Reducing healthcare costs and building trust in biosimilar medicines
Published on 2016/06/29
Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):84-88.
Author byline as per print journal: Niklas Ekman, PhD; Professor Arnold G Vulto, PharmD, PhD; Paul Cornes, MD
|
Abstract: |
Submitted: 19 April 2016; Revised: 1 June 2016; Accepted: 4 June 2016; Published online first: 17 June 2016
Introduction
The 21st Congress of the European Association of Hospital Pharmacists (EAHP) took place on 16–18 March 2016 in Vienna, Austria. The Biosimilar Medicines Group (formerly EBG) held a satellite symposium entitled ‘The facts about biosimilars’ on 17 March 2016 during the conference.
The Biosimilar Medicines Group represents the leading pharmaceutical companies developing, manufacturing and marketing biosimilars across Europe.
It is a sector group of Medicines for Europe (formerly the European Generic medicines Association).
With 10 years of positive patient treatment experience and 20 products successfully launched, use of biosimilars today offers a huge opportunity to deliver significantly improved access to modern therapies for millions of European patients receiving both chronic and acute care.
Increasing costs of biological medicines (‘biologicals’)
Dr Paul Cornes (University Hospitals Bristol, NHS Foundation Trust, Bristol, UK), presenting ‘Biosimilars – can we do without them?’ highlighted the fact that according to the World Health Organization (WHO), cancer has been the world’s top killer since 2010. The disease has the most devastating economic impact of any cause of death in the world. In Europe, 17% of all the ‘healthy’ years lost were due to cancer and 170 million years of ‘healthy life’ were lost due to death and disability from cancer in 2008.
The good news for cancer treatment is that since the 1960s pharmaceutical innovation has enabled the introduction of a whole host of new cancer drugs. The rate of new medicines being introduced is also increasing rapidly. Prior to the 1960s, there were only five cancer medicines available in the US. During the 1960s, only two new cancer medicines were introduced, but in the 30 years between 1970 and 2000 a total of 79 new medicines were introduced and in the five-year period between 2010 and 2015 a staggering 43 new cancer medicines have been introduced. At this rate, more than 100 new cancer medicines could be added in the period between 2010 and 2020, see Figure 1 [1]. This innovation heralds a new era of targeted precision therapy for cancer that could transform the outlook for patients with the world’s most important diseases.
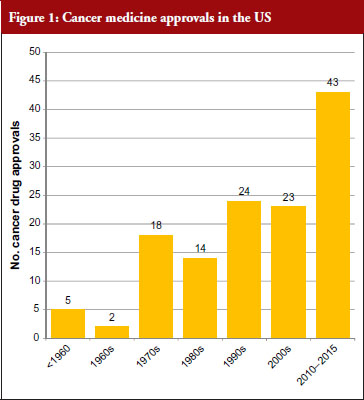
However, there is a widespread belief that this dream may not be affordable – even for the richest nations of the world. Learning howto make this innovation affordable will not just address cancer – but willshow us how to manage the costs of innovation in other diseases as well. One key focus has been on controlling the costs of innovativebiological therapy – as cancer medicine costs are increasing five times faster than any other class of medicine. In fact, eight cancer medicines approved by the US Food and Drug Administration (FDA) in 2015 had six-figure price tags [2].
The problem is not just that these new medicines are expensive, but due to the increasingly ageing population there is ever more cancer to treat. Cancer is often a disease of the elderly, with the peak age for cancer being 70 to 84 years.
The increasing cost of treating the ageing population could therefore lead to healthcare spending growing at faster rate than the gross domestic product (GDP). In the US, healthcare costs were increasing at an annual rate of 7% per year. This rate, if sustained, is forecasted to bankrupt Medicare in nine years and increase the nation’s overall healthcare bill to US$4 trillion in 10 years [3]. Such cost pressures are not just an American issue, as all the world’s developed nations but one have increased health spending in relation to national wealth. It is therefore essential to look at where savings can be made in order to make it possible for the population to age and to still be able to afford innovation in medical treatment.
Simple cuts in the healthcare budget are not the answer. Savings have to be made where they do not put the health of citizens at risk. For example, every 1% decrease in government healthcare spending is associated with a 10.6% rise in maternal mortality in the EU [4]. Debt is therefore a real threat to health.
Given the fact that by 2018, biologicals worth more than US$68 billion in current annual sales will lose patent protection [5], the case for using biosimilars seems clear. Even with only a 20% discount, this could give the world a US$14 billion health innovation fund. Whereas a 30% discount could save US$20 billion and a 40% discount could save US$27 billion.
Dr Cornes highlighted the fact that WHO is clearly an advocate for generics and biosimilars as illustrated by the following WHO statements:
- The leading cause of inefficiency in healthcare is underuse of generic medicines and paying more than necessary for medicines [6]
- A biosimilar medicine is similar in terms of quality, safety and efficacy to an already licensed reference biotherapeutic product [7]
- To use a more expensive version of a medicine is ‘irrational, inappropriate, improper, incorrect’ [8]
He concluded that ‘we have a common interest between patients, physicians, pharmacists, pharmaceutical industry and payers in the success of biosimilar medicines’. He added that ‘biosimilar medicines offer a reward to world health that will be substantial’. By generating savings from within our existing health budgets, on medicines that are equally safe and effective, we can still afford to invest in healthcare innovation even in times of financial crisis.
Information gap for biosimilars
Professor Arnold G Vulto’s (Erasmus University, Rotterdam, The Netherlands) presentation entitled ‘Biosimilars: concerns of prescribers and how to address them as a hospital pharmacist’ highlighted the fact that the total medicine bill will grow exponentially with the introduction of so many blockbuster breakthrough medicines. He too pointed to the ‘savings potential of biosimilar medicines’.
Four biologicals approved in 2014: Keytruda (pembrolizumab), Cyramza (ramucirumab), Opdivo (nivolumab) and Sylvant (siltuximab), are anticipated to be blockbusters by 2019, i.e. medicines that generate annual sales of at least US$1 billion, according to sales forecasts from the Thomson Reuters Cortellis database. When considering all new medicines approved in 2014, 12 are expected to become blockbusters and sales of these medicines are forecast to add US$29 billion per year to the cost of drugs.
Five biologicals newly approved in 2015 are predicted to become blockbusters by 2020 [9]. For all new medicines approved in 2015, 16 are expected to become blockbusters and sales of these medicines are forecast to add more than US$36 billion per year to the cost of drugs. These predictions clearly highlight the problem facing governments when it comes to increasing healthcare costs.
One way to modify these increasing costs would be to increase the use of biosimilars. However, as Professor Vulto pointed out, uptake of biosimilars in the EU varies widely between countries and therapeutic areas; and Europe accounts for 80% of global spending on biosimilars [10].
For example, the human growth hormone (HGH) biosimilar Omnitrope (somatropin) was the first product approved in the EU as a biosimilar back in 2006 [11]. Despite this product being available for 10 years, some countries in the EU, e.g. Greece, Ireland and Slovakia, still have little or no uptake of this biosimilar, see Figure 2. Biosimilars penetration in Europe for erythropoietin (EPO) and granulocyte colony-stimulating factor (G-CSF) also varies, from 0% for EPO in countries such as Belgium to 100% for G-CSF in the Czech Republic, Hungary, Romania and Slovakia. In fact, Eastern Europe is leading the way in biosimilar medicines penetration perhaps driven by economic factors.
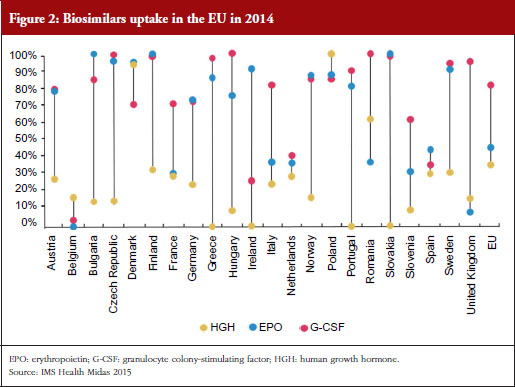
The important issue that needs to be addressed is how to improve physician prescribing of biosimilars. According to Professor Vulto, physicians will prescribe biosimilars when they have sufficient trust in the sameness of the biosimilar. Understanding of the biosimilarity concept is of great importance, especially when there are sufficient incentives to do so.
In the EU, the European Medicines Agency (EMA) assesses the scientific aspects of biosimilars and makes recommendations for a market approval when the biosimilar candidate is assessed to be therapeutically equivalent (comparable quality, safety and efficacy) to the reference biological. Later, based on a scientific appraisal by EMA, decisions on the policy of interchangeability (medical practice) between biosimilars and originator biologicals are made, but not by EMA, rather at the national level. Several EU Member States, such as Finland, Germany and The Netherlands have taken clear positions in support of the interchangeability (a medical practice) of biosimilars, while others, e.g. the UK’s healthcare cost watchdog NICE, have also developed full guidelines [12].
Acceptance of a biosimilar is dependent on the actions of many different stakeholders, such as physicians, patients, pharmacists, third-party payers and policymakers. It is essential to obtain the buy in (‘ownership’) from stakeholders, for example, including prescribers in the production of treatment guidelines. This can help deal with the common misconceptions healthcare professionals may have about biosimilars, such as that they:
- may be of lower quality than the originator medicine
- are poorly supported by research
- have not been researched in all indications
- differ from the originator in potentially relevant aspects
- have been assessed by regulators who are bureaucrats, who have no clinical experience
- used a shortcut in the normally rigorous licensing process.
A clear information gap exists when it comes to biosimilars. One way Professor Vulto suggested to reduce this gap was for regulators to communicate their knowledge actively to medical professionals. In fact, EMA has urged regulators in EU Member States to provide physicians with more information regarding the concepts of comparability and biosimilarity [13]. He also suggested that regulators should also point out that over the past 10 years, since the introduction of biosimilars, there have not been any new or unexpected safety signals; that the assessment system has worked as expected; and that based on what we have learned from their actual use, the mistrust raised against biosimilars was not justified. The Generics and Biosimilars Initiative (GaBI) was also highlighted as being another source of valuable information to build trust in cost-effective treatments. Professor Vulto added that in order to ‘avoid trouble around switching’ it was essential to convince prescribers of the (financial) advantages for society, without compromising quality of treatment, including increased treatment choices and access to medicines for patients.
A regulatory perspective
Dr Niklas Ekman, a senior researcher at the Finnish Medicines Agency (FIMEA, Helsinki, Finland), gave a presentation on ‘Biosimilars from the perspective of an EU regulator’. He highlighted the fact that batch-to-batch variability is inherent for all biologicals, both for originators and biosimilars. Alterations in the quality profile can be introduced through manufacturing process changes. With all changes, whether for an originator or biosimilar, the pre- and post-change version of the medicinal product needs to be demonstrated to be comparable through a comparability exercise. Manufacturers and regulators therefore have extensive experience in assessing the impact of process changes – including in the case of complex biologicals.
The current EU regulatory definition of biosimilars defines a biosimilar as a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product). A biosimilar demonstrates similarity to the reference medicinal product in terms of quality characteristics, biological activity, pharmacokinetic profile, safety and efficacy based on a comprehensive comparability exercise.
A stepwise approach is used to establish biosimilarity. Comprehensive physicochemical and biological characterization should be followed by non-clinical studies, which should include in vitro functional studies and, if needed, in vivo studies. These should be followed by a comparative pharmacokinetic study in a sensitive and homogeneous study population, such as healthy volunteers (if possible/feasible). Finally, efficacy/safety clinical studies to confirm comparable clinical performance of the biosimilar and the reference product should normally be carried out. These studies should be adequately powered, randomized, parallel group (usually equivalence) trials. The study population should be representative of the approved indication(s) and be sensitive for detecting potential differences and the endpoints should be selected with the aim of investigating possible differences, not demonstrating efficacy per se, see Figure 3.
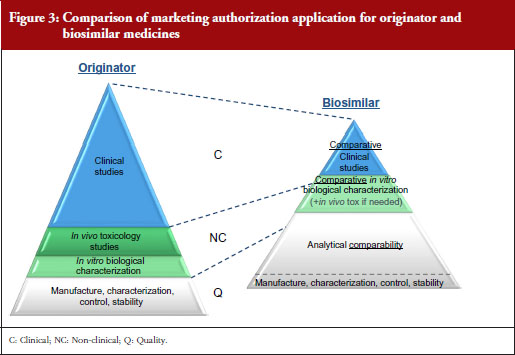
EMA has approved 22 biosimilars to date, although two were withdrawn, leaving a total of 20 biosimilars approved for use in Europe [11]. In March 2016, the agency was also reviewing 12 biosimilars, including adalimumab (2), enoxaparin sodium (2), etanercept (1), infliximab (1), insulin glargine (1), pegfilgrastim (3), rituximab (1) and teriparatide (1).
The success of developing a biosimilar candidate depends on:
- The ability to manufacture a medicinal product containing a version of the active substance of the reference medicinal product in a consistent manner
- The ability to perform thorough physicochemical and biological characterization and to understand the clinical relevance of any differences detected
- The ability to demonstrate bioequivalence
- The availability of suitable clinical models; sensitive endpoints and the possibility to identify relevant comparability margins
In order to prove biosimilarity the amino acid sequence, posology and the route of administration must be the same as the reference biological. In addition, the active substance must be similar in terms of molecular and biological characteristics. Any differences in strength, pharmaceutical form, formulation, excipients or presentation need to be justified. Finally, intended changes to improve efficacy (‘bio-betters’) are not allowed.
Funding of medicines in Finland
The way medicines are funded in Finland results in hospitals leading the adoption of biosimilars due mainly to economic incentives. Prescribers and patients on the other hand have no special interest in biosimilars due to the lack of incentives.
Hospitals
For medicines administered in public hospitals the cost is borne by the community as a whole.
Pharmacies
When prescription medicines are dispensed by pharmacies the costs are covered by the Social Insurance Institution (state). Patients pay an annual maximum of Euros 610 for reimbursed medicines. There are three different levels of reimbursement; 40%, 64% and 100%. Biosimilars have the same reimbursement level as their reference biological. Due to the high price of biologicals, in practice, reimbursement is a prerequisite for the use of any biologicals outside hospitals.
The Finnish Medicines Agency, FIMEA, announced in May 2015 that it considers EU biosimilars interchangeable with their reference biologicals. Automatic substitution at the pharmacy level, however, is not included in the current FIMEA recommendation [14, 15].
The FIMEA position paper on the interchangeability of biosimilars concludes the following:
- Switches between biological products, for example, in the context of hospital tendering processes, are common and usually not problematic
- The clinical crossover studies conducted thus far have given no evidence of adverse effects due to a switch from a reference product to a biosimilar (somatropin, epoetin alfa, filgrastim, insulin glargine, infliximab)
- The theoretical basis of expecting such adverse effects is weak
- The risk of adverse effects can be expected to be similar to the risk associated with changes in the manufacturing process of any biological product
The position of FIMEA is therefore that biosimilars licensed in the EU are interchangeable with their reference products under the supervision of a healthcare professional. As with any biological products, the switch should be documented (including brand name and batch number).
Interchangeability recommendations in other European countries
According to Dr Ekman, similar positions have been adopted by other EU national authorities, including the Medicines Evaluation Board (MEB) in The Netherlands; the Paul Ehrlich Institute (PEI) in Germany; and the Health Products Regulatory Authority (HPRA) in Ireland.
Dutch MEB recommendation
Exchange between biological medicines (regardless of whether they are originator biological products or biosimilar medicinal products) is permitted, but only if adequate clinical monitoring is performed and the patient is properly informed.
German PEI recommendation
Biosimilars can be used in the same way as their reference products to which they have shown equivalence. This implicitly covers both patients who have not yet received biological therapy as well as patients who previously received the originator product.
Irish HPRA recommendation
If it is planned to change the medicine a patient receives from a reference to a biosimilar medicine or vice versa, the treating physician should be involved; this should involve discussion between the prescriber/patient, and prescriber/dispensing pharmacist.
At the end of the session, Medicines for Europe’s Market Access Director Maarten van Baelen concluded that biosimilars today provide a huge opportunity to deliver significantly improved access to existing and future innovative therapies for millions of European patients in both chronic and acute care while supporting the sustainability of our healthcare systems.
Editor’s comment
Medicare is a national social insurance programme, administered by the US federal government since 1966. It provides health insurance for Americans aged 65 and older who have worked and paid into the system. It also provides health insurance to younger people with disabilities.
Minor wording changes have been made to the presentations to clarify the points made.
Acknowledgement
The authors wish to thank Michelle Derbyshire, PhD, GaBI Online Editor, in preparing this meeting report.
Disclosure of financial and competing interests: The satellite symposium was organized by the Biosimilar Medicines Group, a sector group of Medicines for Europe. Fees were paid to the organization for the conference slot, however, none of the speakers received direct payments from Medicines for Europe.
Dr Paul Cornes has received honoraria from Accord Healthcare, Amgen, Bernstein, British Medical Journal, European Generic medicines Association, Hospira, Janssen, Lilly, Merck Serono, Napp, Pharmaceutical Association of Malaysia, Pfizer, Roche, Sandoz and Teva.
Professor Arnold G Vulto declares no personal financial interest in any pharmaceutical business. Any honoraria resulting from his participation in advisory boards or as a speaker at scientific or commercial meetings and any consulting fees received are given to Erasmus University Hospital. Companies/organizations involved are: AbbVie, Amgen, Biogen, European Generic medicines Association, Mundipharma, Pfizer/Hospira, Roche, Sandoz.
Dr Niklas Ekman is employed by a regulatory agency, and has nothing to disclose.
Provenance and peer review: Not commissioned; internally peer reviewed.
Authors
Niklas Ekman, PhD
Senior Researcher
Quality Assessor for biological medicinal products
Member of the Biosimilar Working Party (BMWP), European Medicines Agency (EMA)
Finnish Medicines Agency (FIMEA), 103b Mannerheimintie, PO Box 55, FI-00034 FIMEA, Finland
Professor Arnold G Vulto, PharmD, PhD
Deputy Head Hospital Pharmacy
Professor of Hospital Pharmacy and Practical Therapeutics
Erasmus University Medical Center, PO Box 2040, 230 Gravendijkwal, NL-3015 CE Rotterdam, The Netherlands
Paul Cornes, MD
Clinical Outcomes Group, Bristol Oncology Center, University Hospital Bristol, UK
References
1. Cornes P. Pictogram created from data in – Savage P.Development and economic trends in cancer therapeutic drugs: analysis of modern and historical treatment costs compared to the contemporary GDP per capita. J Clin Oncol. 2014;33(suppl; abstract 17535).
2. Jarvis LM. The year in new drugs. Chemical and Engineering News. 2016;94(5):12-7.
3. Langreth R. Will health costs bankrupt America? Forbes. 23 Feb 2011.
4. Maruthappu M, Ng KY, Williams C, Atun R, Agrawal P, Zeltner T. The association between government healthcare spending and maternal mortality in the European Union, 1981–2010: a retrospective study. BJOG. 2015;122(9):1216-24.
5. Derbyshire M. Patent expiry dates for best-selling biologicals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):178-9. doi:10.5639/gabij.2015.0404.040
6. World Health Organization. The World Health Report 2010. Chapter 4: More health for the money [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://www.who.int/whr/2010/10_chap04_en.pdf
7. World Health Organization. Expert Committee on Biological Standardization. Guidelines on evaluation of similar biotherapeutic products (SBPs). 23 October 2009 [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
8. World Health Organization. The World Medicines Situation Report, 2011 [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://apps.who.int/medicinedocs/documents/s20054en/s20054en.pdf?ua=1
9. Mullard A. 2015 FDA drug approvals. Nat Rev Drug Discovery. 2016;15(2):73-6.
10. GaBI Online – Generics and Biosimilars Initiative. European uptake of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Jun 1]. Available from: www.gabionline.net/Reports/European-uptake-of-biosimilars
11. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Jun 1]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
12. National Institute for Health and Care Excellence. Introducing biosimilar versions of infliximab: Inflectra and Remsima [homepage on the Internet]. [cited 2016 Jun 1]. Available from: http://publications.nice.org.uk/introducing-biosimilar-versions-of-infliximab-inflectra-and-remsima-htta329/insights-from-the-nhs-managing-the-introduction-of-biosimilar-medicines
13. Kurki P. Biosimilars for prescribers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):33-5. doi:10.5639/gabij.2015.0401.008
14. GaBI Online – Generics and Biosimilars Initiative. Finnish drug regulator recommends interchange-ability of biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2016 Jun 1]. Available from: www.gabionline.net/Policies-Legislation/Finnish-drug-regulator-recommends-interchangeability-of-biosimilars
15. FIMEA. Interchangeability of biosimilars – position of Finnish Medicines Agency Fimea. 22 May 2015 [homepage on the Internet]. [cited 2016 Jun 1]. Available from: www.fimea.fi/documents/542809/838272/29197_Biosimilaarien_vaihtokelpoisuus_EN.pdf
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


