its reference product in terms of quality, safety and efficacy. The necessary tests can cover quality, pharmacology, toxicity, pharmacokinetic, pharmacodynamic, and clinical safety (e.g. immunogenicity) and efficacy [5].
Until Spring 2012, MHLW/PMDA has approved two biosimilars: somatropin and epoetin alpha, both in 2009 [6–8].
Safety measures
Safety of medical products is one of the three main interests for PMDA, along with the product evaluation and relief service for those experiencing adverse drug reactions. The Offices of Safety at PMDA work together with the Review and Relief Offices as well as MHLW, to enhance safety measures, including through the MIHARI project and ‘PMDA medi-navi’ service.
The MIHARI project aims at expanding the adverse drug reaction (ADR) information source to include electronic medical information such as claims data and medical records. Knowing ADR incidence together with information on the population taking the drug – such as its size, age distribution – allows quantification and comparison of the incidence across populations, which is often difficult with a system relying on traditional spontaneous ADR reports. With the input from an advisory board consisting of external experts, PMDA has been studying the potential advantages and disadvantages of using electronic databases of insurance claims and medical records stored at hospitals since 2009. PMDA is also developing the methodology for performing quantitative epidemiological analysis on the databases and plans to have the system in operation by 2013 [9].
‘PMDA medi-navi’ is the name of PMDA’s Japanese language email messaging system for healthcare professionals carrying information on the safety of medical products, including regarding the revision of labelling and major product recalls from the market. As of June 2012, the service has more than 60,000 subscribers, about 70% of whom are healthcare professionals in hospitals, clinics, and pharmacies, and 20% involved in manufacturing and marketing of medical products.
Advancing regulatory science
In its day-to-day activities, PMDA is consciously using and trying to advance regulatory science with a view to ensuring that its regulatory judgements are based on well-founded scientific evidence, accurate estimation and evaluation, and the fair balance of many factors including risks and benefits of medical products. PMDA’s efforts not only maintain a high standard for regulatory science, but also contribute to the development of medical products by industry. Reviewers and safety information analysts at PMDA conduct and publish research in their respective fields of expertise [10].
In order to advance regulatory science and foster regulatory scientists both within and outside PMDA, the agency has collaborations with 13 graduate schools across Japan (as of June 2012), in the fields of medicine, pharmaceutical sciences, and other related sciences.
Advancing regulatory science is a major issue not only for PMDA but also for MHLW of Japan. MHLW has set aside Yen 1.2 billion (US$15 million), to promote (a) the establishment of a regulatory science-based evaluation system for the safety and efficacy of innovative drugs and medical devices, and (b) personnel exchange among MHLW, PMDA, National Institute of Health Sciences, universities and other research institutes. In addition, MHLW has set aside a budget of Yen 366 million (US$ 4.6 million) for developing guidelines for speedy regulatory review.
International activities
In this age of globalization, no regulatory authority can fully carry out its duties without cooperating with foreign regulatory agencies. PMDA aims for a ‘win–win’ relationship by providing benefits to international partners.
International vision ‘PMDA EPOCH’
PMDA has developed a vision for international cooperation named ‘PMDA EPOCH Toward 2020’, which illustrates its goal of becoming one of the world’s premier medical products regulatory agencies, comparable to FDA and the European Medicines Agency (EMA), by 2020 [11, 12].
The first two letters of the acronym ‘EPOCH’ stand for Excellence in Performance. PMDA aims to secure the highest level of performance in terms of its product review, safety measures, relief service for those with adverse drug reactions, research in regulatory science, and information transmission. The second and the third letters stand for Partnership with the Orient, or Asia. PMDA is determined to advance cooperation across Asia to improve the level of medical products regulation, and to enhance the worldwide sharing of information and opinions of the Asian community. The letters ‘C’ and ‘H’ stand for Contribution to international Harmonization. As one of the world’s premier regulatory agencies, PMDA will actively contribute to international harmonization of regulations, guidelines and standards. PMDA is now drafting a ‘PMDA Roadmap’ outlining the stages involved in achieving PMDA’s EPOCH vision by 2020.
|
Summary of PMDA EPOCH vision 2020
|
Bilateral and trilateral relationship
PMDA has been cultivating bilateral relationships with Therapeutic Goods Administration (TGA, Australia), the State Food and Drug Administration (SFDA, China), European Medicines Agency (EMA, EU), National Agency of Drug and Food Control (NADFC, Indonesia), The Irish Medicines Board (IMB, Ireland), The Korea Food & Drug Administration (KFDA, Korea), the Health Sciences Authority (HSA, Singapore), the Medicines and Healthcare products Regulatory Agency (MHRA, UK) and Food and Drug Administration (FDA, USA). PMDA has confidentiality arrangements with some of these agencies [13].
Of the various relationships PMDA maintains, the tripartite cooperation among China, Japan and Korea is of particular importance. Since 2007, MHLW and PMDA have been actively involved in tripartite cooperation in clinical research with SFDA and KFDA. With the responsible directors-general of the three agencies meeting every year, and each party coordinating one of the three joint projects – research on ethnic factors (Japan), regulatory systems surrounding good clinical practice implementation (Korea), and joint clinical trials in East Asia (China) – the activities are gathering momentum [14].
Harmonization activities
PMDA has been actively participating in global regulatory harmonisation activities, including the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), the Global Harmonization Task Force (GHTF), the International Cooperation on Cosmetics Regulation (ICCR), and the International Medical Device Regulators Forum (IMDRF).
Information transmission
PMDA provides various types of regulatory information in English on its website. This includes review reports of the approved products, safety information, ‘PMDA Updates’ with monthly news, and ad hoc news releases. PMDA values English language communication for promoting mutual understanding as the basis for effective international cooperation between regulatory authorities of different countries. PMDA is accordingly increasing the size of its workforce engaged in translating Japanese regulatory information into English.
PMDA’s basic policy
In 2008, PMDA published ‘PMDA philosophy’, as the fundamental mindset in carrying out its mission of improving the public health and safety of our nation. With this in mind, PMDA staff are implementing their services in the three major fields of product review, post-market safety measures, and relief for adverse health effects.
Regulatory science and international cooperation are crucial pillars of PMDA’s activities. PMDA believes that it should keep improving the scientific basis of its judgements in order to protect Japanese public health and build public trust.
|
PMDA philosophy
|
Acknowledgement
The authors wish to thank the Offices of Planning and Coordination, OTC/Generic Drugs, Biologics I, Safety I, Review Management,and Regulatory Science of PMDA for their helpful advice.
Competing interests: None.
Provenance and peer review: Commissioned; internally peer reviewed.
Co-authors
Toshiyoshi Tominaga, PhD, Office of International Programs
Tatsuya Kondo, MD, PhD, Chief Executive
Pharmaceuticals and Medical Devices Agency, Shin-Kasumigaseki-Building, 3-3-2 Kasumigaseki, Chiyoda-ku, Tokyo 100-0013, Japan
References
1. Pharmaceuticals and Medical Devices Agency, Japan. The outcomes of the second Science Board Meeting. Press Release [homepage on the Internet]. 31 July 2012 [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/english/international/pdf/arrangements/20120803news_e.pdf
2. Ministry of Health, Labour, and Welfare, Japan. Promotion of the Use of Generic Drugs [homepage on the Internet]. [Cited 2012 Oct. 18]. Available from: http://www.mhlw.go.jp/english/policy_report/2012/09/120921.html
3. Pharmaceuticals and Medical Devices Agency, Japan. Second mid-term plan [homepage on the Internet]. [cited 2012 Oct 18]. Available from: http://reiki.pmda.go.jp/FormLibrary/A_001_Index.aspx
4. Pharmaceuticals and Medical Devices Agency, Japan. MHLW, Guidelines for the quality, safety and efficacy assurance of follow-on biologics [homepage on the Internet]. [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/english/service/pdf/notifi cations/PFSB-ELD-0304007.pdf
5. Yamaguchi T, Arato T. Quality, safety and efficacy of follow-on biologics in Japan. Biologicals. 2011;39(5):328-32.
6. Arato T, Yamaguchi T. Experience of reviewing the follow-on biologics including somatropin and erythropoietin in Japan. Biologicals. 2011;39(5):289-92.
7. GaBI Online – Generics and Biosimilars Initiative. Approval of first biosimilar G-CSF in Japan [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2012 Oct 18]. Available from: www.gabionline.net/Biosimilars/News/Approval-of-first-biosimilar-G-CSF-in-Japan
8. GaBI Online – Generics and Biosimilars Initiative. Japan approves second biosimilar G-CSF [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Mar 8]. Available from: www.gabionline.net/Biosimilars/News/Japan-approves-second-biosimilar-G-CSF
9. Pharmaceuticals and Medical Devices Agency, Japan. MIHARI Project. [homepage on the Internet]. [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/english/service/mihari_project.html
10. Pharmaceuticals and Medical Devices Agency, Japan. Recent publications by PMDA staffs [homepage on the Internet]. [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/regulatory/english_publication.html
11. Pharmaceuticals and Medical Devices Agency, Japan. PMDA International vision [homepage on the Internet]. 2011 Nov [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/english/international/pmda_international_vision.html
12. Tominaga T, Ando Y, Kondo T. International vision and strategy for drug regulatory authority: the PMDA international vision. Clin Pharmacol Ther. 2012;92(3):349-51.
13. Pharmaceuticals and Medical Devices Agency, Japan. International arrangements [homepage on the Internet]. [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/english/international/arrangements.html
14. Pharmaceuticals and Medical Devices Agency, Japan. China/Japan/Korea tripartite cooperation [homepage on the Internet]. 2011 Oct 31 [cited 2012 Oct 18]. Available from: http://www.pmda.go.jp/english/international/cooperation.html
|
Author for correspondence: Yuki Ando, BA, Offi ce of International Programs, Pharmaceuticals and Medical Devices Agency (PMDA), Shin-Kasumigaseki-Building, 3-3-2 Kasumigaseki Chiyoda-ku, Tokyo 100-0013, Japan |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


