Rationalizing FDA guidance on biosimilars—expediting approvals and acceptance
Published on 2018/06/01
Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):84-91.
|
Abstract: |
Submitted: 21 May 2018; Revised: 13 July 2018; Accepted: 17 July 2018; Published online first: 23 July 2018
Background
H.R. 3590 Sec. 7002 [1], commonly known as the Biologics Price Competition and Innovation Act (BPCI Act), a part of the Affordable Healthcare Act, was enacted in 2009 to introduce biosimilars (copies of biological products coming off patent) to the market. Since then, the US Food and Drug Administration (FDA) has licensed 11 products (as of June 2018) [2], comprised of eight molecules: adalimumab, bevacizumab, epoetin, etanercept, filgrastim, infliximab, pegfilgrastim and rituximab. Under the New Drug Application (NDA) filing, insulin glargine products (Lusduna [3] and Basaglar [4]) have also been approved by FDA and from March 2020 all insulin products can be approved as biosimilars [5]. FDA has issued several draft and final guidelines to industry on demonstrating biosimilarity [6], which is the primary determinant for licensing a product, either as a biosimilar or an interchangeable biosimilar. Interchangeable biosimilars are… a separate category of biosimilar products that are additionally tested to demonstrate that automatic substitution of an originator product with the ‘interchangeable biosimilar product’ will not result in reduced efficacy or increased side effects.
The slow entrance and acceptance of biosimilars in the US is the result of high costs and long development times – nearly US$250 million and almost eight years [7] – as well as a gross misunderstanding of the safety of biosimilars among prescribers and the public, principally due to ideas put forward by the products’ originator companies. While FDA has recently launched a campaign to educate stakeholders regarding the safety of biosimilars [8], much work remains to be done in order to simplify and expedite licensing of biosimilars, as emphasized by the author in several publications and a recent citizen petition to FDA [9–11].
The suggestions made in this paper come from decades of experience in developing biosimilar products globally, including through the biosimilars (351(k)) and NDA pathways (505(b)(2)).
The BPCI Act
This review focuses on actions FDA, biosimilar developers and other stakeholders can take, within the boundaries of the statute, to make biosimilars more accessible. In order to first understand what is feasible for FDA and how the relevant guidelines are constructed, a review of the BPCI Act is necessary. Given below are excerpts from the BPCI Act that are relevant to guidelines for the approval of biosimilars by FDA:
(k) Licensure of biological products as biosimilar or interchangeable
- (1) In general
- Any person may submit an application for licensure of a biological product under this subsection.
- (2) Content
- (A) In General
(i) Required Information- An application submitted under this subsection shall include information demonstrating that
- (I) the biological product is biosimilar to a reference product based upon data derived from
- ‘(aa) analytical studies that demonstrate that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components;
- ‘(bb) animal studies (including the assessment of toxicity); and
- ‘(cc) a clinical study or studies (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics) that are sufficient to demonstrate safety, purity, and potency in one or more appropriate conditions of use for which the reference product is licensed and intended to be used and for which licensure is sought for the biological product;
- (II) the biological product and reference product utilize the same mechanism or mechanisms of action for the condition or conditions of use prescribed, recommended, or suggested in the proposed labeling, but only to the extent the mechanism or mechanisms of action are known for the reference product;
- (III) the condition or conditions of use prescribed, recommended, or suggested in the labeling proposed for the biological product have been previously approved for the reference product;
- (IV) the route of administration, the dosage form, and the strength of the biological product are the same as those of the reference product; and
- (V) the facility in which the biological product is manufactured, processed, packed, or held meets standards designed to assure that the biological product continues to be safe, pure, and potent.
- (ii) Determination by Secretary. —The Secretary may determine, in the Secretary’s discretion, that an element described in clause (i)(I) is unnecessary in an application submitted under this subsection.
- (A) In General
- (4) Safety standards for determining interchangeability
- Upon review of an application submitted under this subsection or any supplement to such application, the Secretary shall determine the biological product to be interchangeable with the reference product if the Secretary determines that the information submitted in the application (or a supplement to such application) is sufficient to show that—
(A) the biological product—
(i) is biosimilar to the reference product; and
(ii) can be expected to produce the same clinical result as the reference product in any given patient; and
(B) For a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch.
- Upon review of an application submitted under this subsection or any supplement to such application, the Secretary shall determine the biological product to be interchangeable with the reference product if the Secretary determines that the information submitted in the application (or a supplement to such application) is sufficient to show that—
The statutory requirements provided in section k.2.A.i.I form the basis of biosimilar development. Remarkably, these requirements are left to the discretion of FDA, as shown in k.2.A.ii, leaving only section k.2.A.i.I–V as unchangeable by FDA [1]. Interchangeable licensing has additional legislation, as shown in k.4.A–B. A biosimilar product must demonstrate the same clinical results as the reference product, which can only be shown by patient testing. Studies using a switching-and-alternating protocol, where an originator biological product is switched with a biosimilar product and then back to the originator product, must show no diminished efficacy and no greater risk when compared to the reference product without alteration or switching. This legislation prevents FDA from making any changes to the requirements for interchangeable biosimilars; thus this paper will address issues related to the approval of biosimilars only.
Actions recommended for US FDA
To allow the faster development and adoption of biosimilar products, the following changes in the regulatory approval process are recommended:
- Modify the current requirement for bridging studies between a US-licensed product and a non-US approved comparator, provided the non-US product meets certain specifications, such as same indications, same dosage form and approved using essentially the same dossier as the US-reference product, to establish biosimilarity.
- Present clear scientific evidence to the public and, more particularly, prescribers that a biosimilar product has no ‘clinically meaningful difference’ from the originator product and thus should be acceptable for naïve patients, without involving the legality of substitution issue.
- Encourage the development of in vitro immunogenicity testing methods to reduce exposure to test subjects, which would have ethical advantages and allow comparison of multiple batches of the biosimilar candidate product, improving safety evaluation.
- Replace the current arbitrary comparisons of critical quality attributes, such as protein content or variations in known impurity profiles with clinically-relevant limits and ranges in testing analytical similarity, animal toxicology, pharmacokinetic/pharmacodynamic (PK/PD) immunogenicity, and other safety and efficacy attributes.
- Minimize clinical studies by combining studies intended to establish immunogenicity, efficacy and PK/PD profiles to avoid unnecessary testing on patients.
- Clarify policy on analytical method validation.
- Change the requirement for the use of commercial-scale batches for determination of biosimilarity.
These recommendations are also, in part, the subject of a citizen petition filed by the author to FDA [11].
1. Waiver bridging studies
Developing biosimilars is costly and requires developers to formulate a global strategy where one regulatory dossier can be used to secure regulatory approvals in multiple jurisdictions. Since the BPCI Act requires that a biosimilar be similar to its locally licensed originator (that is, a product approved under Sect. 351(a) of the Public Health Service Act of 1942, as amended) [1], developers are not permitted to use a non-US product as a reference product. As a result, creating a global dossier requires three-way studies, i.e. a US-licensed product, a non-US product, and the biosimilar candidate. To reduce the burden of additional studies, and to reduce unnecessary exposure to humans, several regulatory authorities have established clear policies on bridging studies [12], as shown in Table 1.
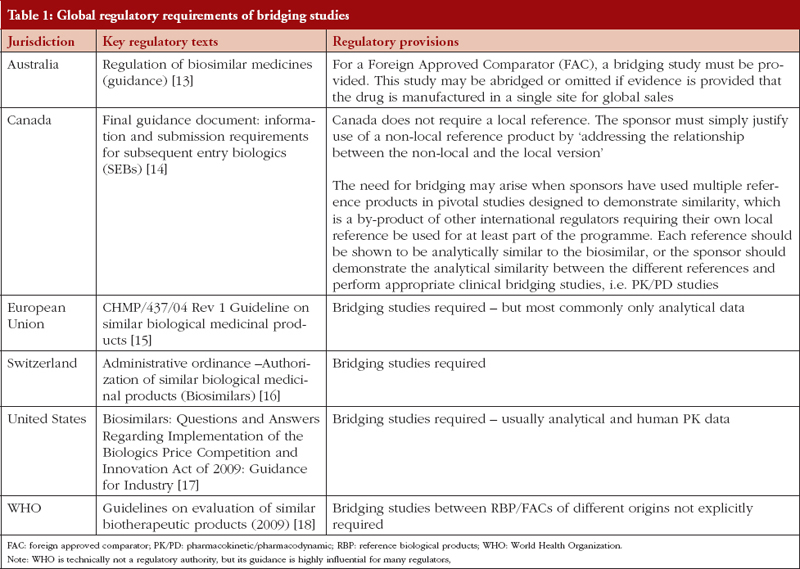
The most stringent requirements are imposed by FDA, whose requirements include analytical similarity and PK/PD studies. It should be noted that FDA requirements for bridging studies are not clearly defined but accepted as the default position of FDA. As such, there is no legal obstacle to FDA changing its position and allowing developers to request a waiver to use a non-US-licensed product as the reference product, provided the conditions, enumerated below, are met:
- The non-US reference product meets all statutory requirements as shown in section k.2.A.i.II–V; and
- The non-US product received approval in its respective jurisdictions by presenting very similar original data, including clinical safety and effectiveness, as the US-licensed reference product; and
- The regulatory filing is not intended to claim interchangeable status for the biosimilar product; or
- The non-US reference product was judged to be equivalent to the US-licensed product in any regulatory filing that presented a bridging study, such as the recent approvals of infliximab [19] and bevacizumab [20].
FDA Commissioner Dr Scott Gottlieb agrees with the suggestions made above, however, there is wider FDA concern that legislative action would be required to make changes to current practice [21]. The author finds no legal reason why this change cannot be made by FDA.
2. Encourage substitution for naïve patients
The BPCI Act creates two categories of biosimilar products: biosimilar and interchangeable biosimilar. The latter classification was intended to allow the automatic substitution of an originator product with a biosimilar product. The labelling of an interchangeable biosimilar requires in patient studies to demonstrate similar efficacy. When a biosimilar product is repeatedly administered, the two products (biosimilar and reference) are alternated to establish that there is no reduction in efficacy or increase in side effects caused by the biosimilar. As a result of the complexity of these studies making them extremely expensive to conduct, developers have been reluctant to file for interchangeable status; and FDA is yet to approve a product as an interchangeable biosimilar. However, there is a need for a strategic approach to allow the substitution of biosimilars based on how FDA characterizes a biosimilar.
‘A biosimilar is a biological product that is highly similar to and has no clinically meaningful differences from an existing the FDA-approved reference product [22]’.
From a scientific and clinical viewpoint, if a product is clinically equivalent, there is no reason why it should not be prescribed to naïve patients. This view is shared by FDA Commissioner Dr Scott Gottlieb who stated that ‘payors can also lead the way in formulary design by making biosimilars the default option for newly diagnosed patients. They can share the savings with patients, maybe by waiving co-insurance [23]’.
The author therefore requests that FDA:
- Declare the official position that a licensed biosimilar product has no clinically meaningful difference and that it can be substituted for the originator product when the originator product is prescribed for a naïve patient.
- Educate prescribers that biosimilars are safe and equally effective, with no risk of additional immunogenicity when used in naïve patients—the most significant barrier to the entry of biosimilars into the US market.
- Motivate and enforce the adoption of biosimilars by payers and make the pricing structure more transparent in order to demonstrate cost savings to patients and prescribers.
3. Allow in vivo immunogenicity study waivers
Immunogenicity is defined as the propensity of biological drugs to generate an immune response to self and related proteins, which may include non-clinical effects and adverse clinical events. Immune responses to biological drugs may hamper their biological activities and result in adverse events, not only by inhibiting the efficacy of the therapeutic element but also by cross-reactions with endogenous protein, leading to loss of its physiological function. For example, neutralizing antibodies to erythropoietin can cause pure red cell aplasia by also neutralizing the endogenous protein. The effects of immunogenicity in biological drug development can be summarized as follows:
- Effects on bio-availability, safety, efficacy and PK, including potential cross-reactivity with endogenous proteins
- Inhibition of the function of endogenous proteins
- Injection site reactions and other systemic reactions, mild or life-threatening
- Formation of anti-drug antibodies, neutralizing antibodies, immune complexes and anti-idiotypic antibodies
Immunogenicity, as stated in FDA guidelines on biological drugs, must be assessed in the target population since animal testing and in vitro models cannot predict immune response in humans [24]. Immunogenicity also has a role in demonstrating product comparability following manufacturing changes. Even minor changes can potentially affect the bioactivity, efficacy or safety of a biological drug. As a result, FDA is making important advances in predicting immunogenicity [25], in particular promoting the use of in vitro immunogenicity assays.
The European Medicines Agency (EMA) provides the following statement regarding use of alternate methods of testing immunogenicity:
‘… ongoing consideration should be given to the use of emerging technologies (novel in silico, in vitro and in vivo models), which might be used as tools during development or for the first estimation of risk for clinical immunogenicity. In vitro assays based on innate and adaptive immune cells could be helpful in revealing cell-mediated responses [26]’.
The characterization and screening of biosimilars for physicochemical determinants or formulation-based factors aid both in the prediction of immunogenicity and in the development of less immunogenic therapeutic agents, considering impurities, heterogeneity, aggregate formation, oxidation and deamidation of the molecule. Moreover, predicting potential immunogenic epitopes in therapeutic biologicals is an important and useful strategy to improve their safety.
Immunogenicity testing however substantially increases the cost and time requirements for drug development and the goal of regulatory guidance should be to minimize human testing where possible. A variety of preclinical immunogenicity assessment strategies are currently used during biological development, as listed in Table 2.
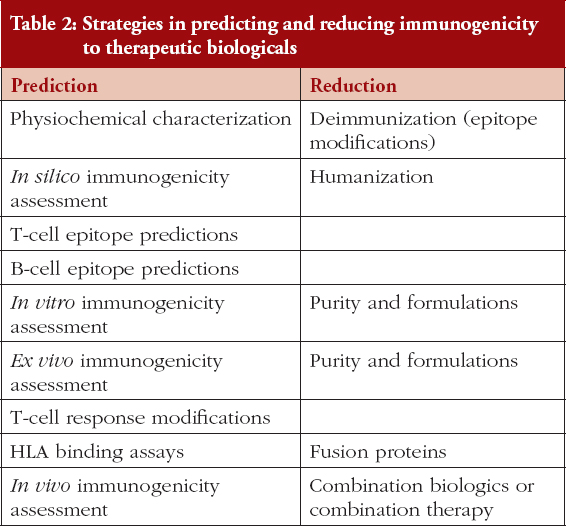
A major advantage of in vitro methods is the ability to test multiple batches for immunogenicity, which is not possible in human subjects. In vitro tests can also be more useful in predicting the difference between a biosimilar product and its reference product.
There are clear ethical complications in testing for immunogenicity in healthy subjects when comparing a reference drug to a biosimilar candidate. To advance the science of in vitro immunogenicity further, FDA should:
- Allow developers to present in vitro, in silico, or novel in vivo test methods and thus request a waiver from clinical immunogenicity testing.
- Continue internal development efforts to find and prescribe testing modalities that reduce the need for clinical testing of immunogenicity.
4. Make pharmacokinetic profiling clinically relevant
Bioequivalence is defined in 21 CFR 320.1 (Hatch-Waxman Act) [27] as ‘the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives become available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study’. Since the site of action is not known in most cases and rarely available for sampling, level in blood was selected as a surrogate to the site of action. The PK profile characterizes two stages, absorption and disposition (distribution and elimination), making it most relevant to generic chemical (small molecule) drugs where disposition is less likely to vary. This makes the PK profile relevant to absorption, and therefore bioavailability, thus providing validation of bioequivalence.
The PK profiling of biosimilars follows the same testing protocols as used for generic drugs. However, extrapolation of testing protocols involves a significant misconception – biosimilar drugs are administered parentally, which means that while differences in absorption are unlikely, differences in disposition are likely (distribution may change due to binding effects for example, and elimination may change due to subtle structural differences). This difference between generics and biosimilars should be addressed in the selection of PK parameters and statistical models applied to demonstrate similarity.
When FDA developed guidance on biosimilars, the term ‘clinical relevance’ was introduced, which is the most crucial aspect of determining biosimilarity and addresses the step-by-step approach [1] of demonstrating: analytical similarity, non-clinical toxicology, PK/PD profile, immunogenicity profile and, if there remains any ‘residual uncertainty’, performing additional clinical studies in healthy subjects. Consideration of ‘clinical relevance’ should therefore also be part of PK/PD analysis.
The author suggests the following changes to the criteria of PK/PD profiling of biosimilars compared to a reference product:
- Waive PK studies where the product is administered by a route (ocular, otic, and possibly others) that does not provide sufficient concentration of the active moiety in blood, such as the intraocular administration of ranibizumab [28]. However, to allow evaluation of disposition kinetics, PK studies involving intravenous administration in an appropriate animal, such as monkeys, should be required for monoclonal antibodies. This could be integrated into the non-clinical toxicology assessment. In most instances, a study population of 10–12 animals should suffice.
- When administered parentally, as most biosimilars are, PK parameters relating to distribution such as distribution volume and parameters relating to elimination such as terminal half-life are more clinically relevant than the area under the curve (AUC) or peak plasma concentration (Cmax). Statistical modelling should include these additional parameters. Distribution volume was introduced as a determinant of clinical efficacy by the author decades ago and finds a new application in the evaluation of biosimilars [29].
- While a confidence interval within 80–125% for the log ratio of Cmax and AUC (and for any other parameters added, as suggested above) has performed well as an acceptance criterion for generic drugs, there is no assurance that these intervals are clinically meaningful for biological drugs. Whether FDA should broaden or narrow the interval of acceptance remains to be determined once the additional parameters suggested above are taken into consideration.
- Encourage the use of scaled average bioequivalence (SABE) testing protocols that allow collection of immunogenicity profiles in a single study. In those instances where the immunogenicity data are required from a patient population, allow PK/PD profiling in patients, reducing the number of clinical studies required.
- Allow PK/PD studies to select a population that is likely to have minimal variation to reduce study sizes. Choosing such populations would help to demonstrate differences between the biosimilar candidate and the reference product.
5. Modify tier testing criteria for analytical similarity
FDA has recently released draft guidance on ‘Statistical Approxaches to Evaluate Analytical Similarity’ for biosimilars [30], one of the most critical components for establishing biosimilarity and a component that determines which additional studies, both clinical and CMC-related, are required. A developer identifies critical quality attributes (CQAs) and tests them using Tier 1, Tier 2, or Tier 3 statistical methods, depending on the nature of data output and the importance of the attribute to the safety and efficacy of a biosimilar product.
For CQAs in Tier 1, equivalence is established by rejecting the interval (null) hypothesis: −1.5 σR ≤ 90% CI of [μT–μR] ≤ 1.5 σ, where μT and μR are the mean responses of the test (the proposed biosimilar product and the reference product lots, respectively). This statistical testing suggests the equivalence acceptance criterion (EAC) = 1.5 × σ, where σ is the variability of the reference product (standard deviation). Statistical justification for the factor of 1.5 [31] follows the idea of the SABE criterion for highly variable generic drug products proposed by FDA. To achieve the desired power for the similarity test, FDA further recommends that an appropriate sample size is selected by evaluating the power using the alternative hypothesis μT − μR = ⅛.
There is no relevance of the factor of 1.5 used in equivalence testing of the most critical CQAs. For example, in the briefing on approval of Sandoz’s filgrastim product [32], FDA stated that one of the CQAs (protein content) initially failed, requiring additional batches to be added to the analyses. While there is a correlation between dose and effect for biological products, a small variation – as observed in the Sandoz data – should not have any clinically meaningful effect, since the release specification provides considerable variability. In essence, a test for analytical similarity may fail, yet such variation is allowed in the commercial product.
The criterion for Tier 1 testing for CQAs can produce misleading results. As an example, 10 batches (a number recommended by FDA) of a biosimilar candidate could be tested against an equal number of reference product batches for a percentage of the labelled quantity of protein. If the variation in the reference product is minimal, approaching a value of zero for σ, then all comparisons will fail, even if there is no clinically meaningful difference. The author has encountered such situations, where an attribute is tightly controlled in the originator product based on decades of manufacturing experience. The question arises if this is a clinically meaningful difference or merely a routine observation. For example, a biosimilar product may be allowed a range of 97–103% or even 95–105% in the Certificate of Analysis (COA) based on the history of manufacturing, yet all samples will fail if the σ of the reference product is minimal. On the other hand, where an attribute has high variability (σ) for the reference product, the product passes the Tier 1 test while failing a Tier 2 test, where 90% of all values fall within 3 × σ. It is for this reason that FDA requires Tier 2 testing for all Tier 1 attributes. To resolve these inconsistencies, the author suggests the following changes to the statistical modelling of CQAs in analytical similarity testing:
- Exclude any quality attributes for testing of analytical similarity that are part of the COA. If FDA accepts the variability as shown in the ranges of acceptance provided in the COA, it is illogical to accept or reject a product based on statistical limits of analytical similarity. The COA is clinically relevant, while the tiered testing of these attributes is not. Critical quality attributes of importance are primary, secondary and tertiary structures, receptor binding and impurity profile of timed samples, in addition to many more that are pertinent to differences in the molecules, albeit subtle.
- Allow developers to identify the CQAs and their range of variability based on clinical meaningfulness rather than using a factor of 1.5 arbitrarily to establish equivalence in a Tier 2 testing.
- If a product fails a Tier 1 test but passes Tier 2 testing, allow this as acceptance of similarity.
6. Clarify analytical testing validation
It is clearly understood that all analytical methods, including bioanalytical methods, must be validated, as provided in a May 2018 final guidance on bioanalytical methods [33]. However, analytical similarity testing requires methods that are often difficult or impossible to validate based on the guidance provided without incurring high cost and time commitments, such as nuclear magnetic resonance techniques or mass spectrometry. While all analytical methods used in the authorization of a biosimilar should be validated, methods used to demonstrate other analytical attributes may be accepted by FDA if they are ‘suitable’, a term often used in FDA guidance but not clearly defined. There is a need for FDA to clearly differentiate between the methods that must be validated and the ones that can be used if found suitable.
7. Encourage development of novel testing methods
Current approaches to evaluating the differences between a biosimilar candidate and a reference product are based on methods for characterizing new molecules; there is a need to develop more sensitive techniques to determine differences in the structure of large molecules, both at steady state and while active within the body. Several new techniques have recently come into practice, including modified capillary electrophoresis, Chip-based (Bioanalyzer) Protein Electrophoresis Assays (CPEA), and many variations of mass spectrometry [34].
FDA defines fingerprint-like similarity as:
‘the results of integrated, multi-parameter approaches that are extremely sensitive in identifying analytical differences (i.e. fingerprint-like analyses) permit a very high level of confidence in the analytical similarity of the proposed biosimilar product and the reference product, and it would be appropriate for the sponsor to use a more targeted and selective approach to conducting animal and/or clinical studies to resolve residual uncertainty and to support a demonstration of biosimilarity [35]’.
The introduction of new methodologies could help to demonstrate clinically meaningful similarity between products that will reduce the number of additional studies required [36, 37].
8. Accept smaller batch sizes
Unlike the development of entirely novel drugs, the development of biosimilars requires commercial-scale batches in order to begin testing for similarity. The rationale for this requirement derives from the assumption that there may not be any in patient or ‘phase III’ studies required that are historically conducted using commercial-scale batches. This requirement generates a huge cost and time burden, preventing smaller developers from entering the market. While FDA has not identified what it considers to be ‘commercial scale’, these issues were highlighted in a Type 2 formal meeting between FDA and sponsors of Biosimilar User Fee Act (BsUFA) products [38]. The author suggests that FDA requires a batch size that is adequate to provide samples for stability, clinical or other required testing, instead of making market projections to justify the size of a commercial batch. Should the developer decide to change the batch size after the product has been approved, the developer may use the Comparability Protocol for Biological Drugs [39] to make this post-approval change. This clarification by FDA would have a significant impact on industry, allowing smaller developers to offer market-ready products using smaller batches and at much lower cost.
9. Minimize clinical studies
The largest contributor to the cost and time requirements of marketing a drug are the clinical studies required to establish biosimilarity. When in patient studies are required, the cost and timeline stretch even further. The current mindset of establishing biosimilarity follows phase I to III testing, which is not relevant to establish the non-inferiority status of a biosimilar candidate with the reference product. As a result, the author makes the following recommendations:
- If it is determined a priori that studies are required in patients, allow developers to conduct PK/PD profiling in patients as well.
- Allow statistical models of PK/PD studies to determine immunogenicity within the same study.
- Allow the use of in vitro models for immunogenicity testing to reduce human exposure.
- Allow the use of animal models to establish differences in the PK (and, where possible, the PD) profile where blood concentrations are not measurable.
Summary
In developing methods for the evaluation of biosimilars, FDA has created highly specific vocabulary, such as ‘no clinically meaningful difference’ and ‘residual uncertainty’. These terms are scientifically important and represent a creative approach to assuring the safety of biosimilars. However, not all FDA guidance adequately takes these two terms into account. To improve this situation, this review makes a number of recommendations:
- Remove the default requirement of conducting bridging studies for non-US reference products, where the reference product meets specific criteria.
- Declare that biosimilars have no clinically meaningful difference from the originator product and, therefore, substitutions for naïve patients should be allowed.
- Remove the default requirement of conducting in vivo immunogenicity testing and allow developers to offer alternative in vitro and in silico testing methods.
- Modify PK/PD protocols and statistical analysis methods to make the outcomes clinically meaningful.
- Modify testing of critical quality attributes by separating them from release specifications to demonstrate analytical similarity.
- Minimize clinical testing by combining studies.
- Clarify the type of validation required for analytical similarity testing.
- Allow the approval of products based on smaller scale studies.
FDA recognizes the need for changes to its guidance. Commissioner Dr Scott Gottlieb [23] has recently expressed a willingness to respond to the urgent need to reinterpret guidelines for the increased approval and adoption of biosimilars.
In June 2018, the FDA withdrew its guidance for Analytical Similarity Testing [40] and a few days later FDA announced a new initiative, Biosimilars Action Plan that includes most of the recommendations made by the author in its citizen petition [41].
Disclaimer
This paper represents solely the views of the author and should not be understood or quoted as being made on behalf of or reflecting the position of any regulatory authority or company.
Competing interests: The author of the paper declared that he is a developer of biosimilar products. The author is founder of Karyo Biologics, LLC and Adello Biologics, which have several biosimilar products at various stages of FDA approval.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. U.S. Food and Drug Administration. Biologics Price Competition and Innovation Act of 2009. Code of Federal Register. H.R. 3590–687. Title VII—Improving access to innovative medical therapies. Subtitle A—Biologics Price Competition and Innovation [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/drugs/ucm216146.pdf
2. U.S. Food and Drug Administration. Biosimilar product information [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/ucm580432.htm
3. U.S. Food and Drug Administration. Lusduna: FDA approved drug products [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=208722
4. U.S. Food and Drug Administration. Basaglar (insulin glargine injection) [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205692Orig1 s000TOC.cfm
5. U.S. Food and Drug Administration. Implementation of the “Deemed to be a License” Provision of the Biologics Price Competition and Innovation Act of 2009 [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM490264.pdf
6. U.S. Food and Drug Administration. Biosimilars guidances [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/General/ucm444891.htm
7. Blackstone Erwin A, Fuhr Joseph P, Jr. The economics of biosimilars. Am Health Drug Benefits. 2013;6(8):469-78.
8. Gottlieb S. FDA taking new steps to better inform physicians about biosimilars through education about these potentially cost-saving options. 2017 Oct 23 [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://blogs.fda.gov/fdavoice/index.php/2017/10/fda-taking-new-steps-to-better-inform-physicians-about-biosimilars-through-education-about-these-potentially-cost-saving-options/
9. Niazi S. Obstacles to success for biosimilars in the US market. European Pharmaceutical Review. 2018 Jan 4.
10. Niazi S. eBook: Challenges facing biosimilar entries into US markets. 2018 Jan 16.
11. Niazi S. Citizen Petition from UIC College of Pharmacy. ID: FDA-2018-P-1876-0001.
12. Webester CJ, Woollett GR. A ‘Global reference’ Comparator for biosimilar development. BioDrugs. 2017;31(4):279-86.
13. Australian Government. Department of Health. Therapeutic Goods Administration. Evaluation of biosimilars products [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.tga.gov.au/sites/default/files/pmargpm-biosimilars-150420_1.pdf
14. Government of Canada. Health Canada. Guidance document: information and submission requirements for biosimilar biologic drugs products [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs-1.html
15. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04 Rev 1. 23 October 2014 [homepage on the Internet]. [cited 2018 Jul 13]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf
16. Swiss Medic. Questions and answers concerning the authorisation of similar biological medicinal products (biosimilars) [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.swissmedic.ch/swissmedic/en/home/legal/legal-basis/administrative-ordinances/questions-and-answers-concerning-the-authorisation-of-similar-bi.html
17. U.S. Food and Drug Administration. Biosimilars: questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009. Guidance for Industry. 2015 April [homepage on the Internet]. [cited 2018 Jul 13]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM444661.pdf
18. World Health Organization. Regulatory evaluation of biosimilars throughout their product life-cycle [homepage on the Internet]. [cited 2018 Jul 13]. Available from: http://www.who.int/bulletin/volumes/96/4/17-206284/en/
19. U.S. Food and Drug Administration. FDA briefing document. Arthritis Advisory Committee Meeting February 09, 2016 BLA 125544 CT-P13, a proposed biosimilar to Remicade® (infliximab) Celltrion [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/%E2%80%A6/UCM484859.pdf
20. U.S. Food and Drug Administration. FDA briefing document. Oncologic Drugs Advisory Committee July 13, 2017 BLA 761028 ABP215, a proposed biosimilar to Avastin (bevacizumab) Amgen Inc [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM566365.pdf
21. Gottlieb: FDA considering an end to biosimilar bridging studies. The Center for Biosimilars. 2018 Feb 16. Available from: http://www.centerforbiosimilars.com/news/gottlieb-fda-considering-an-end-to-biosimilar-bridging-studies-
22. U.S. Food and Drug Administration. Biosimilars and interchangeable products [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580419.htm
23. U.S. Food and Drug Administration. Gottlieb S. Capturing the benefits of competition for patients [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/NewsEvents/Speeches/ucm599833.htm
24. U.S. Food and Drug Administration. Guidance for Industry. Immunogenicity assessment for therapeutic protein products. 2014 Aug [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf
25. U.S. Food and Drug Administration. Immunogenicity of protein-based therapeutics [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/BiologicsBloodVaccines/ScienceResearch/BiologicsResearchAreas/ucm246804.htm
26. European Medicines Agency. Guideline on immunogenicity assessment of therapeutic proteins [homepage on the Internet]. [cited 2018 Jul 13]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/06/WC500228861.pdf
27. U.S. Government Publishing Office. CFR: Public Law 98–417–Sept. 24, 1984. Title I–Abbreviated new drug applications [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.gpo.gov/fdsys/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdf
28. European Medicines Agency. Ranizimumab [homepage on the Internet]. [cited 2018 Jul 13]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000715/WC500043550.pdf
29. Niazi S. Volume of distribution as a function of time. J Pharm Sci. 1976;65(3):452-4.
30. GaBI Online – Generics and Biosimilars Initiative. FDA issues draft guidance on statistical approaches to evaluating similarity for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2018 Jul 13]. Available from: www.gabionline.net/Guidelines/FDA-issues-draft-guidance-on-statistical-approaches-to-evaluating-similarity-for-biosimilars
31. Chow SC, Song F, Bai H. Analytical similarity assessment in biosimilar studies. AAPS J. 2016;18(3):670-7.
32. U.S. Food and Drug Administration. FDA briefing document. Oncologic Drugs Advisory Committee Meeting January 7, 2015 BLA 125553 EP2006, a proposed biosimilar to Neupogen® (filgrastim) Sandoz Inc., a Novartis company [homepage on the Internet]. [cited 2018 Jul 13]. Available from: http://patentdocs.typepad.com/files/briefing-document.pdf
33. U.S. Food and Drug Administration. Bioanalytical method validation. Guidance for Industry. 2018 May [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf
34. Agilent Technologies. Recombinant protein characterization [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.agilent.com/cs/library/primers/Public/5990-8561EN_LO.pdf
35. U.S. Food and Drug Administration. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for Industry. 2016 Dec [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm397017.pdf
36. Niazi SK. Method for comparing structure of one biomolecule with second biomolecule. United States Pub. No. 2018/0024137 A1. 2018 Jan 25.
37. Niazi SK. Single-use bioreactors and mixing vessels. United States patent 9,469.426 B2. 2016 Oct 18.
38. U.S. Food and Drug Administration. Formal meetings between the FDA and sponsors or applicants of BsUFA products. Guidance for Industry. 2018 Jun [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM609662.pdf
39. U.S. Food and Drug Administration. Comparability protocols for human drugs and biologics: chemistry, manufacturing, and controls information. Guidance for Industry. 2016 Apr [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM496611.pdf
40. U.S. Food and Drug Administration. FDA withdraws draft guidance for industry: statistical approaches to evaluate analytical similarity [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/Drugs/DrugSafety/ucm611398.htm
41. U.S. Food and Drug Administration. Biosimilars Action Plan [homepage on the Internet]. [cited 2018 Jul 13]. Available from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm613761.pdf
|
Author: Adjunct Professor Sarfaraz K Niazi, PhD, SI, FRSB, FPAMS, FACB, Adjunct Professor of Biopharmaceutical Sciences, University of Illinois and University of Houston; 20 Riverside Drive, Deerfield, IL 60015, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


