Statistical considerations for the development of biosimilar products
Published on 2013/09/30
Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(1):21-5.
|
Abstract: |
Submitted: 12 August 2013; Revised: 4 November 2013; Accepted: 15 November 2013; Published online first: 29 November 2013
Introduction
A biological medicine is a large molecule derived from living cells. They are often 200 to 1,000 times the size of a small molecule product and are far more complex structurally. They are also highly sensitive, making them more difficult to characterize and produce. Biosimilar products or biosimilars are defined by the World Health Organization (WHO) as ‘a biotherapeutic product which is similar in terms of quality, safety and efficacy to an already licensed reference biotherapeutic product’ (also known as follow-on biologics [FOBs] by the US Food and Drug Administration [FDA] definition). Countries around the world face a growing and aging population and an increase in chronic disease. With expanding demand for high quality health care comes the challenge of controlling healthcare expenditure. The safe and regulated introduction of biosimilars has been predicted to increase access to much needed biological medicines and reduce costs. The patents of a growing number of biological medicines have already expired or are due to expire, which has led to an increased interest from both the biopharmaceutical industry and the regulatory agencies in the development and approval of biosimilars.
In 2004–05, the European Medicines Agency (EMA)/European Commission (EC) was the first major regulatory authority to implement a framework for the marketing authorization of biosimilars. Product-specific guidelines were subsequently provided by the EMA Committee for Medicinal Products for Human Use (CHMP), outlining the data requirements and studies necessary to demonstrate similarity [1–10]. WHO issued a biosimilar guideline in 2009 aimed at providing a consistent scientific standard [11]. FDA issued three draft guidance documents in 2012 on biosimilar product development following the passage of the Affordable Care Act in 2010 [12–15]. Regulation has evolved rapidly worldwide with many countries establishing national guidelines based on WHO and EMA/EC framework. A common message can be synthesized from these guidance documents, unlike generic medicines where the active ingredients are identical, biosimilars are not identical, but only similar to the originator biological. The requirements to demonstrate similarity are therefore not the same as generic drug products. Due to the complex structure of biological products and the sensitive processes involved in production, biosimilars must be shown on the basis of analytical, non-clinical and clinical data to be similar to the originator biological in terms of structural characteristics, safety and efficacy. Minor differences with the active ingredients of the originator biological are expected so long as they are demonstrated not to be clinically meaningful.
The main objective of this paper is to discuss some statistical issues and challenges in development of biosimilars, including criteria for biosimilarity and interchangeability; selection of endpoints and determination of margins to demonstrate biosimilarity; equivalence versus non-inferiority; bridging and regional effect; and how to quantify totality-of-the-evidence.
Criteria for biosimilarity
Similar to WHO, biosimilarity was also defined in the Affordable Care Act as ‘no clinically meaningful differences between the biosimilar product and the reference product in terms of the safety, purity, and potency of the product’ [16].
For small molecule generic drug products, the standards for regulatory approval are well established and have been implemented by many regulatory agencies, such as FDA and EMA. The commonly used method is to assess the average bioequivalence (ABE) in terms of drug absorption between the small molecule drug products through the conduct of bioequivalence (pharmacokinetic) studies. Let T and R be the parameters of interest, e.g. pharmacokinetic response, with means μT and μR, for the test and reference products, respectively. Thus, the interval hypotheses for testing the ABE of two products can be expressed as:
![]()
Where (θL, θU) are the ABE limits. Since ABE criterion only focuses on average bioavailability regardless of the variability of bioavailability between drug products, it is concern that the criterion may not be applicable to the assessment of biosimilarity between biological products given the fundamental differences, e.g. complex structure, sensitive production processes, between biological and chemical drugs. Various investigations have been conducted to look for alternative methods that take variability into consideration, such as scaled average bioequivalence criterion (SABE), similarity test on variability directly, comparison of distributions, and application of a biosimilarity index. These methodologies are briefly discussed below.
Scaled average bioequivalence criterion (SABE)
Scaled average bioequivalence (SABE) criterion is a scaled version of ABE to account for the variability of the reference products. The issue was first brought up in generic drug development and can be illustrated as below [17]:
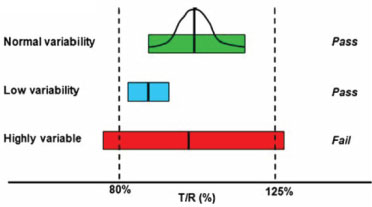
The larger variability the reference drug has the harder it is to achieve bioequivalence (BE) even if the true geometric mean ratio is close to 1. Hence, a scaled average BE approach was recommended for highly variable small molecule drugs [18] where the margin used for ABE criteria is adjusted by:
![]()
where σR measures the within-subject standard deviation of the reference product and σ0 is a pre-specified regulatory constant, e.g. 0.25 by FDA [19] and 0.294 as used by EMA [20]. The criteria can be applied to biosimilar studies with crossover design and would yield a smaller sample size and greater power to conclude bioequivalence. However, for biosimilar products with very long half-life, parallel-group design is often implemented. How SABE criterion can be extended is of interest and has been studied in Zhang and co-authors [21].
Equivalence tests on variabilities
In addition to assessing biosimilarity in terms of comparing averages, it may also be desirable to compare variabilities between the biological products. Such assessment involves tests on the homogeneity of variability between biosimilars and the reference product. A probability-based method [22] examines the probability of the variance ratio falling within pre-specified limits and concludes equivalence in variability if this probability is higher than a pre-specified criterion. Other researchers have adapted the conventional methods, e.g. F-test and non-parametric tests of variances, and modified them to fit into the biosimilarity framework using the concept of two one-sided tests [23, 24]. How an integrated criterion based on both averages and variabilities could be used for assessing biosimilarity remains to be worked out.
Equivalence tests on distributions
Various methodologies have been developed in the general statistical literature to compare two sample distributions, such as the Kolmogorov-Smirnov test [25] and the overlap coefficient test [26]. Though they were thought by some researchers as a potential way of integrated evaluation of both averages and variabilities, when applied to the biosimilar setting, they have the inherent deficiency of not being able to specify a meaningful equivalence margin. Simulations also showed that these two tests are sensitive to group difference, which is undesirable if the difference is not considered relevant [27]. The practical use of these methods hence remains questionable.
Equivalence criteria based on biosimilarity index
Shao and Chow [28] introduced a concept of reproducibility probability as a measure for determining whether it is necessary to require a second trial when the result of the first clinical trial is highly significant. Following the concept, Hsieh and Chow [29] proposed a biosimilarity index as a criterion for the assessment of biosimilarity. The idea is to show that the probability in a study to compare a tested biosimilar product (T) with the reference product (R) (T-R study) is comparable to the reproducibility probability comparing the reference product with the reference product (R-R study). Let us denote reproducibility probability from the R-R study as PRR; and from the T-R study as PTR:

PRR is the probability of concluding average biosimilarity between two applications of the reference product, i.e. of observing its reproducibility, in a future trial, given that average biosimilarity based on a criterion has been established in a first trial:

To conclude the test product is biosimilar to the reference product, PTR should be comparable to PRR. Hence, a criterion for biosimilarity could be PTR > P0 where P0 is a pre-specified acceptable reproducibility probability. For example, if the R-R study suggests the reproducibility probability of PRR = 90%, the criterion of the reproducibility probability for bioequivalence study could be chosen as 80% of PRR which is P0 = 80% × 90% = 72%.
The biosimilarity index has the advantages that (1) the probability of reproducibility will reflect the characteristics of the reference product and the sensitivity of heterogeneity in variance; and (2) it is robust with respect to the selected study endpoint, biosimilarity criteria, and study design. The first property addressed the concern that ABE criteria only focus on average bioavailability regardless whether the variabilities are similar or not. The second property allowed its application to different functional areas (domains) of biological products, such as biological activities, biomarkers, pharmacokinetics (PK), pharmacodynamics (PD), immunogenicity, manufacturing process, clinical efficacy. Consequently, an overall biosimilarity index across domains can be obtained as follows:
Step 1. Obtain pi, the probability of reproducibility for the i-th domain, i = 1,.., K.
Step 2. Define the overall biosimilarity index ![]() , where wi is the weight for the i-th domain.
, where wi is the weight for the i-th domain.
Step 3. Claim global biosimilarity if p > p0 where p0 is a pre-specified acceptable reproducibility probability.
Such integration of information to claim global biosimilarity also reflects FDA’s emphasis on totality-of-the-evidence.
Criteria for interchangeability
The criteria discussed so far focused on biosimilarity intended for new prescriptions. However, for any given patient (especially those with chronic disease and who are already receiving the reference product) who switches from the reference product to a biosimilar, whether efficacy and safety can be maintained is also of interests to patients and physicians. This is referred to as interchangeability, which requires similarity i.e. similar therapeutic and adverse effects as the reference product, within the same patient, and is defined in the US regulation [12] as:
‘(A) The biological product–
(i) is biosimilar to the reference product; and
(ii) can be expected to produce the same clinical result as the reference product in any given patient; and
(B) For a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch.
(C) The term ‘interchangeable’ or ‘interchangeability’, in reference to a biological product that is shown to meet the standards [for biosimilarity], means that the biological product may be substituted for the reference product without the intervention of the healthcare provider who prescribed the reference product.’
Clearly from the definition, results from alternating or switching between the reference and biosimilar products are needed for the evaluation of interchangeability. The parallel design is not adequate to generate such results, as the intra-subject variability and variability due to subject-by-product interaction cannot be estimated in such design. Hence, crossover studies are necessary. Various crossover designs have been proposed and evaluated. They include the traditional two by two crossover design (TR, RT) and higher order designs like (TRT, RTR), (TRR, RTR), (RTR, RRR) and others, where the extra reference (TRR, RTR) design was found to be the most efficient under the individual bioequivalence (IBE) criterion, which is used to assess interchangeability. The IBE criterion is an aggregate measure of differences in means and variances of the reference and biosimilar products, and the subject-by-product interaction [30, 31]. Further research on other statistical criteria and optimal study designs for assessing interchangeability are urgently needed.
Selection of endpoints
Selection of endpoints for biosimilar development is not straightforward. The primary endpoints used in the reference drug development might not be the most adequate for assessing biosimilarity. An example was given in EMA monoclonal antibody guidance: ‘for a new anticancer drug, the preferred endpoint to establish patient benefit would be progression free/disease free survival or overall survival. But these endpoints may not be feasible or sensitive enough for establishing biosimilarity to a reference product since they may be influenced by various factors not attributable to differences between the biosimilar and the reference products themselves, but by factors like tumour burden, performance status, previous lines of treatments, underlying clinical conditions, subsequent lines of treatment (for overall survival), etc.’ [10]. Hence, in general, the most sensitive clinical endpoint that is able to detect product-related differences, if present and, at the same time, to reduce patient and disease-related factors to a minimum in order to increase precision, should be used. Also, although it is often thought that continuous endpoints are more sensitive to detect differences in clinical effects, there could be situations where discrete endpoints are more appropriate. The choice of endpoints should therefore depend on the disease indications and be pre-discussed with the regulatory agencies.
Given an endpoint, should absolute or relative scale being used for comparison is another statistical point to consider. Different quantities in absolute scale, e.g. mean difference for continuous endpoint, or difference in response rates for binary endpoint; or in relative scale, e.g. risk ratio of response rates, can be considered, although they represent different perspectives in thinking about differences. Efficiencies of different endpoint measurement, for given margins, are important considerations for sponsors; while study constancy might be more important for the regulatory agencies.
Available knowledge on the chosen endpoint for the reference product is critical in providing scientific justification for margins and ensuring better understanding and interpretation of the final study results.
Determination of margins
As mentioned above, the clinically meaningful equivalence margin is an important aspect when regulatory agencies review the biosimilar development plan. Sponsors are expected to provide a scientific justification based on their clinical knowledge about the reference product and its therapeutic class when establishing an appropriate equivalence margin that is deemed adequate to enable the detection of clinically meaningful differences in effectiveness and safety between the biosimilar and the reference products. Such choice of the margin is the single greatest challenge in the design and interpretation of biosimilar trials and it has important practical consequences. The smaller the equivalence margin, the narrower the confidence interval must be in order to fall within the margin, and the larger the sample size will be needed. Hence, determining the margin is a critical problem and major focus when designing a biosimilar trial.
The choice of margin and its justification are usually supported by statistical estimation based on historical data of the reference product and by comparison of prior study design, e.g. study population and concomitant therapy, to the current study design to ensure ‘constancy’. Statistical approaches used to determine the non-inferiority (NI) margin for use in NI studies can be adopted to establish a lower (inferiority) margin for equivalence [32, 33]. In FDA’s draft guidance for NI trials, the NI margins of M1 and M2 is recommended where M1 is the entire effect that reference product is presumed to have against placebo and M2 is a margin smaller than M1 based on clinical judgment regarding how much of the M1 active comparator treatment effect can be lost (as a proportion of M1). Typically, M1 is chosen as the lower bound of the 95% CI of a placebo-controlled trial or meta-analysis of trials and M2 is to preserve half of M1. Such choice can be potentially different. For example, as stated in the draft guidance, the choice of M1 could be based on a less extreme boundary of the CI, e.g. 80% instead of 95%, if there exist pharmacologic similarities between the reference and test drugs [32]. Once the lower margin is determined, the upper (superiority) margin can usually be constructed as symmetric to the lower margin.
However, there may be cases in which different upper and lower margins may be appropriate as long as the safety and immunogenicity profile of the biosimilar drug product demonstrates no more risk than the reference product.
Equivalence versus non-inferiority
Equivalence (2-sided) trial designs are typically required for developing biosimilars. However, as asymmetric margins are allowed in some cases discussed above, (one-sided) NI trial designs may also be justifiable in certain circumstances. For example, an important safety issue regarding biologicals is their potential immunogenicity. In clinical trials evaluating immunogenicity or safety, it is generally most important to demonstrate that the biosimilar has no increased risk in terms of safety and immunogenicity compared to the reference product. Hence, one-sided NI setting could be acceptable. Another example quoted from FDA draft guideline [13] is: ‘if doses of the reference product higher than are recommended in its labeling do not create safety concerns, then a one-sided test may be sufficient for comparing the efficacy of certain protein products, e.g. those products that pharmacodynamically saturate the target at some level and are used at or near the maximal level of clinical effect.’ Another situation is when the response rate is extreme high for the reference product, e.g. a response rate of 90%. Under this situation, since superiority is unlikely to occur, it might be justifiable to demonstrate NI to fulfill the equivalence requirement.
Regional effect
In order to support extrapolation from one region to another for global approval, clinical bridging studies, in addition to analytical bridging studies, need to be considered in the early development stage. This may be accomplished by a 3-way comparison to establish PK similarity among the test product (biosimilar), the reference product sourced from EU and the reference product sourced from the US. If regional reference products are only used regionally, the regional effect must be adjusted in the model and appropriate contrasts need to be constructed in order to estimate the treatment differences. This is extremely critical in comparing the reference products when the regional effect is significant. Another statistical issue to consider is multiplicity. Even though no multiplicity adjustment is needed for the co-primary endpoints of AUC and Cmax as is the case in the traditional PK bioequivalence studies, whether an adjustment is needed for the 3-way comparisons among products remains unclear.
Totality-of-the-evidence
FDA emphasized the stepwise approach in developing biosimilars, and totality-of-the-evidence should be considered to address residual uncertainty after each step. The biosimilarity index attempted to combine comparative information from analytical, non-clinical, PK/PD and clinical as a totality-of-the-evidence [29]. Other methodologies to combine information and how to factor early evidence in planning for the next step remain to be of interests to us.
Discussion
Biosimilars will enhance patients’ access to much-needed biological medicines and reduce costs. The requirements for regulatory approval of biosimilars have been delineated in the guidance documents, albeit at a high level. It is up to the regulators and the developers to collaboratively implement development plans for the biosimilar candidates in order to meet these requirements. Sound statistical thinking and planning are indispensible to this endeavor.
Competing interests: The views and opinions discussed here only represent the authors’ position, not necessarily of the author’s company, its affiliates, or anyone else. There is no conflict of interest directly relevant to the content of this manuscript.
Provenance and peer review: Commissioned; externally peer reviewed.
Co-author
Eric Chi, PhD, Director of Biostatistics, Biosimilar Division, Amgen Inc, Thousand Oaks, CA 91320, USA
References
1. European Medicines Agency. Guideline on similar biological medicinal products [homepage on the Internet]. 2005 Nov [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
2. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues [homepage on the Internet]. 2006 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003953.pdf
3. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues [homepage on the Internet]. 2006 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
4. European Medicines Agency. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant granulocyte-colony stimulating factor [homepage on the Internet]. 2006 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003955.pdf
5. European Medicines Agency. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant human soluble insulin [homepage on the Internet]. 2006 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003957.pdf
6. European Medicines Agency. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing somatropin [homepage on the Internet]. 2006 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003956.pdf
7. European Medicines Agency. Draft guideline on similar medicinal products containing recombinant interferon alpha [homepage on the Internet]. 2007 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003931.pdf
8. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins [homepage on the Internet]. 2007 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003947.pdf
9. European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins [homepage on the Internet]. 2009 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003927.pdf
10. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues [homepage on the Internet]. 2012 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
11. World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs) [homepage on the Internet]. 2010 [cited 2013 Nov 4]. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22 APRIL2010.pdf
12. U.S. Food and Drug Administration. Biologics Price Competition and Innovation Act of 2009 [homepage on the Internet]. 2010 [cited 2013 Nov 4]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf
13. U.S. Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Draft guidance [homepage on the Internet]. 2012 [cited 2013 Nov 4]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
14. U.S. Food and Drug Administration. Quality considerations in demonstrating biosimilarity to a reference protein product. Draft guidance [homepage on the Internet]. 2012 [cited 2013 Nov 4]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
15. U.S. Food and Drug Administration. Biosimilars: questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009. Draft guidance [homepage on the Internet]. 2012 [cited 2013 Nov 4]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM273001.pdf
16. U.S. Government Printing Office. The Patient Protection and Affordable Care Act [homepage on the Internet]. 2010 [cited 2013 Nov 4]. Available from: http://www.gpo.gov/fdsys/pkg/BILLS-111 hr3590enr/pdf/BILLS-111 hr3590enr.pdf
17. Davit BM, Conner DP, Fabian-Fritsch B, Haidar SH, Jiang XJ, et al. Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS. 2008;10(1):148-56.
18. Tothfalusi L, Endrenyi L, Arieta AG. Evaluation of bioequivalence for highly variable drugs with scaled average bioequivalence. Clin Pharmacokinet. 2009;48(11):725-43.
19. Haidar SH, Makhlouf F, Schuirmann DJ, Hyslop T, Davit B, Conner D, Yu LX. Evaluation of a scaling approach for the bioequivalence of highly variable drugs. AAPS. 2008;10(3):450-4.
20. European Medicines Agency. Guideline on the investigation of bioequivalence [homepage on the Internet]. 2010 [cited 2013 Nov 4]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
21. Zhang N, Yang J, Chow SC, Endrenyi L, Chi E. Impact of variability on the choice of biosimilarity limits in assessing follow-on biologics. Stat Med. 2013;32(3):424-33.
22. Hsieh TC, Chow SC, Liu JP, Hsiao CF, Chi E. Statistical test for evaluation of biosimilarity in variability of follow-on biologics. J Biopharm Stat. 2010;20(1):75-89.
23. Yang J, Zhang N, Chow SC, Chi E. An adapted F-test for homogeneity of variability in follow-on biologic products. Stat Med. 2013;32(3):415-23.
24. Zhang N, Yang J, Chow SC, Chi E. Nonparametric tests for evaluation of biosimilarity in variability of follow-on biologics. Joint Statistical Meetings; 2010; abstract #306029.
25. Stephens MA. EDF statistics for goodness of fit and some comparisons. J Am Stat Assoc. 1974;69(347):730-7.
26. Mizuno S, Yamaguchi T, Fukushima A, Matsuyama Y, Ohashi Y. Overlap coefficient for assessing the similarity of pharmacokinetic data between ethnically different populations. Clin Trials. 2005;2(2):174-81.
27. Lei L, Olson K. Evaluating statistical methods to establish clinical similarity of two biologics. J Biopharm Stat. 2010;20(1):62-74.
28. Shao J, Chow SC. Reproducibility probability in clinical trials. Stat Med. 2002;21(12):1727-42.
29. Hsieh TC, Chow SC, Yang LY, Chi E. The evaluation of biosimilarity index based on reproducibility probability for assessing follow-on biologics. Stat Med. 2013;32(3):406-14.
30. Hyslop T, Hsuan F, Holder DJ. A small sample confidence interval approach to assess individual bioequivalence. Stat Med. 2000;19(20):2885-97.
31. Chow SC, Shao J, Wang H. Individual bioequivalence testing under 2 × 3 designs. Stat Med. 2002;21:629-48.
32. U.S. Food and Drug Administration. Guidance for industry: non-inferiority clinical trials. Draft guidance [homepage on the Internet]. 2010 [cited 2013 Nov 4]. Available from http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdf
33. European Medicines Agency. Guideline on the choice of the non-inferiority margin [homepage on the Internet]. 2005 [cited 2013 Nov 4]. Available from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003636.pdf
|
Author for correspondence: Nan Zhang, PhD, Biostatistics Senior Manager, Biosimilar Division, Amgen Inc, Mail Stop 24–2-C, One Amgen Center Drive, Thousand Oaks, CA 91320, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2014 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


